Self-Amplifying Replicon RNA Vaccine Delivery to Dendritic Cells by Synthetic Nanoparticles


Abstract
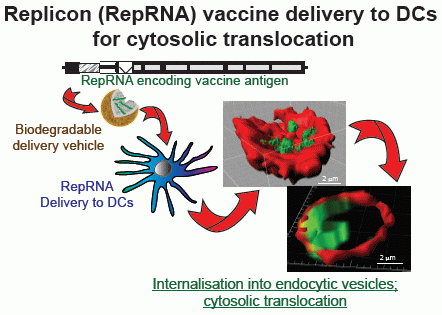
:
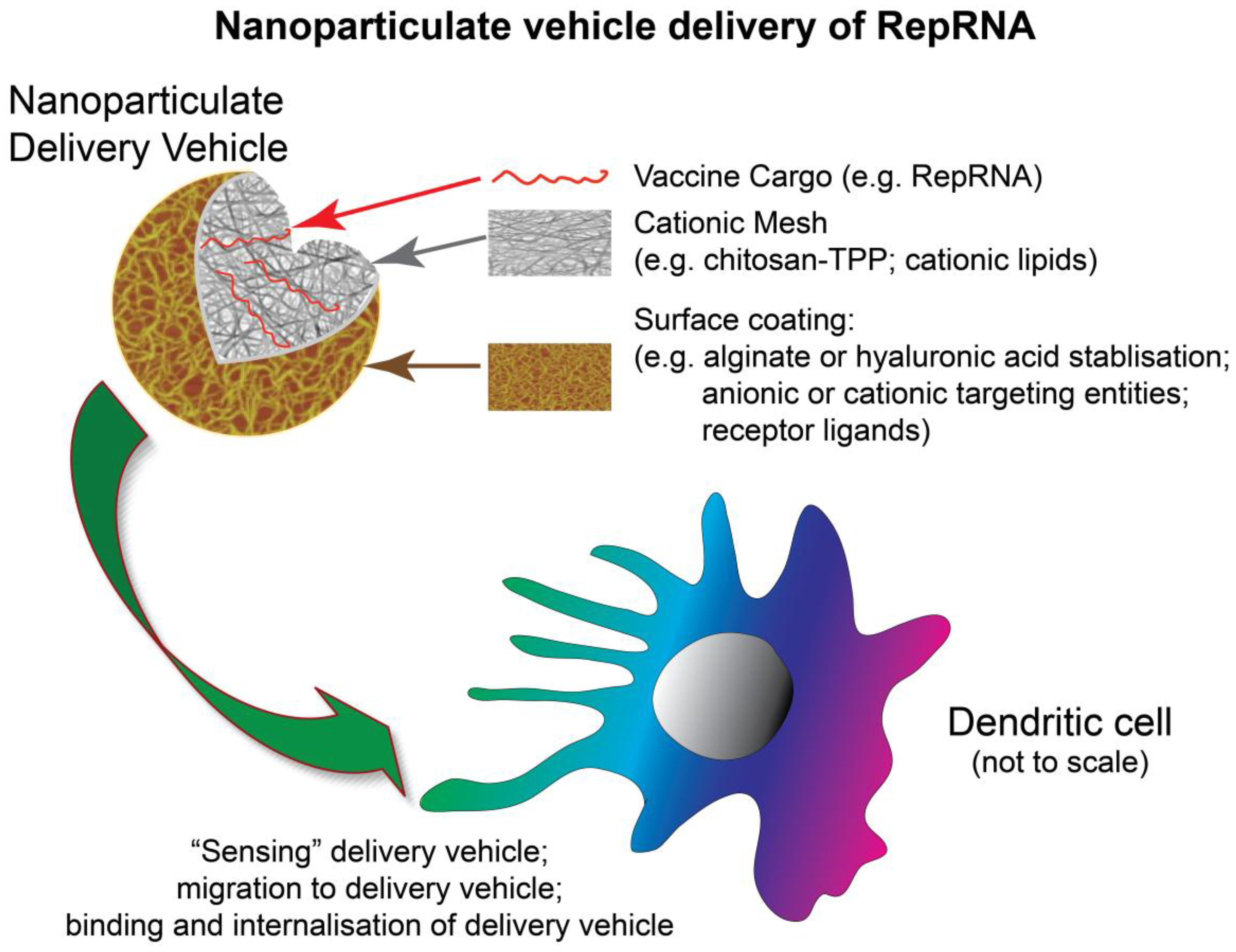
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. The Self-amplifying Replicon RNA
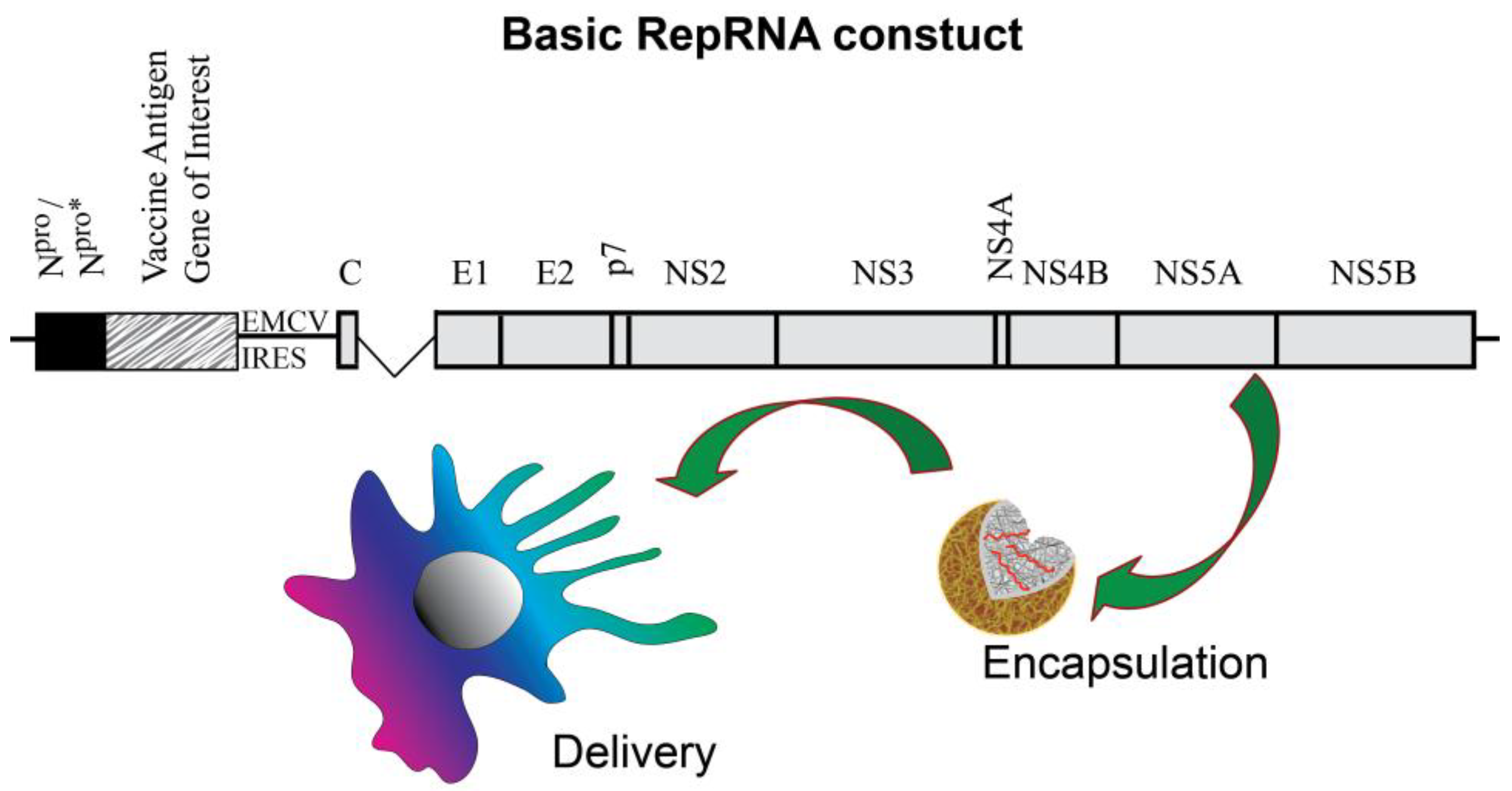
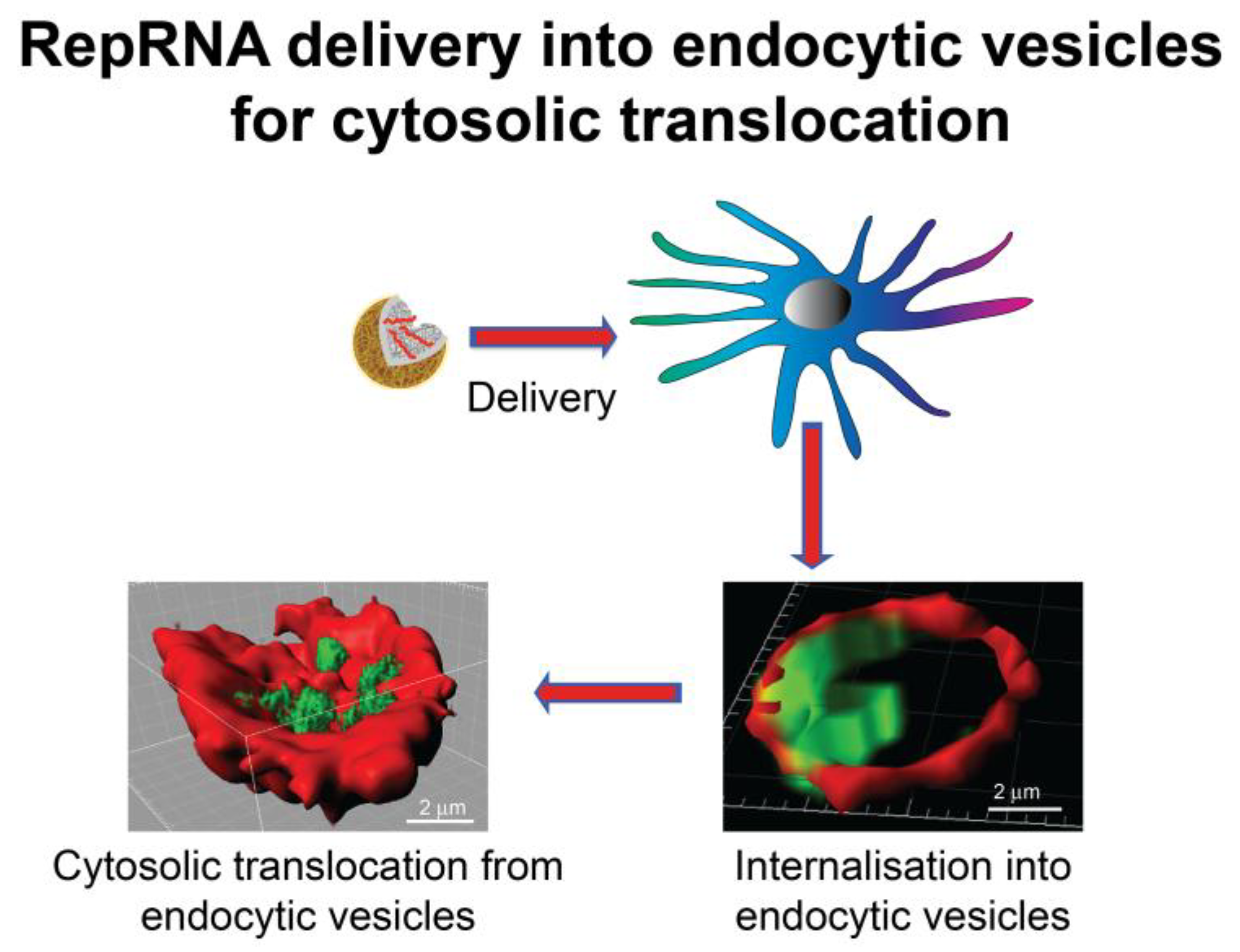
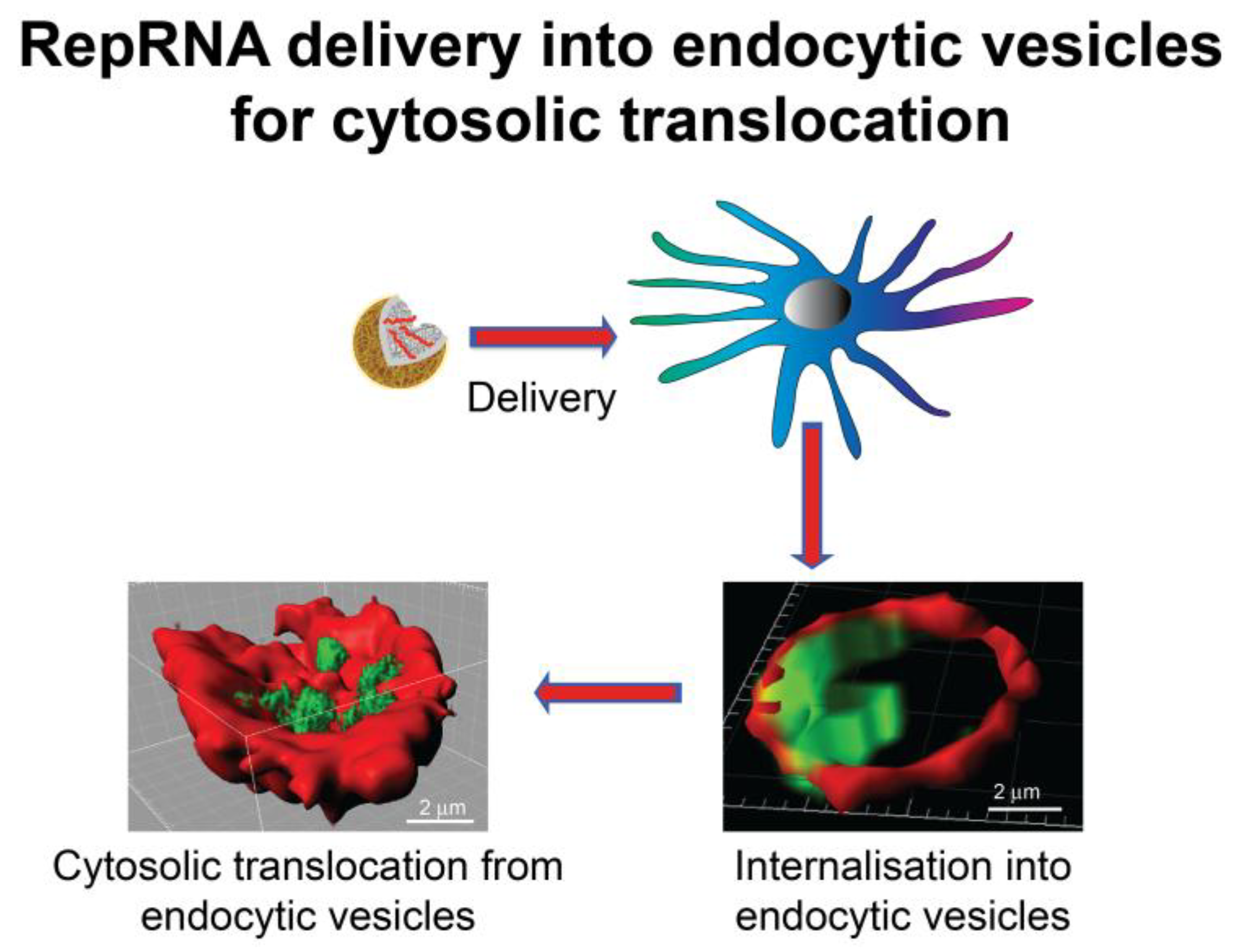
3. Dendritic Cell Handling of Vaccines
4. Endocytic Processing Pathways of Dendritic Cells
5. Learning from Cross-Presentation Pathways in Dendritic Cells
6. Targeting DC with RNA Vaccines
7. DC Vesicular Acidification and Cytosolic Translocation
8. Learning from Cytosolic Translocation of Small RNA Molecules
9. RepRNA Delivery to Dendritic Cells for Encoded Antigen Translation
10. RepRNA Delivery by Nanoparticulate Vehicles Induces Immune Responses in Vivo
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McCullough, K.C.; Bassi, I.; Demoulins, T.; Thomann-Harwood, L.J.; Ruggli, N. Functional RNA delivery targeted to dendritic cells by synthetic nanoparticles. Ther. Deliv. 2012, 3, 1077–1099. [Google Scholar] [CrossRef] [PubMed]
- Thomann-Harwood, L.J.; Kaeuper, P.; Rossi, N.; Milona, P.; Herrmann, B.; McCullough, K.C. Nanogel vaccines targeting dendritic cells: Contributions of the surface decoration and vaccine cargo on cell targeting and activation. J. Control. Release 2013, 166, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D. Happy coupling: Recruiting both antigen and effector function. Nat. Biotechnol. 1997, 15, 618–619. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C. Immunogenicity signals 1,2,3... and 0. Immunol. Today 1989, 10, 283–286. [Google Scholar] [CrossRef]
- Pulendran, B.; Ahmed, R. Translating innate immunity into immunological memory: Implications for vaccine development. Cell 2006, 124, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Atkins, G.J.; Fleeton, M.N.; Sheahan, B.J. Therapeutic and prophylactic applications of alphavirus vectors. Exp. Rev. Mol. Med. 2008, 10, e33. [Google Scholar] [CrossRef]
- Khromykh, A.A. Replicon-based vectors of positive strand RNA viruses. Curr. Opin. Mol. Therapy 2000, 2, 555–569. [Google Scholar]
- Lundstrom, K. Alphavirus-based vaccines. Curr. Opin. Mol. Ther. 2002, 4, 28–34. [Google Scholar] [PubMed]
