
Therapeutic Efficacy of CD34-Derived Allogeneic Dendritic Cells Engineered to Express CD93, CD40L, and CXCL13 in Humanized Mouse Models of Pancreatic Cancer


 ,
, 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells, Reagents, and Antibodies
2.2. CD34+ Human HSC Lentiviral Transduction and Expansion
2.3. Differentiation of CD34+ Human HSCs to DCs
2.4. Lysate Generation
2.5. Antigen Pulsing of DCs
2.6. Flow Cytometry
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Vector Copy Number and RT-qPCR
2.9. Orthotopic Tumor Implantations and Treatment with DCs
2.10. PBMC and CD8+ T Cell Isolation
2.11. DC-CD8+ T Cell Co-Culture and In Vitro T Cell Activation of CD8+ T Cells with DCs
2.12. 51Cr Release Cytotoxicity Assay
2.13. Cytotoxicity Analysis by e-Sight System
2.14. Immunohistochemical Analysis and Digital Pathology
2.15. Statistical Analysis
3. Results
3.1. Derivation of Engineered CD11c+ DCs from CD34+ HSCs

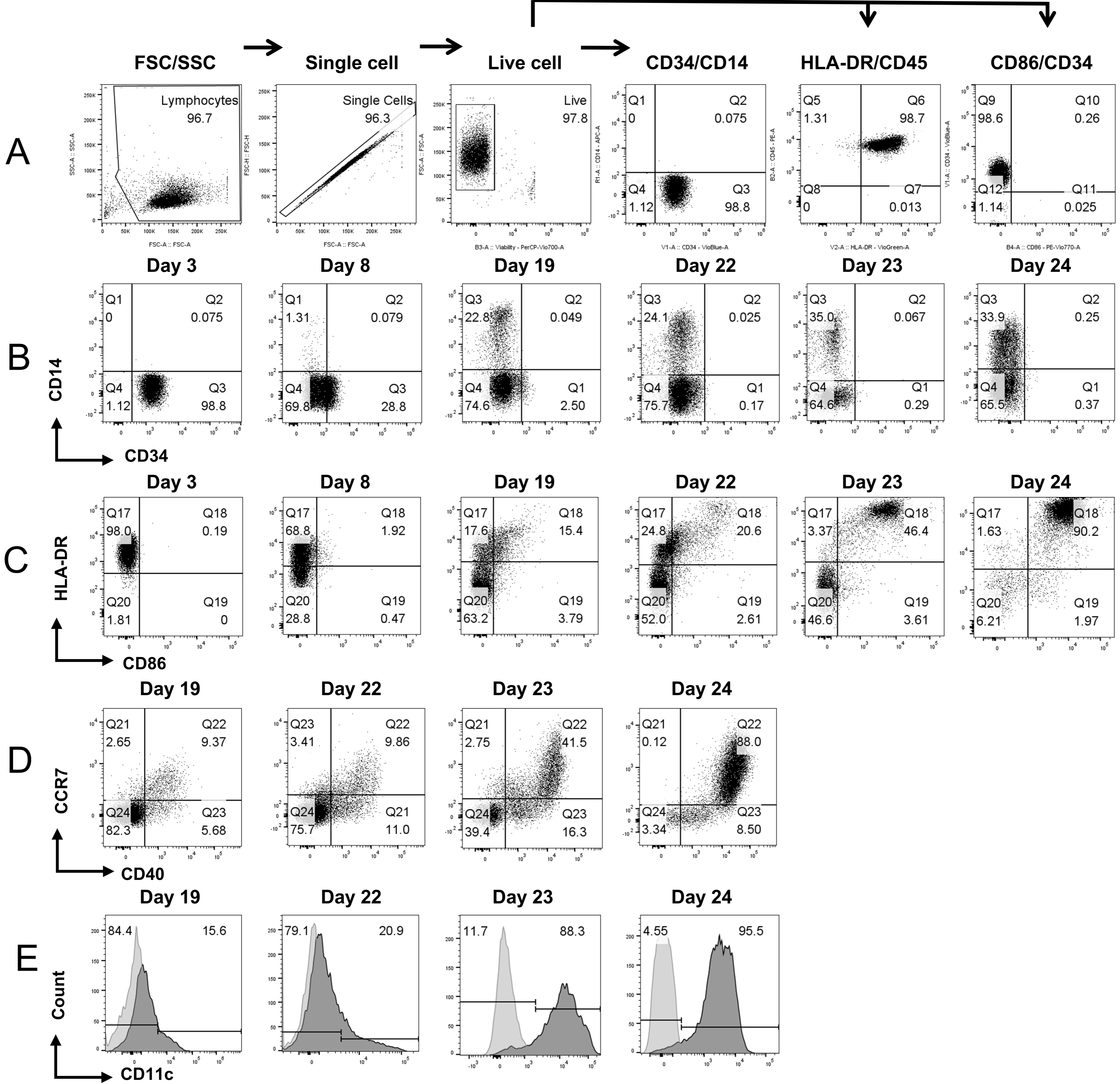
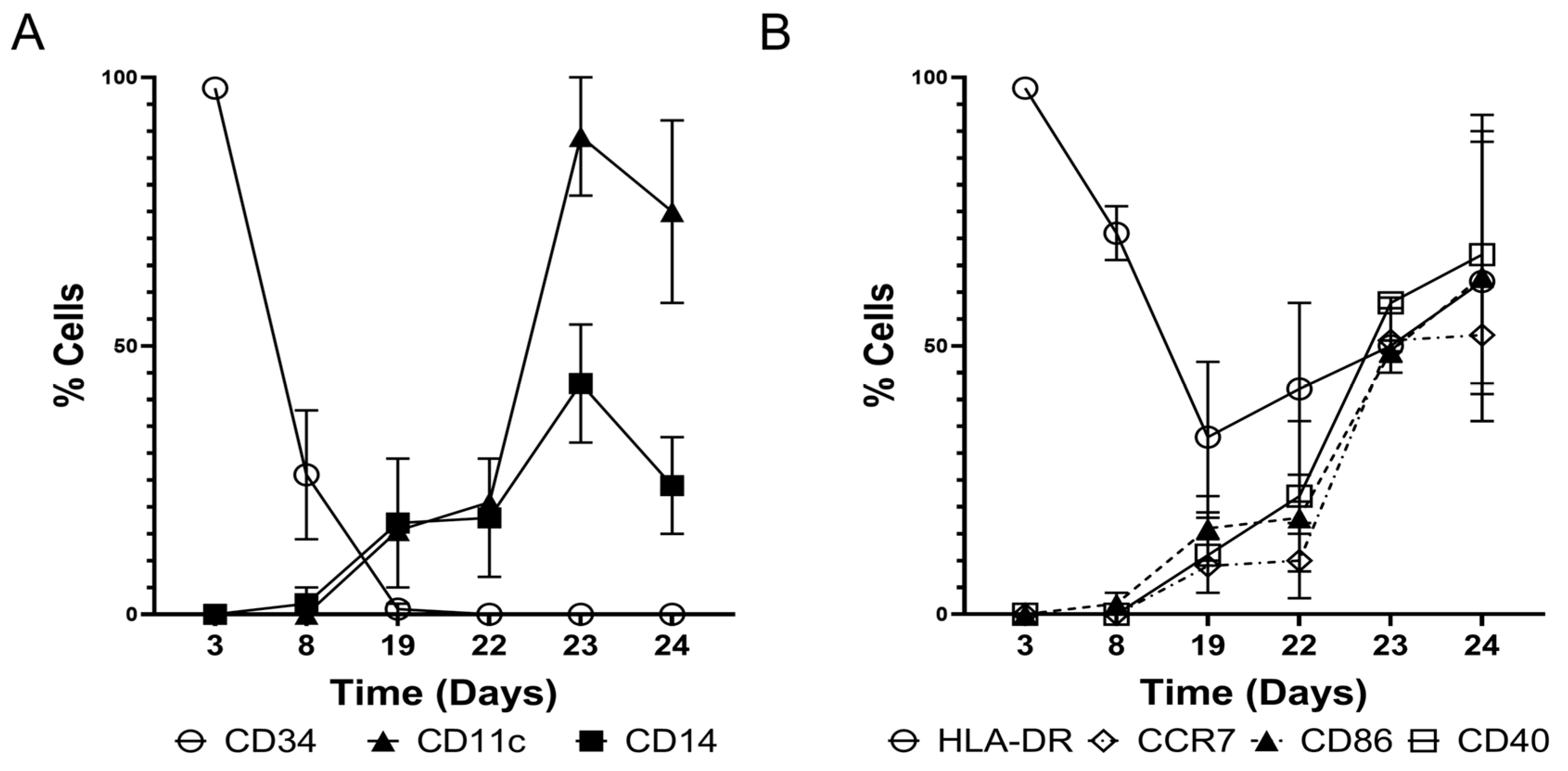
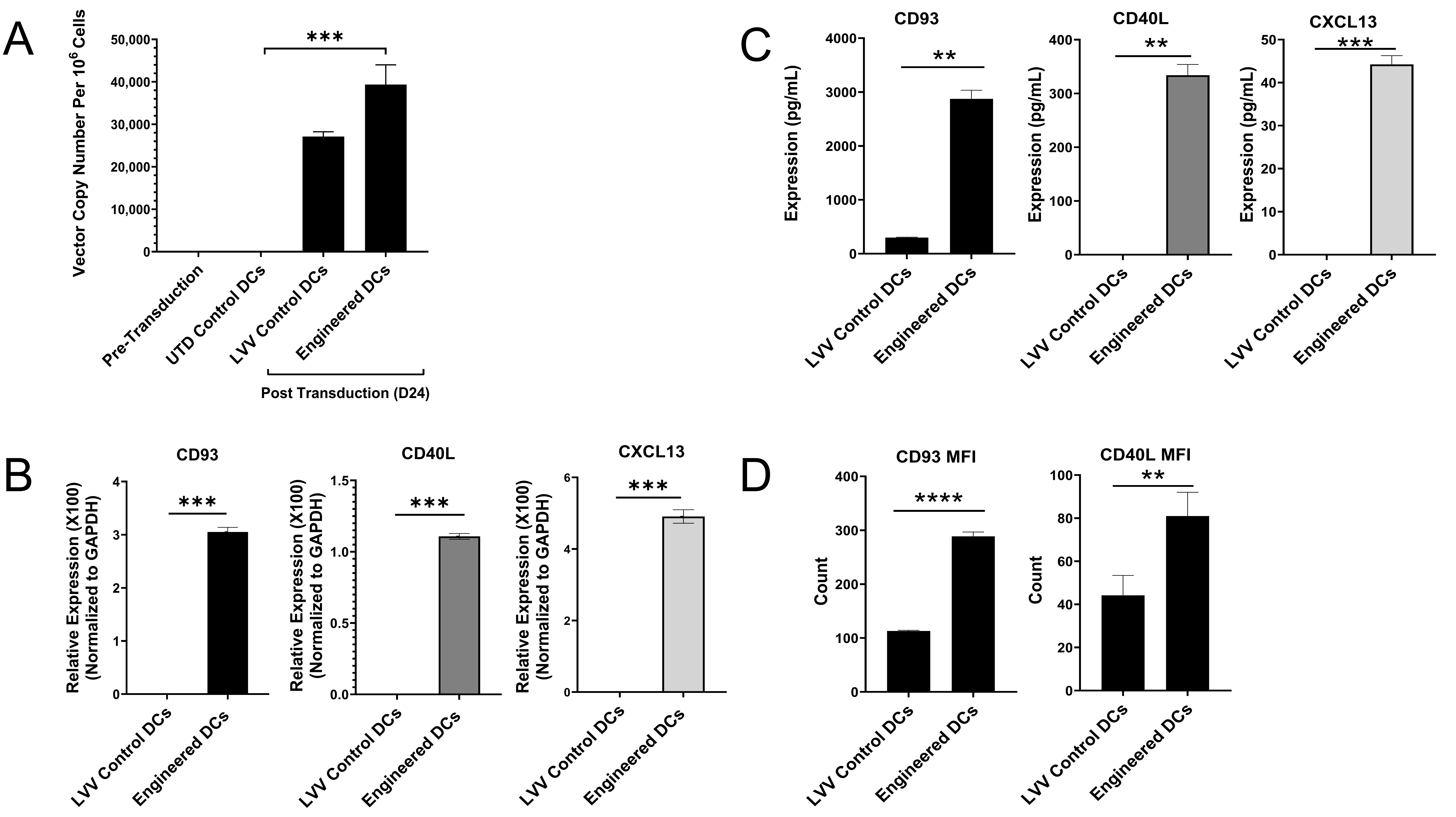
3.2. Engineered DCs Express CD93, CD40L, and CXCL13
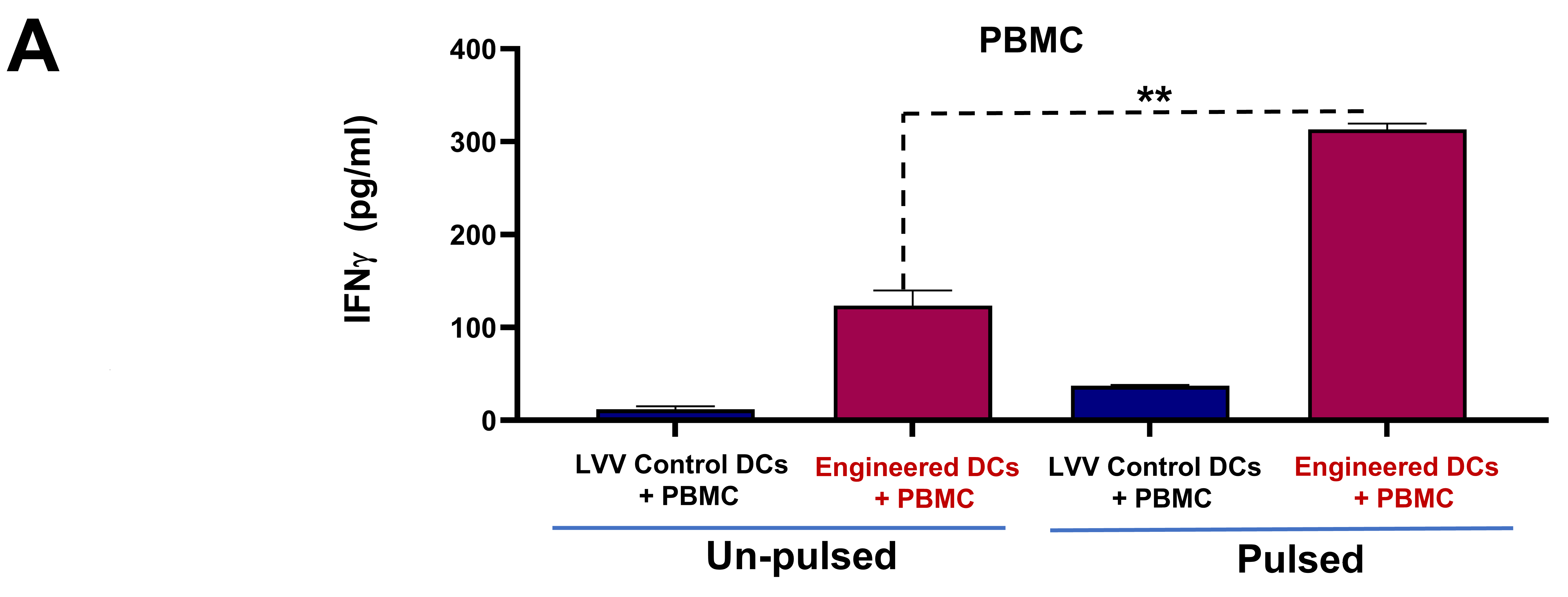
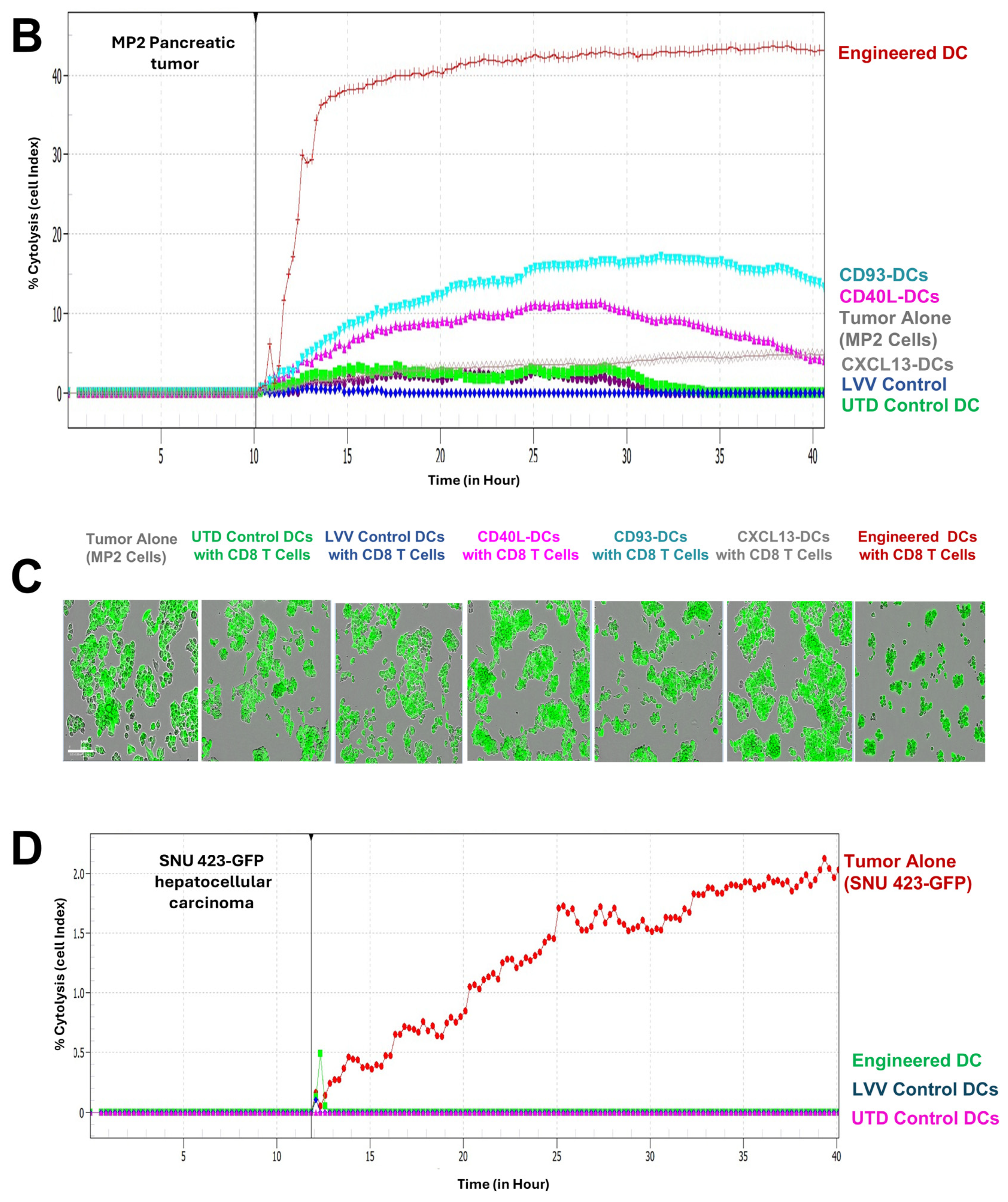
3.3. Engineered DCs Significantly Enhanced Effector Cell Activation In Vitro
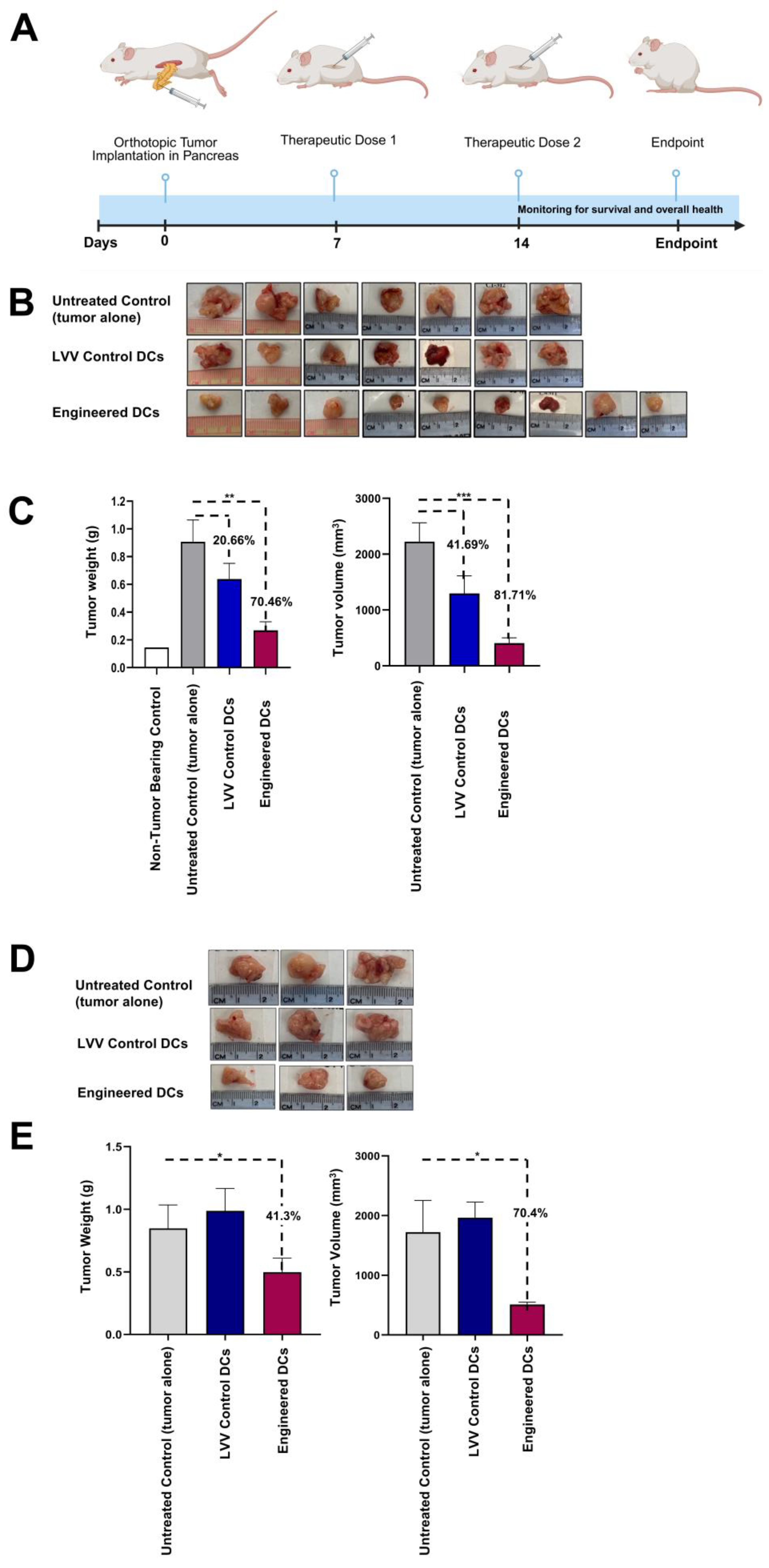
3.4. Treatment with Engineered DCs Leads to Pancreatic Tumor Regression in the hu-BLT Mouse Model
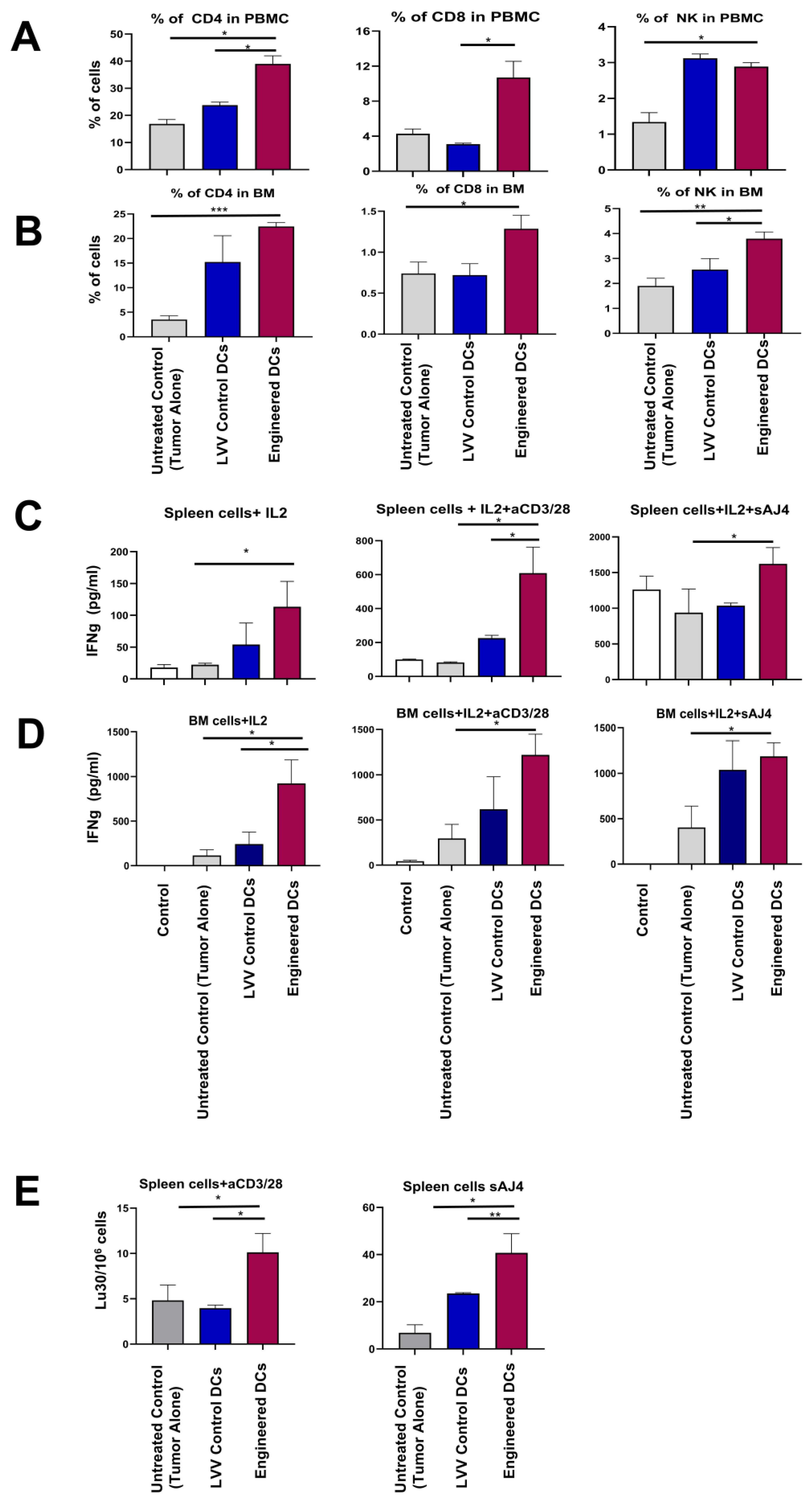
3.5. Engineered DCs Promote Tumor-Targeting Immune Responses In Vivo
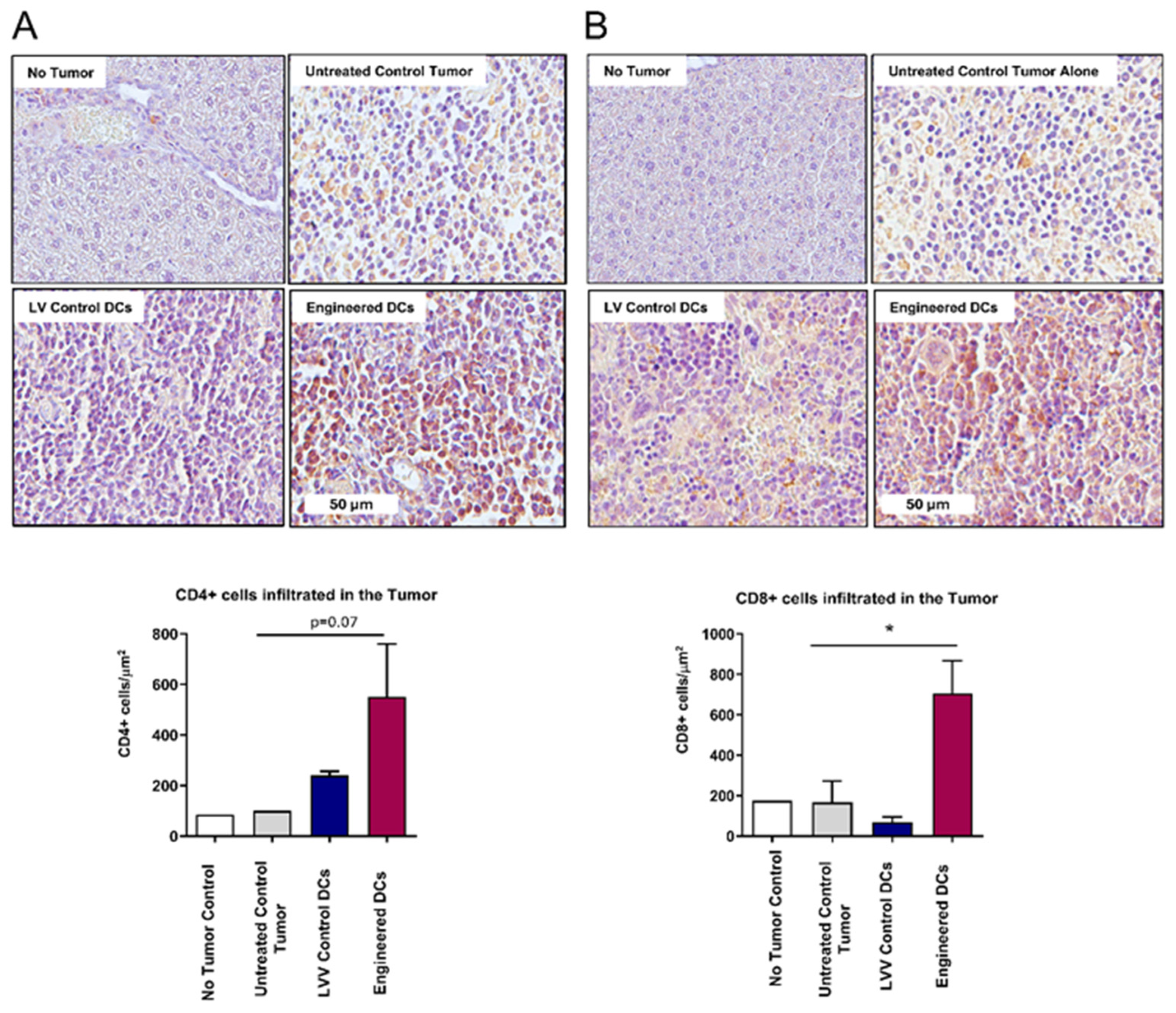
3.6. Enhanced Tumor Infiltration of CD4+ and CD8+ T Cells Following Antigen-Pulsed Engineered DC Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 51Cr | Chromium-51 |
| L | Ligand |
| APC | Antigen Presenting Cells |
| APC-Vio | allophycocyanin-Vio |
| ARC | Animal Research Committee |
| ATCC | American Type Culture Collection |
| BLT | Bone-Liver-Thymus |
| BM | Bone Marrow |
| CD | Cluster of Differentiation |
| CD10 | Cryostor 10 |
| CTL | Cytotoxic T Lymphocytes |
| CXCL-13 | Chemokine (C-X-C motif) ligand-13 |
| DCCT | Dendritic Cell Cancer Therapy |
| DMEM | Dulbecco’s Modified Essential Medium |
| FBS | Fetal Bovine Serum |
| FITC | fluorescein isothiocyanate |
| GEM | Genetically Engineered Mice |
| GFP | Green Fluorescent Protein |
| GMC-SF | Granulocyte Macrophage Colony Stimulating Factor |
