A Pentavalent Epstein-Barr Virus-Like Particle Vaccine Elicits High Titers of Neutralizing Antibodies against Epstein-Barr Virus Infection in Immunized Rabbits

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Methods
2.1. Animals and Ethics Statement
2.2. Cell Lines
2.3. Antibodies
2.4. Virus Production and Purification
2.5. Plasmids
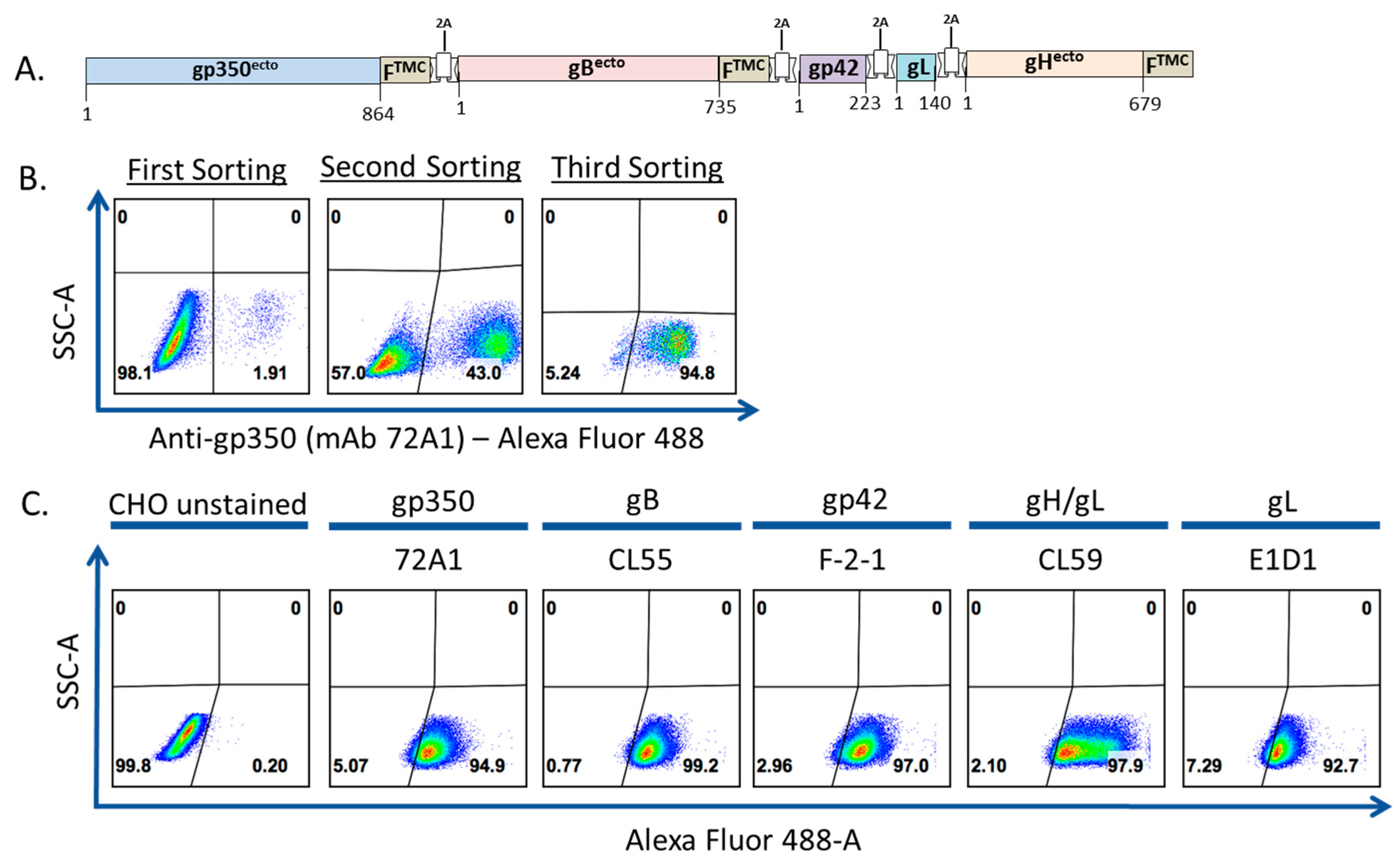
2.6. Transfection of gp350-F-gB-F-gp42-gL-gH-F and Generation of Stable CHO Cell Line
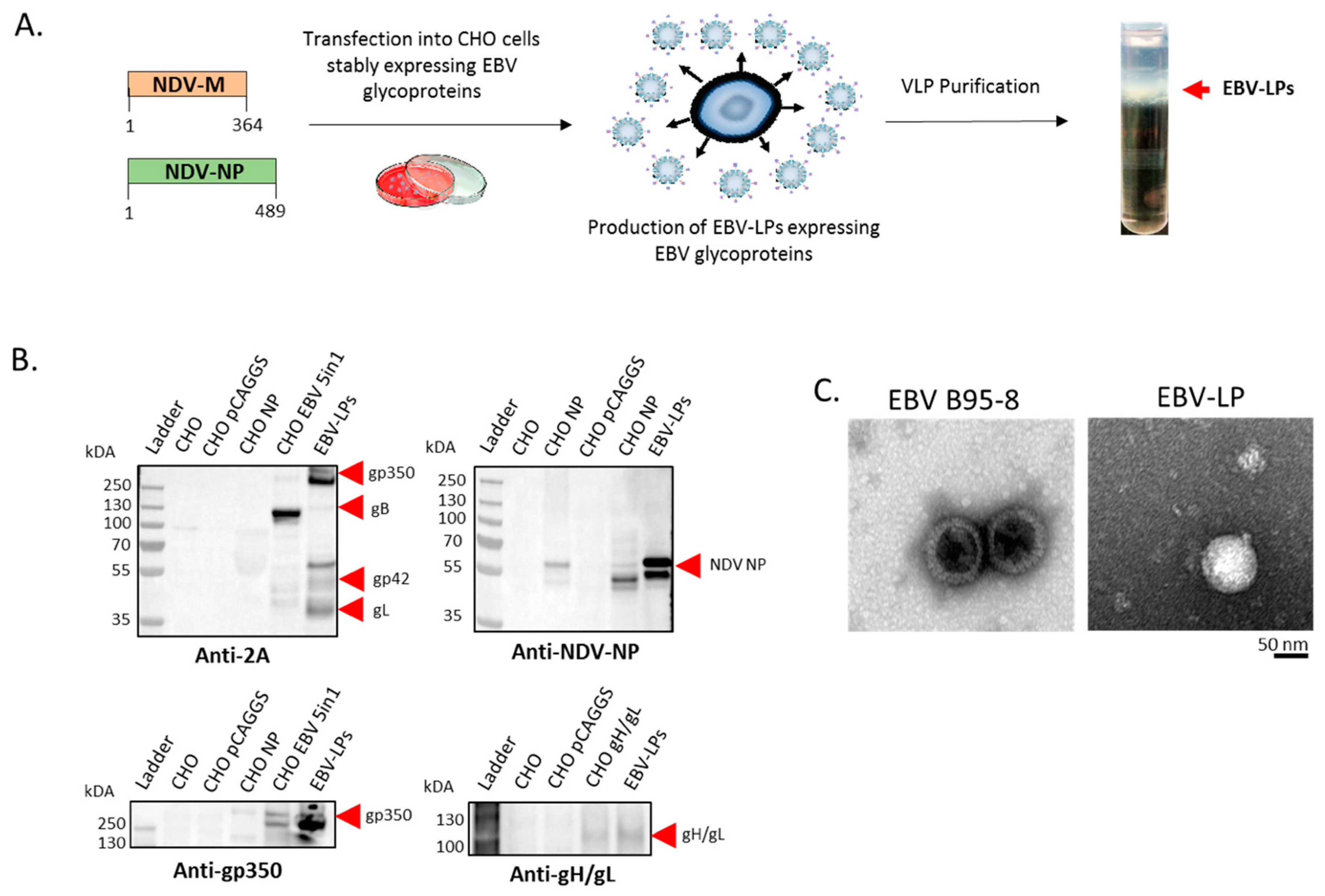
2.7. Transfection, Generation, and Purification of EBV-LPs
2.8. SDS-PAGE, Coomassie Staining, and Immunoblotting
2.9. Transmission Electron Microcopy (TEM)
2.10. Recombinant EBV Proteins
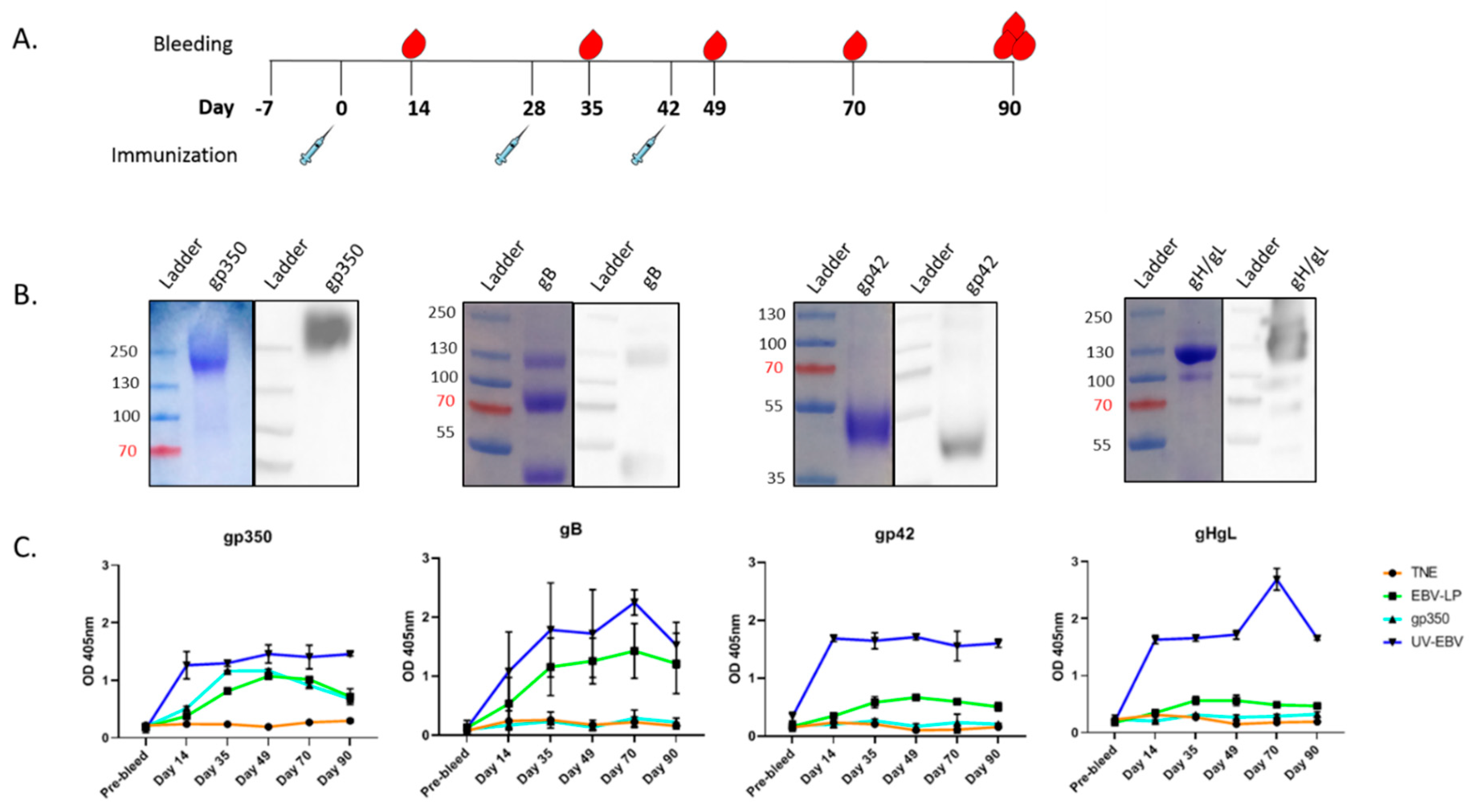
2.11. Immunization of Rabbits
2.12. Determination of Glycoprotein-Specific Antibody Titers in Serum of Immunized Rabbits
2.13. Purification of IgG Antibodies from Rabbit Sera, and Determination of Glycoprotein-Specific Antibody Titers
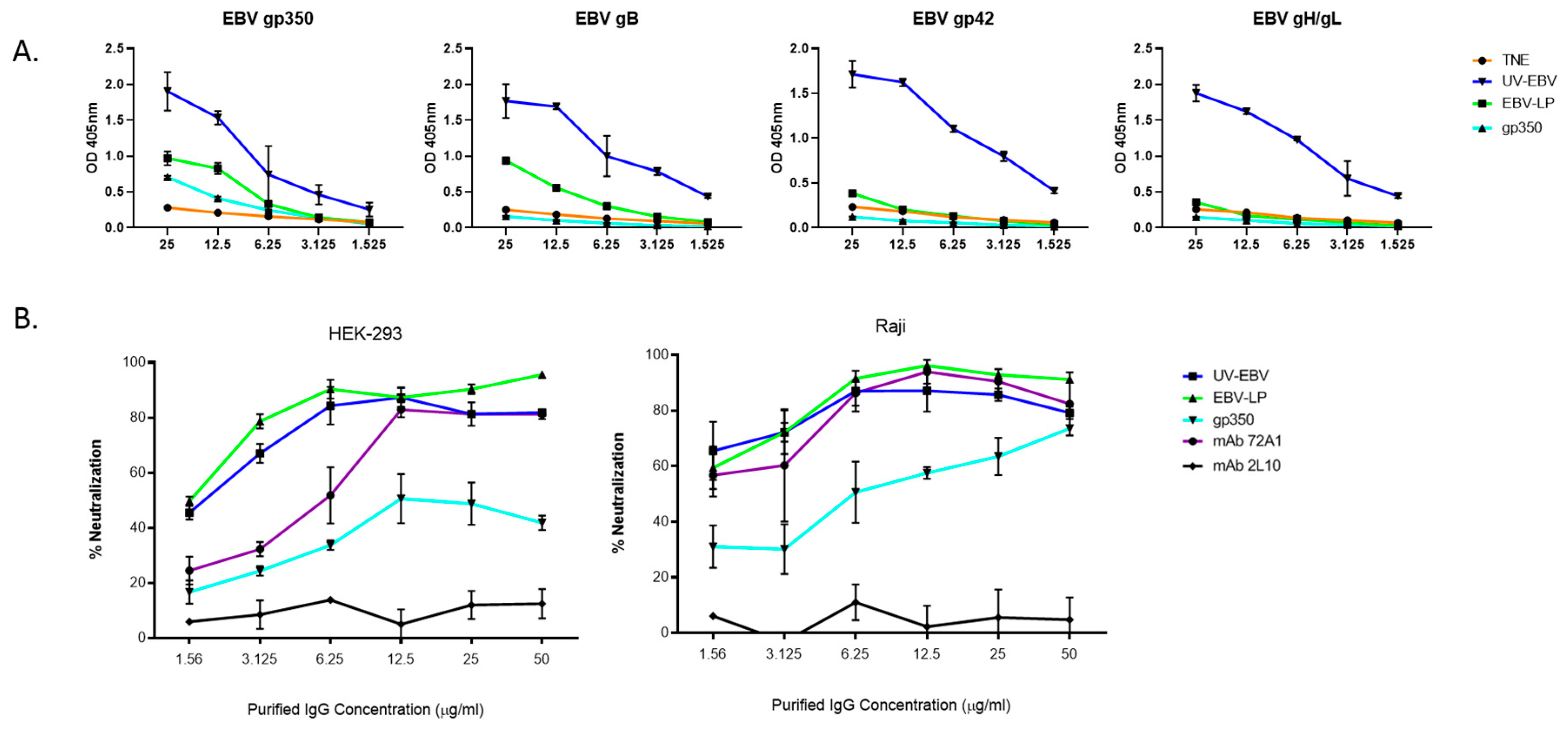
2.14. EBV-eGFP Neutralization Assay in B cells and Epithelial Cells
2.15. Statistical Analysis
3. Results
3.1. Construction, Purification, and Characterization of EBV-LP Vaccine Candidate That Incorporates Five EBV Glycoproteins
3.2. Pentavalent EBV-LPs Are Immunogenic and Elicit EBV-Glycoprotein-Specific Antibodies in Rabbits
3.3. EBV-LPs Produce Robust nAb Responses in Immunized Rabbits That Prevent EBV Infection of Both B Cell and Epithelial Cell Lines
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cohen, J.I.; Mocarski, E.S.; Raab-Traub, N.; Corey, L.; Nabel, G.J. The need and challenges for development of an Epstein-Barr virus vaccine. Vaccine 2013, 31, B194–B196. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med Microbiol. Immunol. 2018, 208, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; De La Cruz-Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr Virus: An Important Vaccine Target for Cancer Prevention. Sci. Transl. Med. 2011, 3, 107fs7. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Levin, L.I.; O’Reilly, E.J.; Falk, K.I.; Ascherio, A. Anti-Epstein–Barr virus antibodies as serological markers of multiple sclerosis: A prospective study among United States military personnel. Mult. Scler. J. 2011, 17, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Handunnetthi, L.; Giovannoni, G.; Ramagopalan, S.V. An Updated Meta-Analysis of Risk of Multiple Sclerosis following Infectious Mononucleosis. PLoS ONE 2010, 5, e12496. [Google Scholar] [CrossRef]
- Cohen, J.I. Vaccine Development for Epstein-Barr Virus. Adv. Exp. Med. Biol. 2018, 1045, 477–493. [Google Scholar]
- Cohen, J.I. Epstein-barr virus vaccines. Clin. Transl. Immunol. 2015, 4, e32. [Google Scholar] [CrossRef]
- Elliott, S.L.; Suhrbier, A.; Miles, J.J.; Lawrence, G.; Pye, S.J.; Le, T.T.; Rosenstengel, A.; Nguyen, T.; Allworth, A.; Burrows, S.; et al. Phase I Trial of a CD8+ T-Cell Peptide Epitope-Based Vaccine for Infectious Mononucleosis. J. Virol. 2007, 82, 1448–1457. [Google Scholar] [CrossRef]