- Pijlman, G.P.; Suhrbier, A.; Khromykh, A.A. Kunjin virus replicons: An RNA-based, non-cytopathic viral vector system for protein production, vaccine and gene therapy applications. Expert Opin. Biol. Ther. 2006, 6, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Rayner, J.O.; Dryga, S.A.; Kamrud, K.I. Alphavirus vectors and vaccination. Rev. Med. Virol. 2002, 12, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T. Pathogen sensors and chemokine receptors in dendritic cell subsets. Vaccine 2012, 30, 7652–7657. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. NY Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Khoo, J.J.; Forster, S.; Mansell, A. Toll-like receptors as interferon-regulated genes and their role in disease. J. Interferon Cytokine Res. 2011, 31, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Summerfield, A.; McCullough, K.C. Dendritic cells in innate and adaptive immune responses against influenza virus. Viruses 2009, 1, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; McCartney, S.A.; Wang, Y.; Colonna, M. TLR7/9 versus TLR3/MDA5 signaling during virus infections and diabetes. J. Leukoc. Biol. 2011, 90, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors and Type I interferons. J. Biol. Chem. 2007, 282, 15319–15323. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, M.; Ruggli, N.; Tache, V.; Gerber, H.; McCullough, K.C.; Summerfield, A. Double-stranded secondary structures on mRNA induce type 1 interferon (IFN α/β) production and maturation of mRNA-transfected monocyte-derived dendritic cells. J. Gene Med. 2005, 7, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Bauhofer, O.; Summerfield, A.; Sakoda, Y.; Tratschin, J.D.; Hofmann, M.A.; Ruggli, N. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol. 2007, 81, 3087–3096. [Google Scholar] [CrossRef] [PubMed]
- Ruggli, N.; Bird, B.H.; Liu, L.; Bauhofer, O.; Tratschin, J.D.; Hofmann, M.A. N(pro) of classical swine fever virus is an antagonist of double-stranded RNA-mediated apoptosis and IFN-alpha/beta induction. Virology 2005, 340, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Ruggli, N.; Summerfield, A.; Fiebach, A.R.; Guzylack-Piriou, L.; Bauhofer, O.; Lamm, C.G.; Waltersperger, S.; Matsuno, K.; Liu, L.; Gerber, M.; et al. Classical swine fever virus can remain virulent after specific elimination of the interferon regulatory factor 3-degrading function of Npro. J. Virol. 2009, 83, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Bauhofer, O.; Summerfield, A.; McCullough, K.C.; Ruggli, N. Role of double-stranded RNA and Npro of classical swine fever virus in the activation of monocyte-derived dendritic cells. Virology 2005, 343, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Suter, R.; Summerfield, A.; Thomann-Harwood, L.J.; McCullough, K.C.; Tratschin, J.D.; Ruggli, N. Immunogenic and replicative properties of classical swine fever virus replicon