| GMP | Good Manufacturing Practice |
| GVHD | Graft-versus-host disease |
| h | Hours |
| HSC | Hematopoietic Stem Cells |
| Hu- | Humanized |
| i.d. | Intradermal Injection |
| IFN-γ | Interferon-gamma |
| IL | Interleukin |
| IMDM | Iscove’s Modified Dulbeccos’ Medium |
| IRB/REB | Institutional Review Board/Research Ethics Board |
| imDC | Immature Dendritic Cells |
| LVV | Lentiviral Vector |
| LNP | Lipid Nanoparticles |
| LU | Lytic Units |
| min | Minutes |
| MC-SF | Macrophage Colony Stimulating Factor |
| mAbs | Monoclonal Antibodies |
| MDSCs | Myeloid-derived suppressor cells |
| mDC | Mature Dendritic Cells |
| MEM | Modified Essential Medium |
| MLR | Mixed Lymphocyte Reaction |
| MoDCs | Monocyte-Derived Dendritic Cells |
| MP2 | MiaPaca-2 |
| MUC1 | Mucin-1 |
| NK | Natural Killer |
| NSG | NOD SCID GAMMA |
| OSCSCs | Osteocarcinoma Cells |
| PC | Pancreatic Cancer |
| PBMC | Peripheral Blood Mononucleated Cells |
| PE | phycoerythrin |
| RTCA | Real-Time Cell Analysis |
| s | Seconds |
| SCF | Stem Cell Factor |
| SCGM | Stem Cell Growth Medium |
| TC | Tissue Culture |
| TDLN | Tumor-Draining Lymph Node |
| TME | Tumor Microenvironment |
| Tregs | T Regulatory Cells |
| UCLA | University of California, Los Angeles |
| UTD | Untransduced |
| VB | Vio blue |
| VCN | Vector Copy Number |
References
- Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E.; Malvezzi, M. European cancer mortality predictions for the year 2020 with a focus on prostate cancer. Ann. Oncol. 2020, 31, 650–658. [Google Scholar] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar]
- Matsumoto, I.; Murakami, Y.; Shinzeki, M.; Asari, S.; Goto, T.; Tani, M.; Motoi, F.; Uemura, K.; Sho, M.; Satoi, S.; et al. Proposed preoperative risk factors for early recurrence in patients with resectable pancreatic ductal adenocarcinoma after surgical resection: A multi-center retrospective study. Pancreatology 2015, 15, 674–680. [Google Scholar]
- Paniccia, A.; Hosokawa, P.; Henderson, W.; Schulick, R.D.; Edil, B.H.; McCarter, M.D.; Gajdos, C. Characteristics of 10-Year Survivors of Pancreatic Ductal Adenocarcinoma. JAMA Surg. 2015, 150, 701–710. [Google Scholar]
- Nishio, K.; Kimura, K.; Amano, R.; Yamazoe, S.; Ohrira, G.; Nakata, B.; Hirakawa, K.; Ohira, M. Preoperative predictors for early recurrence of resectable pancreatic cancer. World J. Surg. Oncol. 2017, 15, 16. [Google Scholar] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar]