- Gu, S.Y.; Huang, T.M.; Ruan, L.; Miao, Y.H.; Lu, H.; Chu, C.M.; Motz, M.; Wolf, H. First EBV vaccine trial in humans using recombinant vaccinia virus expressing the major membrane antigen. Dev. Boil. Stand. 1995, 84, 171–177. [Google Scholar]
- Moutschen, M.; Leonard, P.; Sokal, E.M.; Smets, F.; Haumont, M.; Mazzu, P.; Bollen, A.; Denamur, F.; Peeters, P.; Dubin, G.; et al. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein–Barr virus vaccine in healthy adults. Vaccine 2007, 25, 4697–4705. [Google Scholar] [CrossRef]
- Sokal, E.M.; Hoppenbrouwers, K.; Vandermeulen, C.; Moutschen, M.; Léonard, P.; Moreels, A.; Haumont, M.; Bollen, A.; Smets, F.; Denis, M. Recombinant gp350 Vaccine for Infectious Mononucleosis: A Phase 2, Randomized, Double-Blind, Placebo-Controlled Trial to Evaluate the Safety, Immunogenicity, and Efficacy of an Epstein-Barr Virus Vaccine in Healthy Young Adults. J. Infect. Dis. 2007, 196, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Rees, L.; Tizard, E.J.; Morgan, A.J.; Cubitt, W.D.; Finerty, S.; Oyewole-Eletu, T.A.; Owen, K.; Royed, C.; Stevens, S.J.C.; Shroff, R.; et al. A Phase I Trial of Epstein-Barr Virus Gp350 Vaccine for Children With Chronic Kidney Disease Awaiting Transplantation. Transplant 2009, 88, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Meckiff, B.J.; Taylor, G. The T-cell Response to Epstein-Barr Virus-New Tricks from an Old Dog. Front. Immunol. 2019, 10, 2193. [Google Scholar] [CrossRef] [PubMed]
- Kaeuferle, T.; Krauss, R.; Blaeschke, F.; Willier, S.; Feuchtinger, T. Strategies of adoptive T -cell transfer to treat refractory viral infections post allogeneic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, L.P.; Bollard, C.M.; Keller, M.D. Adoptive T Cell Therapy for Epstein–Barr Virus Complications in Patients With Primary Immunodeficiency Disorders. Front. Immunol. 2018, 9, 556. [Google Scholar] [CrossRef] [PubMed]
- Van Zyl, D.G.; Mautner, J.; Delecluse, H.-J. Progress in EBV Vaccines. Front. Oncol. 2019, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A. Exploiting Mucosal Immunity for Antiviral Vaccines. Annu. Rev. Immunol. 2016, 34, 575–608. [Google Scholar] [CrossRef]
- Awasthi, S.; Friedman, H.M. A paradigm shift: Vaccine-induced antibodies as an immune correlate of protection against herpes simplex virus type 1 genital herpes. J. Infect. Dis. 2013, 209, 813–815. [Google Scholar] [CrossRef]
- Gilbert, P.B.; Gabriel, E.; Miao, X.; Li, X.; Su, S.-C.; Parrino, J.; Chan, I.S.F. Fold rise in antibody titers by measured by glycoprotein-based enzyme-linked immunosorbent assay is an excellent correlate of protection for a herpes zoster vaccine, demonstrated via the vaccine efficacy curve. J. Infect. Dis. 2014, 210, 1573–1581. [Google Scholar] [CrossRef]
