
Elucidating the Role of Host Long Non-Coding RNA during Viral Infection: Challenges and Paths Forward


Abstract
1. Introduction
2. Functional Diversity of lncRNAs and Their Involvement in Viral Infections
2.1. Epigenetic Regulation and Promotion of Viral Latency
2.2. Scaffolding and Nuclear Localization
2.3. Transcriptional Regulation of mRNA via miRNA Sponges
2.4. Alternative Splicing
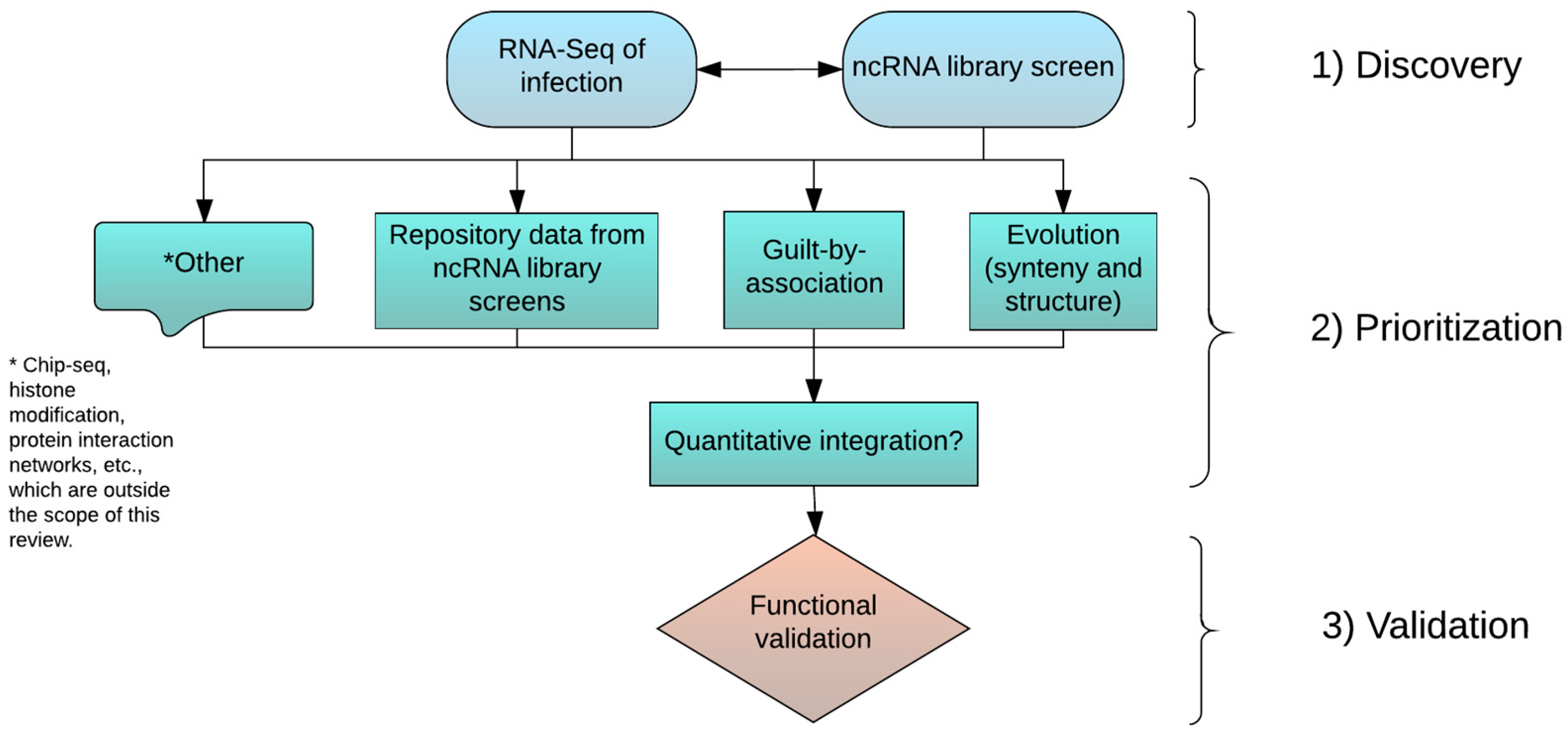
3. Discovery, Prioritization, and Validation of lncRNAs
3.1. Discovering Viral Infection-Related lncRNAs: The Different Flavors of Transcriptome Deep Sequencing
3.1.1. Total RNA vs. mRNA
3.1.2. Computational Considerations for Identifying lncRNAs from RNA-seq Data
3.1.3. Singling Out Cells in Lieu of Bulk Analysis
3.2. Prioritization of Infection-Related lncRNAs by Computational Prediction
3.2.1. Prediction of lncRNA Function by “Guilt-by-Association”
3.2.2. Prediction of lncRNA Function Based on Local Genomic Context
3.3. Identifying Functional lncRNAs Using Evolutionary Analysis
3.3.1. Incorporating Synteny in Sequence Homology Searches
3.3.2. Using Structural Conservation to Functionally Annotate lncRNAs
3.4. Large Scale Perturbation Studies for Probing lncRNA Functions
3.4.1. Gain-Of-Function Library Screen
3.4.2. New Developments: Multiplexed Library Screen and Single cell Library Screen
3.5. Considerations for Experimentally Validating Specific lncRNAs
4. Conclusions and Future Direction
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peng, X.; Gralinski, L.; Armour, C.D.; Ferris, M.T.; Thomas, M.J.; Proll, S.; Bradel-Tretheway, B.G.; Korth, M.J.; Castle, J.C.; Biery, M.C.; et al. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. mBio 2010, 1, e00206-10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, C.Y.; Yedavalli, V.S.; Jeang, K.T. Neat1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. mBio 2013, 4, e00596-12. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Sova, P.; Green, R.R.; Thomas, M.J.; Korth, M.J.; Proll, S.; Xu, J.; Cheng, Y.; Yi, K.; Chen, L.; et al. Deep sequencing of HIV-infected cells: Insights into nascent transcription and host-directed therapy. J. Virol. 2014, 88, 8768–8782. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Peng, X.; Xie, T.; Lu, X.; Liu, F.; Wu, H.; Yang, Z.; Wang, J.; Cheng, L.; Wu, N. Detection of the long noncoding RNAs nuclear-enriched autosomal transcript 1 (NEAT1) and metastasis associated lung adenocarcinoma transcript 1 in the peripheral blood of HIV-1-infected patients. HIV Med. 2016, 17, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Winterling, C.; Koch, M.; Koeppel, M.; Garcia-Alcalde, F.; Karlas, A.; Meyer, T.F. Evidence for a crucial role of a host non-coding RNA in influenza a virus replication. RNA Biol. 2014, 11, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Mumtaz, P.T.; Bhat, S.A.; Ahmad, S.M.; Dar, M.A.; Ahmed, R.; Urwat, U.; Ayaz, A.; Shrivastava, D.; Shah, R.A.; Ganai, N.A. LncRNAs and immunity: Watchdogs for host pathogen interactions. Biol. Proced. Online 2017, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Valadkhan, S.; Gunawardane, L.S. LncRNA-mediated regulation of the interferon response. Virus Res. 2016, 212, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ding, C. Roles of lncRNAs in viral infections. Front. Cell. Infect. Microbiol. 2017, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, A.; Stadler, P.F. Evolutionary clues in lncRNAs. Wiley Interdiscip. Rev. RNA 2017, 8, e1376. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Bohmdorfer, G.; Wierzbicki, A.T. Control of chromatin structure by long noncoding RNA. Trends Cell Biol. 2015, 25, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Plath, K.; Mlynarczyk-Evans, S.; Nusinow, D.A.; Panning, B. Xist RNA and the mechanism of X chromosome inactivation. Annu. Rev. Genet. 2002, 36, 233–278. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A. Xist function: Bridging chromatin and stem cells. Trends Genet. TIG 2007, 23, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Senner, C.E.; Brockdorff, N. Xist gene regulation at the onset of X inactivation. Curr. Opin. Genet. Dev. 2009, 19, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Mira-Bontenbal, H.; Gribnau, J. New xist-interacting proteins in X-chromosome inactivation. Curr. Biol. 2016, 26, R338–R342. [Google Scholar] [CrossRef] [PubMed]
- Beckedorff, F.C.; Amaral, M.S.; Deocesano-Pereira, C.; Verjovski-Almeida, S. Long non-coding RNAs and their implications in cancer epigenetics. Biosci. Rep. 2013, 33, e00061. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, H.; Wang, H. Long noncoding RNAs in DNA methylation: New players stepping into the old game. Cell Biosci. 2016, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Betancur, J.G. Pervasive lncRNA binding by epigenetic modifying complexes—The challenges ahead. Biochim. Biophys. Acta 2016, 1859, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.L.; Luo, G.; Wise, J.A.; Lou, H. Regulation of alternative splicing by local histone modifications: Potential roles for RNA-guided mechanisms. Nucleic Acids Res. 2014, 42, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Pruszko, M.; Milano, E.; Forcato, M.; Donzelli, S.; Ganci, F.; Di Agostino, S.; De Panfilis, S.; Fazi, F.; Bates, D.O.; Bicciato, S.; et al. The mutant p53-ID4 complex controls VEGFA isoforms by recruiting lncRNA MALAT1. EMBO Rep. 2017, 18, 1331–1351. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.Z.; Zhang, Z.W.; Liu, Y.L.; Shi, C.X.; Zhang, J.; Zhang, Y.G. Relationship of long noncoding RNA and viruses. Genomics 2016, 107, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Han, P.; Ye, W.; Chen, H.; Zheng, X.; Cheng, L.; Zhang, L.; Yu, L.; Wu, X.; Xu, Z.; et al. The long noncoding RNA NEAT1 exerts antihantaviral effects by acting as positive feedback for RIG-I signaling. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Imam, H.; Bano, A.S.; Patel, P.; Holla, P.; Jameel, S. The lncRNA NRON modulates HIV-1 replication in a NFAT-dependent manner and is differentially regulated by early and late viral proteins. Sci. Rep. 2015, 5, 8639. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, C.; Ma, X.; Geng, G.; Liu, B.; Zhang, Y.; Zhang, S.; Zhong, F.; Liu, C.; Yin, Y.; et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 2016, 7, 11730. [Google Scholar] [CrossRef] [PubMed]
- Vigneau, S.; Rohrlich, P.S.; Brahic, M.; Bureau, J.F. Tmevpg1, a candidate gene for the control of Theiler’s virus persistence, could be implicated in the regulation of gamma interferon. J. Virol. 2003, 77, 5632–5638. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zhu, X.; Chen, Y.; Wei, H.; Chen, Q.; Chi, X.; Qi, B.; Zhang, L.; Zhao, Y.; Gao, G.F.; et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 2014, 16, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.; Ackley, A.; Turner, A.W.; Famiglietti, M.; Bosque, A.; Clemson, M.; Planelles, V.; Morris, K.V. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol. Ther. 2014, 22, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wang, H.; Herndier, B.; Ganem, D. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 1996, 93, 6641–6646. [Google Scholar] [CrossRef] [PubMed]