particles modified to induce IFN-alpha/beta and carry foreign genes. Vaccine 2011, 29, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, V.; Behrens, S.E.; Aliberti, J.; Rehermann, B. Dendritic cells transfected with cytopathic self-replicating RNA induce crosspriming of CD8+ T cells and antiviral immunity. Immunity 2004, 20, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, S.; Haddadi, A.; Hung, R.W.; Lavasanifar, A. Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations. Adv. Drug Del. Rev. 2011, 63, 943–955. [Google Scholar] [CrossRef]
- Klippstein, R.; Pozo, D. Nanotechnology-based manipulation of dendritic cells for enhanced immunotherapy strategies. Nanomedicine 2010, 6, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R.M.; Aberle, J.H.; Aberle, S.W.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Mimicking live flavivirus immunization with a noninfectious RNA vaccine. Proc. Natl. Acad. Sci. USA 2004, 101, 1951–1956. [Google Scholar] [CrossRef] [PubMed]
- De Haes, W.; Rejman, J.; Pollard, C.; Merlin, C.; Vekemans, M.; Florence, E.; de Smedt, S.C.; Grooten, J.; Vanham, G.; de Koker, S.; et al. Lipoplexes carrying mRNA encoding Gag protein modulate dendritic cells to stimulate HIV-specific immune responses. Nanomedicine (Lond.) 2013, 8, 77–87. [Google Scholar] [CrossRef]
- De Haes, W.; van Mol, G.; Merlin, C.; de Smedt, S.C.; Vanham, G.; Rejman, J. Internalization of mRNA lipoplexes by dendritic cells. Mol. Pharm. 2012, 9, 2942–2949. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.C.; Basta, S.; Knotig, S.; Gerber, H.; Schaffner, R.; Kim, Y.B.; Saalmuller, A.; Summerfield, A. Intermediate stages in monocyte-macrophage differentiation modulate phenotype and susceptibility to virus infection. Immunology 1999, 98, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Benvegnu, T.; Berchel, M.; Lebegue, L.; Pichon, C.; Jaffrès, P.-A.; Midoux, P. Enhancement of dendritic cells transfection in vivo and of vaccintion against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 2011, 7, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Gosset, D.; Mevel, M.; Miramon, M.L.; Yaouanc, J.J.; Pichon, C.; Benvegnu, T.; Jaffres, P.A.; Midoux, P. Selective gene delivery in dendritic cells with mannosylated and histidylated lipopolyplexes. J. Drug Target 2011, 19, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Pichon, C.; Midoux, P. Mannosylated and histidylated LPR technology for vaccination with tumor antigen mRNA. Methods Mol. Biol. 2013, 969, 247–274. [Google Scholar] [PubMed]