- Belshe, R.B.; Leone, P.A.; Bernstein, D.I.; Wald, A.; Levin, M.J.; Stapleton, J.T.; Gorfinkel, I.; Morrow, R.L.A.; Ewell, M.G.; Stokes-Riner, A.; et al. Efficacy results of a trial of a herpes simplex vaccine. New Engl. J. Med. 2012, 366, 34–43. [Google Scholar] [CrossRef] [PubMed]
- A Thorley-Lawson, D.; A Poodry, C. Identification and isolation of the main component (gp350-gp220) of Epstein-Barr virus responsible for generating neutralizing antibodies in vivo. J. Virol. 1982, 43, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Janz, A.; Oezel, M.; Kurzeder, C.; Mautner, J.; Pich, D.; Kost, M.; Hammerschmidt, W.; Delecluse, H.-J. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 2000, 74, 10142–10152. [Google Scholar] [CrossRef] [PubMed]
- Sathiyamoorthy, K.; Jiang, J.; Hu, Y.X.; Rowe, C.L.; Möhl, B.S.; Chen, J.; Jiang, W.; Mellins, E.D.; Longnecker, R.; Zhou, Z.H.; et al. Assembly and Architecture of the EBV B Cell Entry Triggering Complex. PLoS Pathog. 2014, 10, e1004309. [Google Scholar] [CrossRef]
- Sathiyamoorthy, K.; Hu, Y.X.; Möhl, B.S.; Chen, J.; Longnecker, R.; Jardetzky, T.S. Structural basis for Epstein–Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nat. Commun. 2016, 7, 13557. [Google Scholar] [CrossRef]
- Fingeroth, J.D.; Weis, J.J.; Tedder, T.F.; Strominger, J.L.; Biro, P.A.; Fearon, D.T. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc. Natl. Acad. Sci. USA 1984, 81, 4510–4514. [Google Scholar] [CrossRef]
- Ogembo, J.G.; Kannan, L.; Ghiran, I.; Nicholson-Weller, A.; Finberg, R.W.; Tsokos, G.C.; Fingeroth, J.D. Human complement receptor type 1/CD35 is an Epstein-Barr Virus receptor. Cell Rep. 2013, 3, 371–385. [Google Scholar] [CrossRef]
- Tanner, J.; Weis, J.; Fearon, D.; Whang, Y.; Kieff, E. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 1987, 50, 203–213. [Google Scholar] [CrossRef]
- Li, Q.; Spriggs, M.K.; Kovats, S.; Turk, S.M.; Comeau, M.R.; Nepom, B.; Hutt-Fletcher, L.M. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 1997, 71, 4657–4662. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Genet. 2011, 9, 369–381. [Google Scholar] [CrossRef]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; Perez-White, B.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Wang, H.-B.; Zhang, A.; Chen, M.-L.; Fang, Z.-X.; Dong, X.-D.; Li, S.-B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein–Barr virus entry. Nat. Microbiol. 2018, 3, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt-Fletcher, L.M. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. USA 2009, 106, 20464–20469. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, L.S.; Hutt-Fletcher, L.M. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. J. Virol. 2011, 85, 13214–13223. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L.M. EBV glycoproteins: Where are we now? Futur. Virol. 2015, 10, 1155–1162. [Google Scholar] [CrossRef]
- Wang, L.-H.; Yang, Y.-J.; Cheng, W.-C.; Wang, W.-M.; Lin, S.-H.; Shieh, C.-C. Higher Risk for Hematological Malignancies in Inflammatory Bowel Disease: A Nationwide Population-based Study in Taiwan. Am. J. Gastroenterol. 2016, 111, 1313–1319. [Google Scholar] [CrossRef]