- Truong, L.H.; Pauklin, S. Pancreatic Cancer Microenvironment and Cellular Composition: Current Understandings and Therapeutic Approaches. Cancers 2021, 13, 5028. [Google Scholar] [CrossRef]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [PubMed]
- Hegde, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020, 37, 289–307.e9. [Google Scholar]
- Kunk, P.R.; Bauer, T.W.; Slingluff, C.L.; Rahma, O.E. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J. Immunother. Cancer 2016, 4, 14. [Google Scholar]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar]
- Stromnes, I.M.; Burrack, A.L.; Hulbert, A.; Bonson, P.; Black, C.; Brockenbrough, J.S.; Raynor, J.F.; Spartz, E.J.; Pierce, R.H.; Greenberg, P.D.; et al. Differential Effects of Depleting versus Programming Tumor-Associated Macrophages on Engineered T Cells in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2019, 7, 977–989. [Google Scholar]
- Aerts, J.G.; de Goeje, P.L.; Cornelissen, R.; Kaijen-Lambers, M.E.; Bezemer, K.; van der Leest, C.H.; Mahaweni, N.M.; Kunert, A.; Eskens, F.A.; Waasdorp, C.; et al. Autologous Dendritic Cells Pulsed with Allogeneic Tumor Cell Lysate in Mesothelioma: From Mouse to Human. Clin. Cancer Res. 2018, 24, 766–776. [Google Scholar] [PubMed]
- Lepisto, A.J.; Moser, A.J.; Zeh, H.; Lee, K.; Bartlett, D.; McKolanis, J.R.; Geller, B.A.; Schmotzer, A.; Potter, D.P.; Whiteside, T.; et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008, 6, 955–964. [Google Scholar]
- Shindo, Y.; Hazama, S.; Maeda, Y.; Matsui, H.; Iida, M.; Suzuki, N.; Yoshimura, K.; Ueno, T.; Yoshino, S.; Sakai, K.; et al. Adoptive immunotherapy with MUC1-mRNA transfected dendritic cells and cytotoxic lymphocytes plus gemcitabine for unresectable pancreatic cancer. J. Transl. Med. 2014, 12, 175. [Google Scholar] [PubMed]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar]
- Hauber, I.; Beschorner, N.; Schrödel, S.; Chemnitz, J.; Kröger, N.; Hauber, J.; Thirion, C. Improving Lentiviral Transduction of CD34(+) Hematopoietic Stem and Progenitor Cells. Hum. Gene Ther. Methods 2018, 29, 104–113. [Google Scholar]
- Satoh, T.; Manel, N. Gene transduction in human monocyte-derived dendritic cells using lentiviral vectors. Methods Mol. Biol. 2013, 960, 401–409. [Google Scholar]