- Merry, C.R.; Forrest, M.E.; Sabers, J.N.; Beard, L.; Gao, X.H.; Hatzoglou, M.; Jackson, M.W.; Wang, Z.; Markowitz, S.D.; Khalil, A.M. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 2015, 24, 6240–6253. [Google Scholar] [CrossRef] [PubMed]
- Di Ruscio, A.; Ebralidze, A.K.; Benoukraf, T.; Amabile, G.; Goff, L.A.; Terragni, J.; Figueroa, M.E.; De Figueiredo Pontes, L.L.; Alberich-Jorda, M.; Zhang, P.; et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013, 503, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Savell, K.E.; Gallus, N.V.; Simon, R.C.; Brown, J.A.; Revanna, J.S.; Osborn, M.K.; Song, E.Y.; O’Malley, J.J.; Stackhouse, C.T.; Norvil, A.; et al. Extra-coding RNAs regulate neuronal DNA methylation dynamics. Nat. Commun. 2016, 7, 12091. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, Y.; Bao, X.; Zhu, X.; Kwok, Y.K.; Sun, K.; Chen, X.; Huang, Y.; Jauch, R.; Esteban, M.A.; et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. 2015, 25, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Shi, H.; Li, H.; Li, L.; Fang, R.; Cai, X.; Liu, B.; Zhang, X.; Ye, L. HBXIP and LSD1 scaffolded by lncRNA hotair mediate transcriptional activation by c-Myc. Cancer Res. 2016, 76, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Wu, Z.; Liao, K.; Zhang, S. Long noncoding RNA HOTAIR can serve as a common molecular marker for lymph node metastasis: A meta-analysis. Tumour Biol. 2014, 35, 8445–8450. [Google Scholar] [CrossRef] [PubMed]
- Hajjari, M.; Khoshnevisan, A.; Shin, Y.K. Molecular function and regulation of long non-coding RNAs: Paradigms with potential roles in cancer. Tumour Biol. 2014, 35, 10645–10663. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.C.; Campilongo, F.; Barclay, R.A.; DeMarino, C.; Iglesias-Ussel, M.D.; Kashanchi, F.; Romerio, F. The Human Immunodeficiency Virus 1 ASP RNA promotes viral latency by recruiting the Polycomb Repressor Complex 2 and promoting nucleosome assembly. Virology 2017, 506, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.D.; Sung, S. Long noncoding RNA: Unveiling hidden layer of gene regulatory networks. Trends Plant Sci. 2012, 17, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.S.; Fox, A.H. Paraspeckles: Nuclear bodies built on long noncoding RNA. J. Cell Biol. 2009, 186, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. Linking long noncoding RNA localization and function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Hirose, T. Paraspeckle nuclear bodies—Useful uselessness? Cell. Mol. Life Sci. 2012, 69, 3027–3036. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E. Long noncoding RNAs: Re-writing dogmas of RNA processing and stability. Biochim. Biophys. Acta 2016, 1859, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Li, J.; Zhou, K.; Liang, T. Competing endogenous RNA: A novel posttranscriptional regulatory dimension associated with the progression of cancer. Oncol. Lett. 2015, 10, 2683–2690. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Dinger, M.E. Endogenous microRNA sponges: Evidence and controversy. Nat. Rev. Genet. 2016, 17, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Thierry-Mieg, J.; Thierry-Mieg, D.; Nishida, A.; Pipes, L.; Bozinoski, M.; Thomas, M.J.; Kelly, S.; Weiss, J.M.; Raveendran, M.; et al. Tissue-specific transcriptome sequencing analysis expands the non-human primate reference transcriptome resource (NHPRTR). Nucleic Acids Res. 2015, 43, D737–D742. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.L.; Xiang, Y.Y.; Ji, L.J.; Lu, X.J. Competing endogenous RNA interplay in cancer: Mechanism, methodology, and perspectives. Tumour Biol. 2015, 36, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejias, A.; Tay, Y. Competing endogenous RNA networks: Tying the essential knots for cancer biology and therapeutics. J. Hematol. Oncol. 2015, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Cazalla, D.; Yario, T.; Steitz, J.A. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 2010, 328, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.E.; Oei, T.; Steitz, J.A. Herpesvirus saimiri microRNAs preferentially target host cell cycle regulators. J. Virol. 2015, 89, 10901–10911. [Google Scholar] [CrossRef] [PubMed]
- Tavanez, J.P.; Quina, A.S.; Cunha, C. Virus and noncoding RNAs: Stars in the host-virus interaction game. Future Virol. 2014, 9, 1077–1087. [Google Scholar] [CrossRef]
- Hu, S.; Wang, X.; Shan, G. Insertion of an Alu element in a lncRNA leads to primate-specific modulation of alternative splicing. Nat. Struct. Mol. Biol. 2016, 23, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hammerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. (Berl.) 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zheng, J.; Deng, J.; Zhang, L.; Li, N.; Li, W.; Li, F.; Lu, J.; Zhou, Y. LincRNA-uc002yug.2 involves in alternative splicing of RUNX1 and serves as a predictor for esophageal cancer and prognosis. Oncogene 2015, 34, 4723–4734. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.; Briggs, J.A.; Vanichkina, D.P.; Poth, E.M.; Beveridge, N.J.; Ratnu, V.S.; Nayler, S.P.; Nones, K.; Hu, J.; Bredy, T.W.; et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 2014, 19, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Fullwood, M.J. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Josset, L.; Tchitchek, N.; Gralinski, L.E.; Ferris, M.T.; Eisfeld, A.J.; Green, R.R.; Thomas, M.J.; Tisoncik-Go, J.; Schroth, G.P.; Kawaoka, Y.; et al. Annotation of long non-coding RNAs expressed in collaborative cross founder mice in response to respiratory virus infection reveals a new class of interferon-stimulated transcripts. RNA Biol. 2014, 11, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Clark, M.B.; Crawford, J.; Brunck, M.E.; Gerhardt, D.J.; Taft, R.J.; Nielsen, L.K.; Dinger, M.E.; Mattick, J.S. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nat. Protoc. 2014, 9, 989–1009. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. Tophat: Discovering splice junctions with RNA-seq. Bioinformatics (Oxf. Engl.) 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal RNA-seq aligner. Bioinformatics (Oxf. Engl.) 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Lu, M.; Wang, D.; Li, H.; He, X. Genome-wide identification of long noncoding RNAs in human intervertebral disc degeneration by RNA sequencing. BioMed Res. Int. 2016, 2016, 3684875. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.F.; Jungreis, I.; Kellis, M. Phylocsf: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics (Oxf. Engl.) 2011, 27, i275–i282. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Pian, C.; Zhang, G.; Chen, Z.; Chen, Y.; Zhang, J.; Yang, T.; Zhang, L. LncRNApred: Classification of long non-coding RNAs and protein-coding transcripts by the ensemble algorithm with a new hybrid feature. PLoS ONE 2016, 11, e0154567. [Google Scholar] [CrossRef] [PubMed]