- Molesworth, S.J.; Lake, C.M.; Borza, C.M.; Turk, S.M.; Hutt-Fletcher, L.M. Epstein-Barr Virus gH Is Essential for Penetration of B Cells but Also Plays a Role in Attachment of Virus to Epithelial Cells. J. Virol. 2000, 74, 6324–6332. [Google Scholar] [CrossRef]
- Sashihara, J.; Burbelo, P.D.; Savoldo, B.; Pierson, T.C.; Cohen, J.I. Human antibody titers to Epstein-Barr Virus (EBV) gp350 correlate with neutralization of infectivity better than antibody titers to EBV gp42 using a rapid flow cytometry-based EBV neutralization assay. Virology 2009, 391, 249–256. [Google Scholar] [CrossRef]
- Bu, W.; Joyce, M.G.; Nguyen, H.; Banh, D.V.; Aguilar, F.; Tariq, Z. Immunization with Components of the Viral Fusion Apparatus Elicits Antibodies That Neutralize Epstein-Barr Virus in B Cells and Epithelial Cells. Immunity 2019, 50, 1305–1316. [Google Scholar] [CrossRef]
- Strnad, B.C.; Schuster, T.; Klein, R.; Hopkins, R.F.; Witmer, T.; Neubauer, R.H.; Rabin, H. Production and characterization of monoclonal antibodies against the Epstein-Barr virus membrane antigen. J. Virol. 1982, 41, 258–264. [Google Scholar] [CrossRef]
- Balachandran, N.; Oba, D.E.; Hutt-Fletcher, L.M. Antigenic cross-reactions among herpes simplex virus types 1 and 2, Epstein-Barr virus, and cytomegalovirus. J. Virol. 1987, 61, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Cao, Z.; Chen, Q.; Arjunaraja, S.; Snow, A.; Snapper, C.M. Rabbits immunized with Epstein-Barr virus gH/gL or gB recombinant proteins elicit higher serum virus neutralizing activity than gp350. Vaccine 2016, 34, 4050–4055. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Turk, S.M.; Hutt-Fletcher, L.M. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J. Virol. 1995, 69, 3987–3994. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.; Foley, J.; Tison, T.; Silva, R.; Ogembo, J.G. Novel Epstein-Barr virus-like particles incorporating gH/gL-EBNA1 or gB-LMP2 induce high neutralizing antibody titers and EBV-specific T-cell responses in immunized mice. Oncotarget 2016, 8, 19255–19273. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, M.; Bu, W.; Joyce, M.G.; Meng, G.; Whittle, J.R.; Baxa, U.; Yamamoto, T.; Narpala, S.; Todd, J.-P.; Rao, S.S.; et al. Rational Design of an Epstein-Barr Virus Vaccine Targeting the Receptor-Binding Site. Cell 2015, 162, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Snijder, J.; Ortego, M.S.; Weidle, C.; Stuart, A.B.; Gray, M.D.; McElrath, M.J.; Pancera, M.; Veesler, D.; McGuire, A.T. An Antibody Targeting the Fusion Machinery Neutralizes Dual-Tropic Infection and Defines a Site of Vulnerability on Epstein-Barr Virus. Immunity 2018, 48, 799–811. [Google Scholar] [CrossRef]
- Bootz, A.; Karbach, A.; Spindler, J.; Kropff, B.; Reuter, N.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog. 2017, 13, e1006601. [Google Scholar] [CrossRef]