- Triozzi, P.L.; Aldrich, W. Phenotypic and functional differences between human dendritic cells derived in vitro from hematopoietic progenitors and from monocytes/macrophages. J. Leukoc. Biol. 1997, 61, 600–608. [Google Scholar]
- Tossetta, G.; Piani, F.; Borghi, C.; Marzioni, D. Role of CD93 in Health and Disease. Cells 2023, 12, 1778. [Google Scholar] [CrossRef]
- Barbera, S.; Raucci, L.; Lugano, R.; Tosi, G.M.; Dimberg, A.; Santucci, A.; Galvagni, F.; Orlandini, M. CD93 Signaling via Rho Proteins Drives Cytoskeletal Remodeling in Spreading Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 12417. [Google Scholar]
- Langenkamp, E.; Zhang, L.; Lugano, R.; Huang, H.; Abu Elhassan, T.E.; Georganaki, M.; Bazzar, W.; Lööf, J.; Trendelenburg, G.; Essand, M.; et al. Elevated expression of the C-type lectin CD93 in the glioblastoma vasculature regulates cytoskeletal rearrangements that enhance vessel function and reduce host survival. Cancer Res. 2015, 75, 4504–4516. [Google Scholar]
- Steinberger, P.; Szekeres, A.; Wille, S.; Stöckl, J.; Selenko, N.; Prager, E.; Staffler, G.; Madic, O.; Stockinger, H.; Knapp, W. Identification of human CD93 as the phagocytic C1q receptor (C1qRp) by expression cloning. J. Leukoc. Biol. 2002, 71, 133–140. [Google Scholar]
- Li, D.-K.; Wang, W. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies (Review). Oncol. Lett. 2020, 20, 176. [Google Scholar] [PubMed]
- Banchereau, J.; Bazan, F.; Blanchard, D.; Brière, F.; Galizzi, J.P.; van Kooten, C.; Liu, Y.J.; Rousset, F.; Saeland, S. The CD40 antigen and its ligand. Annu. Rev. Immunol. 1994, 12, 881–922. [Google Scholar] [PubMed]
- Johnson, S.; Zhan, Y.; Sutherland, R.M.; Mount, A.M.; Bedoui, S.; Brady, J.L.; Carrington, E.M.; Brown, L.E.; Belz, G.T.; Heath, W.R.; et al. Selected Toll-like receptor ligands and viruses promote helper-independent cytotoxic T cell priming by upregulating CD40L on dendritic cells. Immunity 2009, 30, 218–227. [Google Scholar]
- Salgado, C.G.; Nakamura, K.; Sugaya, M.; Tada, Y.; Asahina, A.; Koyama, Y.-I.; Irie, S.; Tamaki, K. Functional CD40 ligand is expressed on epidermal Langerhans cells. J. Leukoc. Biol. 1999, 66, 281–285. [Google Scholar] [PubMed]
- Korniotis, S.; Saichi, M.; Trichot, C.; Hoffmann, C.; Amblard, E.; Viguier, A.; Grondin, S.; Noel, F.; Mattoo, H.; Soumelis, V. GM-CSF-activated human dendritic cells promote type 1 T follicular helper cell polarization in a CD40-dependent manner. J. Cell Sci. 2022, 135, jcs260298. [Google Scholar]