- Ounzain, S.; Burdet, F.; Ibberson, M.; Pedrazzini, T. Discovery and functional characterization of cardiovascular long noncoding RNAs. J. Mol. Cell. Cardiol. 2015, 89, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Trapnell, C. Single-cell transcriptome sequencing: Recent advances and remaining challenges. F1000Research 2016, 5, 182. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Amaral, P.P.; Mercer, T.R.; Pang, K.C.; Bruce, S.J.; Gardiner, B.B.; Askarian-Amiri, M.E.; Ru, K.; Solda, G.; Simons, C.; et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 2008, 18, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Signal, B.; Gloss, B.S.; Dinger, M.E. Computational approaches for functional prediction and characterisation of long noncoding RNAs. Trends Genet. TIG 2016, 32, 620–637. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.D.; Diaz, A.; Nellore, A.; Delgado, R.N.; Park, K.Y.; Gonzales-Roybal, G.; Oldham, M.C.; Song, J.S.; Lim, D.A. Integration of genome-wide approaches identifies lncRNAs of adult neural stem cells and their progeny in vivo. Cell Stem Cell 2013, 12, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Marinov, G.K.; Pepke, S.; Singer, Z.S.; He, P.; Williams, B.; Schroth, G.P.; Elowitz, M.B.; Wold, B.J. Single-cell transcriptome analysis reveals dynamic changes in lncRNA expression during reprogramming. Cell Stem Cell 2015, 16, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, J.; Oliver, P.L.; Lunter, G.; Ponting, C.P. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS Genet. 2009, 5, e1000617. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Li, Y.; Wang, Z.; Zhou, S.; Sun, H.; Yu, G. The evolution and expression pattern of human overlapping lncRNA and protein-coding gene pairs. Sci. Rep. 2017, 7, 42775. [Google Scholar] [CrossRef] [PubMed]
- Chodroff, R.A.; Goodstadt, L.; Sirey, T.M.; Oliver, P.L.; Davies, K.E.; Green, E.D.; Molnar, Z.; Ponting, C.P. Long noncoding RNA genes: Conservation of sequence and brain expression among diverse amniotes. Genome Biol. 2010, 11, R72. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Gao, L.; Wang, Y.; Chiu, D.K.; Wang, T.; Deng, Y. Advances in long noncoding RNAs: Identification, structure prediction and function annotation. Brief. Funct. Genom. 2016, 15, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Mohammadin, S.; Edger, P.P.; Pires, J.C.; Schranz, M.E. Positionally-conserved but sequence-diverged: Identification of long non-coding RNAs in the Brassicaceae and Cleomaceae. BMC Plant Biol. 2015, 15, 217. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shishkin, A.A.; Zhu, X.; Kadri, S.; Maza, I.; Guttman, M.; Hanna, J.H.; Regev, A.; Garber, M. Evolutionary analysis across mammals reveals distinct classes of long non-coding RNAs. Genome Biol. 2016, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.D.L.; Devisetty, U.K.; Palos, K.; Haug-Baltzell, A.K.; Lyons, E.; Beilstein, M.A. Evolinc: A tool for the identification and evolutionary comparison of long intergenic non-coding RNAs. Front. Genet. 2017, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Arfat, Y.; Li, D.; Zhao, F.; Chen, Z.; Yin, C.; Sun, Y.; Hu, L.; Yang, T.; Qian, A. Structure prediction: New insights into decrypting long noncoding RNAS. Int. J. Mol. Sci. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, B.; Hein, J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 2003, 31, 3423–3428. [Google Scholar] [CrossRef] [PubMed]