- Fouts, A.E.; Comps-Agrar, L.; Stengel, K.F.; Ellerman, D.; Schoeffler, A.J.; Warming, S.; Eaton, D.L.; Feierbach, B. Mechanism for neutralizing activity by the anti-CMV gH/gL monoclonal antibody MSL-109. Proc. Natl. Acad. Sci. USA 2014, 111, 8209–8214. [Google Scholar] [CrossRef]
- Cairns, T.M.; Huang, Z.-Y.; Gallagher, J.R.; Lin, Y.; Lou, H.; Whitbeck, J.C.; Wald, A.; Cohen, G.H.; Eisenberg, R.J. Patient-Specific Neutralizing Antibody Responses to Herpes Simplex Virus Are Attributed to Epitopes on gD, gB, or Both and Can Be Type Specific. J. Virol. 2015, 89, 9213–9231. [Google Scholar] [CrossRef]
- Peng, T.; Ponce-De-Leon, M.; Jiang, H.; Dubin, G.; Lubinski, J.M.; Eisenberg, R.J.; Cohen, G.H. The gH-gL Complex of Herpes Simplex Virus (HSV) Stimulates Neutralizing Antibody and Protects Mice against HSV Type 1 Challenge. J. Virol. 1998, 72, 65–72. [Google Scholar] [CrossRef]
- Ogembo, J.G.; Muraswki, M.R.; McGinnes, L.W.; Parcharidou, A.; Sutiwisesak, R.; Tison, T.; Avendano, J.; Agnani, D.; Finberg, R.W.; Morrison, T.G.; et al. A chimeric EBV gp350/220-based VLP replicates the virion B-cell attachment mechanism and elicits long-lasting neutralizing antibodies in mice. J. Transl. Med. 2015, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A.; Geilinger, K. Monoclonal antibodies against the major glycoprotein (gp350/220) of Epstein-Barr virus neutralize infectivity. Proc. Natl. Acad. Sci. USA 1980, 77, 5307–5311. [Google Scholar] [CrossRef] [PubMed]
- Mutsvunguma, L.Z.; Rodriguez, E.; Escalante, G.M.; Muniraju, M.; Williams, J.C.; Warden, C.; Qin, H.; Wang, J.; Wu, X.; Barasa, A.; et al. Identification of multiple potent neutralizing and non-neutralizing antibodies against Epstein-Barr virus gp350 protein with potential for clinical application and as reagents for mapping immunodominant epitopes. Virology 2019, 536, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jun-Ichi, M.; Satoshi, T.; Kimi, A.; Fumi, T.; Akira, T.; Kiyoshi, T.; Ken-Ichi, Y. Expression vector system based on the chicken β-actin promoter directs efficient production of interleukin-5. Gene 1989, 79, 269–277. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.-R.; Li, L.-H.; Park, H.-J.; Park, J.-H.; Lee, K.Y.; Kim, M.-K.; Shin, B.A.; Choi, S.-Y. High Cleavage Efficiency of a 2A Peptide Derived from Porcine Teschovirus-1 in Human Cell Lines, Zebrafish and Mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef] [PubMed]
- Chiuppesi, F.; Nguyen, J.; Park, S.; Contreras, H.; Kha, M.; Meng, Z.; Kaltcheva, T.; Iniguez, A.; Martinez, J.; La Rosa, C.; et al. Multiantigenic Modified Vaccinia Virus Ankara Vaccine Vectors To Elicit Potent Humoral and Cellular Immune Reponses against Human Cytomegalovirus in Mice. J. Virol. 2018, 92, e01012-18. [Google Scholar] [CrossRef]
- Pantua, H.D.; McGinnes, L.W.; Peeples, M.E.; Morrison, T. Requirements for the Assembly and Release of Newcastle Disease Virus-Like Particles. J. Virol. 2006, 80, 11062–11073. [Google Scholar] [CrossRef]