- Peng, X.; Remacle, J.E.; Kasran, A.; Huylebroeck, D.; Ceuppens, J.L. IL-12 up-regulates CD40 ligand (CD154) expression on human T cells. J. Immunol. 1998, 160, 1166–1172. [Google Scholar]
- Gunn, M.D.; Ngo, V.N.; Ansel, K.M.; Ekland, E.H.; Cyster, J.G.; Williams, L.T. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature 1998, 391, 799–803. [Google Scholar]
- Förster, R.; Mattis, A.E.; Kremmer, E.; Wolf, E.; Brem, G.; Lipp, M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 1996, 87, 1037–1047. [Google Scholar] [PubMed]
- Gao, S.H.; Liu, S.Z.; Wang, G.Z.; Zhou, G.B. CXCL13 in Cancer and Other Diseases: Biological Functions, Clinical Significance, and Therapeutic Opportunities. Life 2021, 11, 1282. [Google Scholar] [CrossRef]
- Kumari, R.; Feuer, G.; Bourré, L. Humanized Mouse Models for Immuno-oncology Drug Discovery. Curr. Protoc. 2023, 3, e852. [Google Scholar]
- Mian, S.A.; Anjos-Afonso, F.; Bonnet, D. Advances in Human Immune System Mouse Models for Studying Human Hematopoiesis and Cancer Immunotherapy. Front. Immunol. 2020, 11, 619236. [Google Scholar]
- Liljenfeldt, L.; Gkirtzimanaki, K.; Vyrla, D.; Svensson, E.; Loskog, A.S.; Eliopoulos, A.G. Enhanced therapeutic anti-tumor immunity induced by co-administration of 5-fluorouracil and adenovirus expressing CD40 ligand. Cancer Immunol. Immunother. 2014, 63, 273–282. [Google Scholar] [PubMed]
- Herzog, G.I.; Solgi, G.; Wiegmann, D.S.; Nienhaus, C.; Schrezenmeier, H.; Yildiz, T.; Lotfi, R. Quality of tumor lysates used for pulsing dendritic cells is influenced by the method used to harvest adherent tumor cells. BMC Res. Notes 2011, 4, 153. [Google Scholar]
- Alaniz, L.; Rizzo, M.M.; Mazzolini, G. Pulsing dendritic cells with whole tumor cell lysates. Methods Mol. Biol. 2014, 1139, 27–31. [Google Scholar] [PubMed]
- Chiang, J.; Chen, P.-C.; Pham, J.; Nguyen, C.-Q.; Kaur, K.; Raman, S.S.; Jewett, A. Characterizing hepatocellular carcinoma stem markers and their corresponding susceptibility to NK-cell based immunotherapy. Front. Immunol. 2023, 14, 1284669. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar]