- Sundfeld, D.; Havgaard, J.H.; de Melo, A.C.; Gorodkin, J. Foldalign 2.5: Multithreaded implementation for pairwise structural RNA alignment. Bioinformatics (Oxf. Engl.) 2016, 32, 1238–1240. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Weinberg, Z.; Ruzzo, W.L. Cmfinder—A covariance model based RNA motif finding algorithm. Bioinformatics (Oxf. Engl.) 2006, 22, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Seemann, S.E.; Mirza, A.H.; Hansen, C.; Bang-Berthelsen, C.H.; Garde, C.; Christensen-Dalsgaard, M.; Torarinsson, E.; Yao, Z.; Workman, C.T.; Pociot, F.; et al. The identification and functional annotation of RNA structures conserved in vertebrates. Genome Res. 2017, 27, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Konig, R.; Chiang, C.Y.; Tu, B.P.; Yan, S.F.; DeJesus, P.D.; Romero, A.; Bergauer, T.; Orth, A.; Krueger, U.; Zhou, Y.; et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat. Methods 2007, 4, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. LincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Lennox, K.A.; Behlke, M.A. Cellular localization of long non-coding RNAs affects silencing by rnai more than by antisense oligonucleotides. Nucleic Acids Res. 2016, 44, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Autuoro, J.M.; Pirnie, S.P.; Carmichael, G.G. Long noncoding RNAs in imprinting and X chromosome inactivation. Biomolecules 2014, 4, 76–100. [Google Scholar] [CrossRef] [PubMed]
- Sigoillot, F.D.; Lyman, S.; Huckins, J.F.; Adamson, B.; Chung, E.; Quattrochi, B.; King, R.W. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat. Methods 2012, 9, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR toolbox to study virus-host interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, M.; Bassik, M.C.; Weissman, J.S. Functional genomics platform for pooled screening and generation of mammalian genetic interaction maps. Nat. Protoc. 2014, 9, 1825–1847. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Koirala, P.; Huang, J.; Ho, T.T.; Wu, F.; Ding, X.; Mo, Y.Y. LncRNA AK023948 is a positive regulator of AKT. Nat. Commun. 2017, 8, 14422. [Google Scholar] [CrossRef] [PubMed]
- Bassik, M.C.; Kampmann, M.; Lebbink, R.J.; Wang, S.; Hein, M.Y.; Poser, I.; Weibezahn, J.; Horlbeck, M.A.; Chen, S.; Mann, M.; et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 2013, 152, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nunez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y.; et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 2016, 167. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; Tian, R.; Blau, J.; Markegard, E.; Wu, D.; Biton, A.; Zaitlen, N.; McCormick, F.; Kampmann, M.; McManus, M.T. Decoding directional genetic dependencies through orthogonal CRISPR/Cas screens. 2017. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Weiner, A.; Yofe, I.; Lara-Astiaso, D.; Keren-Shaul, H.; David, E.; Salame, T.M.; Tanay, A.; van Oudenaarden, A.; Amit, I. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 2016, 167. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Duan, J.; Li, B.; Zhou, P.; Hon, G.C. Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Mol. Cell 2017, 66. [Google Scholar] [CrossRef] [PubMed]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.E.; Klein, A.M. Genetic screening enters the single-cell era. Nat. Methods 2017, 14, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Sun, T.; Hacisuleyman, E.; Fei, T.; Wang, X.; Brown, M.; Rinn, J.L.; Lee, M.G.; Chen, Y.; Kantoff, P.W.; et al. Integrative analyses reveal a long noncoding RNA-mediated sponge regulatory network in prostate cancer. Nat. Commun. 2016, 7, 10982. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Adriaens, C.; Standaert, L.; Barra, J.; Latil, M.; Verfaillie, A.; Kalev, P.; Boeckx, B.; Wijnhoven, P.W.; Radaelli, E.; Vermi, W.; et al. P53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med. 2016, 22, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Myacheva, K.; Gross, M.; Klingenberg, M.; Duran Arque, B.; Diederichs, S. Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 2017, 45, e12. [Google Scholar] [CrossRef] [PubMed]