- Mulama, D.H.; Mutsvunguma, L.Z.; Totonchy, J.; Ye, P.; Foley, J.; Escalante, G.M.; Rodriguez, E.; Nabiee, R.; Muniraju, M.; Wussow, F.; et al. A multivalent Kaposi sarcoma-associated herpesvirus-like particle vaccine capable of eliciting high titers of neutralizing antibodies in immunized rabbits. Vaccine 2019, 37, 4184–4194. [Google Scholar] [CrossRef]
- Barasa, A.; Ye, P.; Phelps, M.; Ganapathiram, A.; Tison, T.; Ogembo, J.G. BALB/c mice immunized with a combination of virus-like particles incorporating Kaposi sarcomaassociated herpesvirus (KSHV) envelope glycoproteins gpK8.1, gB, and gH/gL induced comparable serum neutralizing antibody activity to UV-inactivated KSHV. Oncotarget 2017, 8, 34481–34497. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, O.M.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Vaseghi, H.R.; Qian, L.; Liu, J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Bozzacco, L.; Keeffe, J.R.; Khouri, R.; Olsen, P.C.; Gazumyan, A.; Schaefer-Babajew, D.; Ávila-Ríos, S.; Nogueira, L.; Patel, R.; et al. Recurrent Potent Human Neutralizing Antibodies to Zika Virus in Brazil and Mexico. Cell 2017, 169, 597–609.e11. [Google Scholar] [CrossRef] [PubMed]
- Cheru, L.; Wu, Y.; Diouf, A.; Moretz, S.E.; Muratova, O.V.; Song, G.; Fay, M.P.; Miller, L.H.; Long, C.A.; Miura, K. The IC50 of anti-Pfs25 antibody in membrane-feeding assay varies among species. Vaccine 2010, 28, 4423–4429. [Google Scholar] [CrossRef] [PubMed]
- Ruiss, R.; Jochum, S.; Wanner, G.; Reisbach, G.; Hammerschmidt, W.; Zeidler, R. A Virus-Like Particle-Based Epstein-Barr Virus Vaccine. J. Virol. 2011, 85, 13105–13113. [Google Scholar] [CrossRef]
- Van Zyl, D.; Tsai, M.-H.; Shumilov, A.; Schneidt, V.; Poirey, R.; Schlehe, B.; Fluhr, H.; Mautner, J.; Delecluse, H.-J. Immunogenic particles with a broad antigenic spectrum stimulate cytolytic T cells and offer increased protection against EBV infection ex vivo and in mice. PLoS Pathog. 2018, 14, e1007464. [Google Scholar] [CrossRef] [PubMed]
- Fuenmayor, J.; Gòdia, F.; Cervera, L. Production of virus-like particles for vaccines. New Biotechnol. 2017, 39, 174–180. [Google Scholar] [CrossRef]
- Domi, A.; Feldmann, F.; Basu, R.; McCurley, N.; Shifflett, K.; Emanuel, J.; Hellerstein, M.S.; Guirakhoo, F.; Orlandi, C.; Flinko, R.; et al. A Single Dose of Modified Vaccinia Ankara expressing Ebola Virus Like Particles Protects Nonhuman Primates from Lethal Ebola Virus Challenge. Sci. Rep. 2018, 8, 864. [Google Scholar] [CrossRef]
- Lázaro-Frías, A.; Gómez-Medina, S.; Sánchez-Sampedro, L.; Ljungberg, K.; Ustav, M.; Liljestrom, P.; Munoz-Fontela, C.; Esteban, M.; García-Arriaza, J. Distinct Immunogenicity and Efficacy of Poxvirus-Based Vaccine Candidates against Ebola Virus Expressing GP and VP40 Proteins. J. Virol. 2018, 92, e00363-18. [Google Scholar] [CrossRef]