- Kaur, K.; Kozlowska, A.K.; Topchyan, P.; Ko, M.-W.; Ohanian, N.; Chiang, J.; Cook, J.; Maung, P.O.; Park, S.-H.; Cacalano, N.; et al. Probiotic-Treated Super-Charged NK Cells Efficiently Clear Poorly Differentiated Pancreatic Tumors in Hu-BLT Mice. Cancers 2019, 12, 63. [Google Scholar]
- Seyfizadeh, N.; Muthuswamy, R.; Mitchell, D.A.; Nierkens, S.; Seyfizadeh, N. Migration of dendritic cells to regional lymph nodes in a vaccination trial. Cancer Cell Int. 2004, 4, S15. [Google Scholar]
- Jewett, A.; Wang, M.Y.; Teruel, A.; Poupak, Z.; Bostanian, Z.; Park, N.H. Cytokine dependent inverse regulation of CD54 (ICAM1) and major histocompatibility complex class I antigens by nuclear factor kappaB in HEp2 tumor cell line: Effect on the function of natural killer cells. Hum. Immunol. 2003, 64, 505–520. [Google Scholar]
- Song, G.; Hsiao, H.; Wang, J.L.; Mannion, C.; Stojadinovic, A.; Avital, I.; Fu, S.W.; Mason, J.; Chen, W.; Jewett, A.; et al. Differential impact of tumor-infiltrating immune cells on basal and luminal cells: Implications for tumor invasion and metastasis. Anticancer. Res. 2014, 34, 6363–6380. [Google Scholar] [PubMed]
- Bazeed, A.Y.; Day, C.M.; Garg, S. Pancreatic Cancer: Challenges and Opportunities in Locoregional Therapies. Cancers 2022, 14, 4257. [Google Scholar] [CrossRef]
- Ghislat, G.; Lawrence, T. Autophagy in dendritic cells. Cell Mol. Immunol. 2018, 15, 944–952. [Google Scholar] [PubMed]
- Yu, J.; Sun, H.; Cao, W.; Song, Y.; Jiang, Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp. Hematol. Oncol. 2022, 11, 3. [Google Scholar] [PubMed]
- Uchida, N.; Nassehi, T.; Drysdale, C.M.; Gamer, J.; Yapundich, M.; Demirci, S.; Haro-Mora, J.J.; Leonard, A.; Hsieh, M.M.; Tisdale, J.F. High-Efficiency Lentiviral Transduction of Human CD34(+) Cells in High-Density Culture with Poloxamer and Prostaglandin E2. Mol. Ther. Methods Clin. Dev. 2019, 13, 187–196. [Google Scholar]
- Ferlazzo, G.; Wesa, A.; Wei, W.-Z.; Galy, A. Dendritic Cells Generated Either from CD34+ Progenitor Cells or from Monocytes Differ in Their Ability to Activate Antigen-Specific CD8+ T Cells1. J. Immunol. 1999, 163, 3597–3604. [Google Scholar]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010, 328, 749–752. [Google Scholar]
- Balan, S.; Arnold-Schrauf, C.; Abbas, A.; Couespel, N.; Savoret, J.; Imperatore, F.; Villani, A.-C.; Manh, T.-P.V.; Bhardwaj, N.; Dalod, M. Large-Scale Human Dendritic Cell Differentiation Revealing Notch-Dependent Lineage Bifurcation and Heterogeneity. Cell Rep. 2018, 24, 1902–1915.e6. [Google Scholar] [PubMed]