- Pulido-Quetglas, C.; Aparicio-Prat, E.; Arnan, C.; Polidori, T.; Hermoso, T.; Palumbo, E.; Ponomarenko, J.; Guigo, R.; Johnson, R. Scalable design of paired CRISPR guide RNAs for genomic deletion. PLoS Comput. Biol. 2017, 13, e1005341. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dominguez, J.R.; Bai, Z.; Xu, D.; Yuan, B.; Lo, K.A.; Yoon, M.J.; Lim, Y.C.; Knoll, M.; Slavov, N.; Chen, S.; et al. De novo reconstruction of adipose tissue transcriptomes reveals long non-coding RNA regulators of brown adipocyte development. Cell Metab. 2015, 21, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Huang, H.T.; Liang, Y.; Trimarchi, T.; Aifantis, I.; Tsirigos, A. LncRNA-screen: An interactive platform for computationally screening long non-coding RNAs in large genomics datasets. BMC Genom. 2017, 18, 434. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| lncRNA | Encoding Organism | General Function | Specific Function | Infection Type | ID Method | Citation |
|---|---|---|---|---|---|---|
| NEAT1 | Host | Scaffold | Nuclear localization, paraspeckle formation | HIV-1, HTNV | 1 of 83 lncRNAs profiled in HIV-1-infected Jurkat and MT4 cells | [2,4,24] |
| NRON | Host | Scaffold | Latency via inhibition of NFAT nuclear translocation | HIV-1 | 1 of 90 lncRNAs profiled in two human T cell lines | [25,26] |
| Tmevpg1 (NeST, IfngAS1) | Host | Epigenetics | IFN-gamma-mediated regulation of adaptive immunity | Theiler’s murine encephalo myelitis (TMEV) | Candidate gene from Tmevp3 locus | [27] and reviews by [6,23] |
| NRAV | Host | Epigenetics | Modulates transcription of ISGs, promotes IAV replication | Influenza A Virus (IAV) | 1 of 907 differentially expressed lncRNAs from microarray analysis | [28] and reviews by [6,8] |
| HIV-expressed antisense lncRNA (ASP-L) | Pathogen | Epigenetics | Epigenetic transcriptional regulation | HIV-1 | qPCR | [29] |
| PAN RNA | Pathogen | Epigenetics | Required for KSHV gene expression, repression of IFN-alpha, IFN-gamma, ISGs | Kaposi’s Sarcoma-associated Herpes-virus (KSHV) | Northern Blot | [30] and reviews by [7,8] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemler, D.J.; Brochu, H.N.; Yang, F.; Harrell, E.A.; Peng, X. Elucidating the Role of Host Long Non-Coding RNA during Viral Infection: Challenges and Paths Forward. Vaccines 2017, 5, 37. https://doi.org/10.3390/vaccines5040037
Lemler DJ, Brochu HN, Yang F, Harrell EA, Peng X. Elucidating the Role of Host Long Non-Coding RNA during Viral Infection: Challenges and Paths Forward. Vaccines. 2017; 5(4):37. https://doi.org/10.3390/vaccines5040037
Chicago/Turabian StyleLemler, David J., Hayden N. Brochu, Fang Yang, Erin A. Harrell, and Xinxia Peng. 2017. "Elucidating the Role of Host Long Non-Coding RNA during Viral Infection: Challenges and Paths Forward" Vaccines 5, no. 4: 37. https://doi.org/10.3390/vaccines5040037
APA StyleLemler, D. J., Brochu, H. N., Yang, F., Harrell, E. A., & Peng, X. (2017). Elucidating the Role of Host Long Non-Coding RNA during Viral Infection: Challenges and Paths Forward. Vaccines, 5(4), 37. https://doi.org/10.3390/vaccines5040037