- McCullough, K.C.; Bassi, I.; Milona, P.; Suter, R.; Thomann-Harwood, L.; Englezou, P.; Démoulins, T.; Ruggli, N. Self-replicating replicon-RNA delivery to dendritic cells by chitosan-nanoparticles for translation in vitro and in vivo. Mol. Ther. Nucleic Acids 2014, 3, e173. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, S.; Kurts, C. Endocytosis mechanisms and the cell biology of antigen. Curr. Opin. Immunol. 2008, 20, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.E. Recent advances in antigen processing and presentation. Nat. Immunol. 2007, 8, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.; Neefjes, J. MHC class II molecules on the move for successful antigen presentation. EMBO J. 2008, 27, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Amigorena, S. The cell biology of antigen presentation in dendritic cells. Curr. Opin. Immunol. 2001, 13, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Wubbolts, R.; Stoorvogel, W. Endosomal sorting of MHC class II determines antigen presentation by dendritic cells. Curr. Opin. Cell Biol. 2008, 20, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, D.; le Bon, A.; Dumas, A.; Taleb, K.; Sachse, M.; Sikora, R.; Julithe, M.; Benmerah, A.; Bismuth, G.; Niedergang, F. Antigen stored in dendritic cells after macropinocytosis is released unprocessed from late endosomes to target B cells. Blood 2011, 119, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Wykes, M.; Pombo, A.; Jenkins, C.; MacPherson, G.G. Dendritic cells interact directly with naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J. Immunol. 1998, 161, 1313–1319. [Google Scholar] [PubMed]
- Ackerman, A.L.; Kyritsis, C.; Tampé, R.; Cresswell, P. Access of soluble antigens to the endoplasmic reticulum can explain cross-presentation by dendritic cells. Nat. Immunol. 2004, 6, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Amigorena, S.; Savina, A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr. Opin. Immunol. 2010, 22, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Heath, W.; Belz, G.; Behrens, G.M.N.; Smith, C.M.; Forehan, S.P.; Parish, I.A.; Davey, G.M.; Wilson, N.S.; Carbone, F.R.; Villadangos, J.A. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol. Rev. 2004, 199, 9–26. [Google Scholar] [PubMed]
- Monu, N.; Trombetta, E.S. Cross-talk between the endocytic pathway and the endoplasmic reticulum in cross-presentationby MHC class I molecules. Curr. Opin. Immunol. 2007, 19, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Vyas, J.M.; van der Veen, A.G.; Ploegh, H.L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008, 8, 607–618. [Google Scholar] [CrossRef]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: Multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef] [PubMed]