- Schweneker, M.; Laimbacher, A.S.; Zimmer, G.; Wagner, S.; Schraner, E.M.; Wolferstätter, M.; Klingenberg, M.; Dirmeier, U.; Steigerwald, R.; Lauterbach, H.; et al. Recombinant Modified Vaccinia Virus Ankara Generating Ebola Virus-Like Particles. J. Virol. 2017, 91, e00343-17. [Google Scholar] [CrossRef]
- Tapia, M.D.; O Sow, S.; E Lyke, K.; Haidara, F.C.; Diallo, F.; Doumbia, M.; Traore, A.; Coulibaly, F.; Kodio, M.; Onwuchekwa, U.; et al. Use of ChAd3-EBO-Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: A phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial, and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 2015, 16, 31–42. [Google Scholar]
- Levine, M.M.; Tapia, M.; Hill, A.V.; Sow, S.O. How the current West African Ebola virus disease epidemic is altering views on the need for vaccines and is galvanizing a global effort to field-test leading candidate vaccines. J. Infect. Dis. 2014, 211, 504–507. [Google Scholar] [CrossRef][Green Version]
- Anywaine, Z.; Whitworth, H.; Kaleebu, P.; PrayGod, G.; Shukarev, G.; Manno, D.; Kapiga, S.; Grosskurth, H.; Kalluvya, S.; Bockstal, V.; et al. Safety and Immunogenicity of a 2-Dose Heterologous Vaccination Regimen With Ad26.ZEBOV and MVA-BN-Filo Ebola Vaccines: 12-Month Data From a Phase 1 Randomized Clinical Trial in Uganda and Tanzania. J. Infect. Dis. 2019, 220, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Milligan, I.D.; Gibani, M.M.; Sewell, R.; Clutterbuck, E.A.; Campbell, D.; Plested, E.; Nuthall, E.; Voysey, M.; Silva-Reyes, L.; McElrath, M.J.; et al. Safety and Immunogenicity of Novel Adenovirus Type 26– and Modified Vaccinia Ankara–Vectored Ebola Vaccines. JAMA 2016, 315, 1610. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Fast, P.E.; Modjarrad, K.; Clarke, D.K.; Martin, B.K.; Fusco, J.; Nichols, R.; Heppner, D.G.; Simon, J.K.; Dubey, S.; et al. rVSVDeltaG-ZEBOV-GP (also designated V920) recombinant vesicular stomatitis virus pseudotyped with Ebola Zaire Glycoprotein: Standardized template with key considerations for a risk/benefit assessment. Vaccine X 2019, 1, 100009. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Bharadwaj, M.; Tellam, J.; Connolly, G.; Cooper, L.; Moss, D.; Thomson, S.; Yotnda, P.; Khanna, R. Induction of therapeutic T-cell responses to subdominant tumor-associated viral oncogene after immunization with replication-incompetent polyepitope adenovirus vaccine. Cancer Res. 2004, 64, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.P.; Taylor, G.S.; Jia, H.; Ma, B.; Chan, S.; Ho, R.; Wong, W.-L.; Wilson, S.; Johnson, B.; Edwards, C.; et al. Phase I trial of recombinant modified vaccinia ankara encoding Epstein-Barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013, 73, 1676–1688. [Google Scholar] [CrossRef]
- Taylor, G.S.; Jia, H.; Harrington, K.; Lee, L.W.; Turner, J.E.; Ladell, K.; Price, D.A.; Tanday, M.; Matthews, J.; Roberts, C.; et al. A recombinant modified vaccinia ankara vaccine encoding Epstein-Barr Virus (EBV) target antigens: A phase I trial in UK patients with EBV-positive cancer. Clin. Cancer Res. 2014, 20, 5009–5022. [Google Scholar] [CrossRef]