- Kirkling, M.E.; Cytlak, U.; Lau, C.M.; Lewis, K.L.; Resteu, A.; Khodadadi-Jamayran, A.; Siebel, C.W.; Salmon, H.; Merad, M.; Tsirigos, A.; et al. Notch Signaling Facilitates In Vitro Generation of Cross-Presenting Classical Dendritic Cells. Cell Rep. 2018, 23, 3658–3672.e6. [Google Scholar]
- Guo, A.; Zhang, J.; Tian, Y.; Peng, Y.; Luo, P.; Zhang, J.; Liu, Z.; Wu, W.; Zhang, H.; Cheng, Q. Identify the immune characteristics and immunotherapy value of CD93 in the pan-cancer based on the public data sets. Front. Immunol. 2022, 13, 907182. [Google Scholar]
- Luke, G.A.; Ryan, M.D. Using the 2A Protein Coexpression System: Multicistronic 2A Vectors Expressing Gene(s) of Interest and Reporter Proteins. Methods Mol. Biol. 2018, 1755, 31–48. [Google Scholar]
- Liu, Z.; Chen, O.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Vaseghi, H.R.; Qian, L.; Liu, J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7, 2193. [Google Scholar]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [PubMed]
- Ridge, J.P.; Di Rosa, F.; Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998, 393, 474–478. [Google Scholar]
- Hughes, P.D.; Belz, G.T.; Fortner, K.A.; Budd, R.C.; Strasser, A.; Bouillet, P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity 2008, 28, 197–205. [Google Scholar]
- Ansel, K.M.; Ngo, V.N.; Hyman, P.L.; Luther, S.A.; Förster, R.; Sedgwick, J.D.; Browning, J.L.; Lipp, M.; Cyster, J.G. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 2000, 406, 309–314. [Google Scholar]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [PubMed]
- Chuprin, J.; Buettner, H.; Seedhom, M.O.; Greiner, D.L.; Keck, J.G.; Ishikawa, F.; Shultz, L.D.; Brehm, M.A. Humanized mouse models for immuno-oncology research. Nat. Rev. Clin. Oncol. 2023, 20, 192–206. [Google Scholar]
- Ma, Q.; Chen, Y.; Qin, Q.; Guo, F.; Wang, Y.S.; Li, D. CXCL13 expression in mouse 4T1 breast cancer microenvironment elicits antitumor immune response by regulating immune cell infiltration. Precis. Clin. Med. 2021, 4, 155–167. [Google Scholar]
- Sun, Y.; Chen, W.; Torphy, R.J.; Yao, S.; Zhu, G.; Lin, R.; Lugano, R.; Miller, E.N.; Fujiwara, Y.; Bian, L.; et al. Blockade of the CD93 pathway normalizes tumor vasculature to facilitate drug delivery and immunotherapy. Sci. Transl. Med. 2021, 13, eabc8922. [Google Scholar]