- Platta, H.W.; Stenmark, H. Endocytosis and signaling. Curr. Opin. Cell Biol. 2011, 23, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Pust, S.; Skotland, T.; van Deurs, B. Clathrin-independent endocytosis: Mechanisms and function. Curr. Opin. Cell Biol. 2011, 23, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Belizaire, R.; Unanue, E.R. Targeting proteins to distinct subcellular compartments reveals unique requirements for MHC class I and II presentation. Proc. Natl. Acad. Sci. USA 2009, 106, 17463–17468. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Kartenbeck, J.; Helenius, A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001, 3, 473–483. [Google Scholar] [CrossRef]
- Vazquez-Calvo, A.; Saiz, J.C.; McCullough, K.C.; Sobrino, F.; Martin-Acebes, M.A. Acid-dependent viral entry. Virus Res. 2012, 167, 125–137. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.C.; Summerfield, A. Targeting the porcine immune system-Particulate vaccines in the 21st century. Dev. Comp. Immunol. 2009, 33, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Schalk, J.A.; Mooi, F.R.; Berbers, G.A.; van Aerts, L.A.; Ovelgönne, H.; Kimman, T.G. Preclinical and clinical safety studies on DNA vaccines. Hum. Vaccine 2006, 2, 45–53. [Google Scholar] [CrossRef]
- Gao, W.; Xiao, Z.; Radovic-Moreno, A.; Shi, J.; Langer, R.; Farokzad, O.C. Progress in siRNA delivery using multifunctional nanoparticles. Methods Mol. Biol. 2010, 629, 53–67. [Google Scholar] [PubMed]
- Garcia-Gaumont, C.; Seksek, O.; Grzybowska, J.; Borowski, E.; Bolard, J. Delivery systems for antisense oligonucleotides. Pharmacol. Ther. 2000, 87, 255–277. [Google Scholar] [CrossRef] [PubMed]
- Howard, K.A. Delivery of RNA interference therapeutics using polycation-based nanoparticles. Adv. Drug Del. Rev. 2009, 61, 710–720. [Google Scholar] [CrossRef]
- Katas, H.; Alpar, H.O. Development and characterisation of chitosan nanoparticles for siRNA delivery. J. Control. Release 2006, 115, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Landesman-Milo, D.; Peer, D. Altering the immune response with lipid-based nanoparticles. J. Control. Release 2012, 161, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Nagasaki, Y. Smart siRNA delivery systems based on polymeric nanoassemblies and nanoparticles. Nanomedicine 2010, 5, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Tavernier, G.; Andries, O.; Demeester, J.; Sanders, N.N.; de Smedt, S.C.; Rejman, J. mRNA as gene therapeutic: How to control protein expression. J. Control. Release 2011, 150, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Naquib, S.; Wu, Z. Recent advances of siRNA delivery by nanoparticles. Expert Opin. Drug Deliv. 