- Rühl, J.; Citterio, C.; Engelmann, C.; Haigh, T.A.; Dzionek, A.; Dreyer, J.H.; Khanna, R.; Taylor, G.S.; Wilson, J.B.; Leung, C.S.; et al. Heterologous prime-boost vaccination protects against EBV antigen–expressing lymphomas. J. Clin. Investig. 2019, 129, 2071–2087. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2A Position | Nucleotide Sequence | Amino Acid Sequence |
|---|---|---|
| gp350-gB | GCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTGGACC | A T N F S L L K Q A G D V E E N P G P |
| gB-g42 | GCCACCAATTTCTCGTTACTTAAACAAGCGGGTGACGTTGAAGAGAATCCGGGACCT | A T N F S L L K Q A G D V E E N P G P |
| g42-gL | GCGACTAACTTCTCATTGTTGAAACAGGCAGGAGATGTCGAAGAGAACCCTGGTCCA | A T N F S L L K Q A G D V E E N P G P |
| gL-gH | GCAACGAATTTCTCCCTTCTAAAGCAAGCCGGTGACGTGGAGGAGAATCCCGGACCC | A T N F S L L K Q A G D V E E N P G P |
| Primer Name | Primer Sequence (5’-3’) |
|---|---|
| Cloning Primers | |
| EBV gH-gL-Fc-His FWD | AAAAAGCGGCCGCGCCACCATGCGTGCTGTTGGTGTATTTC |
| EBV gH-gL-Fc-His REV | AAAAAACTAGTGTGTGCTCTTTCTTCATACAGG |
| Sequencing primers | |
| FC-Hisseqprimer4 | GCTTTAATAAGATCTCTAG |
| FC-Hisseqprimer5 | TGCTGGGCACGGTGGGCATG |
| FC-Hisseqprimer6 | GGGTCTTTTCTGCAGAAGCTTG |
| IC50 *(µg/mL) | |||||
|---|---|---|---|---|---|
| Cell-Line | gp350 | UV-EBV | EBV-LPs | mAb 72A1 | mAb 2L10 |
| HEK-293 | 5.67 | 3.11 | 2.85 | 6.25 | nd |
| Raji | 8.97 | 3.42 | 3.71 | 4.81 | nd |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escalante, G.M.; Foley, J.; Mutsvunguma, L.Z.; Rodriguez, E.; Mulama, D.H.; Muniraju, M.; Ye, P.; Barasa, A.K.; Ogembo, J.G. A Pentavalent Epstein-Barr Virus-Like Particle Vaccine Elicits High Titers of Neutralizing Antibodies against Epstein-Barr Virus Infection in Immunized Rabbits. Vaccines 2020, 8, 169. https://doi.org/10.3390/vaccines8020169
Escalante GM, Foley J, Mutsvunguma LZ, Rodriguez E, Mulama DH, Muniraju M, Ye P, Barasa AK, Ogembo JG. A Pentavalent Epstein-Barr Virus-Like Particle Vaccine Elicits High Titers of Neutralizing Antibodies against Epstein-Barr Virus Infection in Immunized Rabbits. Vaccines. 2020; 8(2):169. https://doi.org/10.3390/vaccines8020169
Chicago/Turabian StyleEscalante, Gabriela M., Joslyn Foley, Lorraine Z. Mutsvunguma, Esther Rodriguez, David H. Mulama, Murali Muniraju, Peng Ye, Anne K. Barasa, and Javier Gordon Ogembo. 2020. "A Pentavalent Epstein-Barr Virus-Like Particle Vaccine Elicits High Titers of Neutralizing Antibodies against Epstein-Barr Virus Infection in Immunized Rabbits" Vaccines 8, no. 2: 169. https://doi.org/10.3390/vaccines8020169
APA StyleEscalante, G. M., Foley, J., Mutsvunguma, L. Z., Rodriguez, E., Mulama, D. H., Muniraju, M., Ye, P., Barasa, A. K., & Ogembo, J. G. (2020). A Pentavalent Epstein-Barr Virus-Like Particle Vaccine Elicits High Titers of Neutralizing Antibodies against Epstein-Barr Virus Infection in Immunized Rabbits. Vaccines, 8(2), 169. https://doi.org/10.3390/vaccines8020169