2011, 8, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Wasungu, L.; Hoekstra, D. Cationic lipids, lipoplexes and intracellular delivery of genes. J. Control. Release 2006, 116, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Hassane, F.S.; Saleh, A.F.; Abes, R.; Gait, M.J.; Lebleu, B. Cell penetrating peptides: Overview and applications to the delivery of oligonucleotides. Cell Mol. Life Sci. 2010, 67, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Pichon, C.; Yaouanc, J.-J.; Jaffrès, P.-A. Chemical vectors for gene delivery: A current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br. J. Pharmacol. 2009, 157, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Korsholm, K.S.; Andersen, P.; Perrie, Y.; Christensen, D. Liposomal vaccine delivery systems. Exp. Opin. Drug Del. 2011, 8, 505–519. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, M.L.; Rabasco, A.M. Charged liposomes as carriers to enhance the permeation through the skin. Exp. Opin. Drug Del. 2011, 8, 857–871. [Google Scholar] [CrossRef]
- Jiang, H.-L.; Kim, T.H.; Kim, Y.-K.; Park, I.-Y.; Cho, M.-H.; Cho, C.S. Efficient gene delivery using chitosan-polyethylenimine hybrid systems. Biomed. Mater. 2008, 3. [Google Scholar] [CrossRef]
- Won, Y.-W.; Lim, K.S.; Kim, Y.-H. Intracellular organelle-targeted non-viral gene delivery systems. J. Control. Release 2011, 152, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Kobayashi, S.; Futaki, S. Endosome-disruptive peptides for improving cytosolic delivery of bioactive macromolecules. Peptide Sci. 2010, 94, 763–770. [Google Scholar] [CrossRef]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Ruan, L.; Ma, L.; Cui, Y.; Wang, J.M.; Le, Y. RNA interference as a novel and powerful tool in immunopharmacological research. Int. Immunopharm. 2007, 7, 417–426. [Google Scholar] [CrossRef]
- Davidson, B.L.; McCray, P.B. Current prospects for RNA interference-based therapies. Nat. Rev. Gen. 2011, 12, 329–340. [Google Scholar] [CrossRef]
- Kim, H.-K.; Wei, H.; Kulkarni, A.; Pogranichniy, R.N.; Thompson, D.H. Effective targeted gene delivery to dendritic cells via synergetic interaction of mannosylated lipid with DOPE and BCAT. Biomacromolecules 2012, 13, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Mockey, M.; Bourseau, E.; Chandrashekhar, V.; Chaudhuri, A.; Lafosse, S.; le Cam, E.; Quesniaux, V.F.J.; Ryffel, B.; Pichon, C.; Midoux, P. mRNA-based cancer vaccine: Prevention of B16 melanoma progression and metastasis by systemic injection of MART 1 mRNA histidylated lipopolyplexes. Cancer Gene Ther. 2007, 14, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Markov, O.O.; Mironova, N.L.; Maslov, M.A.; Petukhov, I.A.; Morosova, N.G.; Vlassov, V.V.; Zenkova, M.A. Novel cationic liposomes provide highly efficient delivery of DNA and RNA into dendritic cell progenitors and their immature offsets. J. Control. Release 2012, 160, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Debus, H.; Baumhof, P.; Probst, J.; Kissel, T. Delivery of messenger RNA using poly(ethylene imine)-poly(ethylene glycol)-copolymer blends for polyplex formation: Biophysical characterization and in vitro transfection properties. J. Control. Release 2010, 148, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Tavernier, G.; Bavarsad, N.; Demeester, J.; de Smedt, S.C. mRNA transfection of cervical carcinoma and mesenchymal stem cells mediated by cationic carriers. J. Control. Release 2010, 147, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Tratschin, J.D.; Ruggli, N.; McCullough, K.C. Pestivirus Replicons Providing an RNA-Based Viral Vector System. U.S. Patent US 20110189224 A1, 4th June 2008. [Google Scholar]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.; Chan, V.; Mao, H.Q.; Leong, K.W. Interactions of phospholipid bilayer with chitosan: Effect of molecular weight and pH. Biomacromolecules 2001, 2, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Kogure, K.; Moriguchi, R.; Nakamura, Y.; Higashi, T.; Nakamura, T.; Serada, S.; Fujimoto, M.; Naka, T.; Futaki, S.; Harashima, H. nanoparticles for ex vivo siRNA delivery to dendritic cells for cancer vaccines: Programmed endosomal escape and dissociation. J. Control. Release 2010, 143, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 2014. [Google Scholar] [CrossRef]
- Sharma, R.; Ghasparian, A.; Robinson, J.A.; McCullough, K.C. Synthetic virus-like particles target dendritic cell lipid rafts for rapid endocytosis primarily but not exclusively by macropinocytosis. PLoS One 2012, 7, e43248. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stoter, M.; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Rehman, Z.U.; Hoekstra, D.; Zuhorn, I.S. Mechanism of polyplex- and lipoplex-mediated delivery of nucleic acids: Real-time visualization of transient membrane destabilization without endosomal lysis. ACS Nano 2013, 7, 3767–3777. [Google Scholar] [CrossRef] [PubMed]
- Suter, R.; Ruggli, N.; Institute of Virology and Immunology, Mittelhäusern, Switzerland. Personal communication. 2012.
- Kuhn, A.N.; Diken, M.; Kreiter, S.; Selmi, A.; Kowalska, J.; Jemielity, J.; Darzynkiewicz, E.; Huber, C.; Tureci, O.; Sahin, U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010, 17, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, S. Messenger RNA-based vaccines. Expert Opin. Biol. Ther. 2004, 4, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. MicroBiol. Rev. 1994, 58, 491–562. [Google Scholar] [PubMed]
- Berben-Bloemheuvel, G.; Kasperaitis, M.A.; van Heugten, H.; Thomas, A.A.; van Steeg, H.; Voorma, H.O. Interaction of initiation factors with the cap structure of chimaeric mRNA containing the 5'-untranslated regions of Semliki Forest virus RNA is related to translational efficiency. Eur. J. BioChem. 1992, 208, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Van Duijn, L.P.; Kasperaitis, M.; Ameling, C.; Voorma, H.O. Additional methylation at the N(2)-position of the cap of 26S Semliki Forest virus late mRNA and initiation of translation. Virus Res. 1986, 5, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Maruggi, G.; Shaw, C.A.; Otten, G.R.; Mason, P.W.; Beard, C.W. Engineered alphavirus replicon vaccines based on known attenuated viral mutants show limited effects on immunogenicity. Virology 2013, 447, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.L.; Zhao, Q.; O’Donnell, V.K.; Mason, P.W. Adaptation of West Nile virus replicons to cells in culture and use of replicon-bearing cells to probe antiviral action. Virology 2005, 331, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Kolupaeva, V.G.; Pestova, T.V.; Hellen, C.U. Ribosomal binding to the internal ribosomal entry site of classical swine fever virus. RNA 2000, 6, 1791–1807. [Google Scholar] [CrossRef] [PubMed]
- Rijnbrand, R.; van der Straaten, T.; van Rijn, P.A.; Spaan, W.J.; Bredenbeek, P.J. Internal entry of ribosomes is directed by the 5' noncoding region of classical swine fever virus and is dependent on the presence of an RNA pseudoknot upstream of the initiation codon. J. Virol. 1997, 71, 451–457. [Google Scholar] [PubMed]
- Xu, J.; Luft, J.C.; Yi, X.; Tian, S.; Owens, G.; Wang, J.; Johnson, A.; Berglund, P.; Smith, J.; Napier, M.E.; et al. RNA replicon delivery via lipid-complexed PRINT protein particles. Mol. Pharm. 2013, 10, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCullough, K.C.; Milona, P.; Thomann-Harwood, L.; Démoulins, T.; Englezou, P.; Suter, R.; Ruggli, N. Self-Amplifying Replicon RNA Vaccine Delivery to Dendritic Cells by Synthetic Nanoparticles. Vaccines 2014, 2, 735-754. https://doi.org/10.3390/vaccines2040735
McCullough KC, Milona P, Thomann-Harwood L, Démoulins T, Englezou P, Suter R, Ruggli N. Self-Amplifying Replicon RNA Vaccine Delivery to Dendritic Cells by Synthetic Nanoparticles. Vaccines. 2014; 2(4):735-754. https://doi.org/10.3390/vaccines2040735
Chicago/Turabian StyleMcCullough, Kenneth C., Panagiota Milona, Lisa Thomann-Harwood, Thomas Démoulins, Pavlos Englezou, Rolf Suter, and Nicolas Ruggli. 2014. "Self-Amplifying Replicon RNA Vaccine Delivery to Dendritic Cells by Synthetic Nanoparticles" Vaccines 2, no. 4: 735-754. https://doi.org/10.3390/vaccines2040735
APA StyleMcCullough, K. C., Milona, P., Thomann-Harwood, L., Démoulins, T., Englezou, P., Suter, R., & Ruggli, N. (2014). Self-Amplifying Replicon RNA Vaccine Delivery to Dendritic Cells by Synthetic Nanoparticles. Vaccines, 2(4), 735-754. https://doi.org/10.3390/vaccines2040735