- Kim, J.T.; Bresson-Tan, G.; Zack, J.A. Current Advances in Humanized Mouse Models for Studying NK Cells and HIV Infection. Microorganisms 2023, 11, 1984. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, M.B.; Vrbanac, V.; Tivey, T.; Tsang, K.; Tager, A.M.; Aliprantis, A.O. Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. PLoS ONE 2012, 7, e44664. [Google Scholar]
- Fransen, M.F.; Schoonderwoerd, M.; Knopf, P.; Camps, M.G.; Hawinkels, L.J.; Kneilling, M.; van Hall, T.; Ossendorp, F. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight 2018, 3, e124507. [Google Scholar] [PubMed]
- Lucarini, V.; Ziccheddu, G.; Macchia, I.; La Sorsa, V.; Peschiaroli, F.; Buccione, C.; Sistigu, A.; Sanchez, M.; Andreone, S.; D’URso, M.T.; et al. IL-33 restricts tumor growth and inhibits pulmonary metastasis in melanoma-bearing mice through eosinophils. Oncoimmunology 2017, 6, e1317420. [Google Scholar]
- Fu, C.; Zhou, L.; Mi, Q.S.; Jiang, A. DC-Based Vaccines for Cancer Immunotherapy. Vaccines 2020, 8, 706. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [PubMed]
- Van Gool, S.W.; Makalowski, J.; Kampers, L.F.C.; Van de Vliet, P.; Sprenger, T.; Schirrmacher, V.; Stücker, W. Dendritic cell vaccination for glioblastoma multiforme patients: Has a new milestone been reached? Transl. Cancer Res. 2023, 12, 2224–2228. [Google Scholar] [PubMed]
- Lee, K.-W.; Yam, J.W.P.; Mao, X. Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations. Cells 2023, 12, 2147. [Google Scholar] [CrossRef] [PubMed]
- Hannani, D.; Leplus, E.; Laulagnier, K.; Chaperot, L.; Plumas, J. Leveraging a powerful allogeneic dendritic cell line towards neoantigen-based cancer vaccines. Genes. Cancer 2023, 14, 3–11. [Google Scholar]
- van Gulijk, M.; Dammeijer, F.; Aerts, J.; Vroman, H. Combination Strategies to Optimize Efficacy of Dendritic Cell-Based Immunotherapy. Front. Immunol. 2018, 9, 2759. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huerta-Yepez, S.; Gonzalez, J.D.; Sheik, N.; Beraki, S.; Kathirvel, E.; Rodriguez-Frandsen, A.; Chen, P.-C.; Sargsyan, T.; Mahammad, S.; Dybul, M.R.; et al. Therapeutic Efficacy of CD34-Derived Allogeneic Dendritic Cells Engineered to Express CD93, CD40L, and CXCL13 in Humanized Mouse Models of Pancreatic Cancer. Vaccines 2025, 13, 749. https://doi.org/10.3390/vaccines13070749
Huerta-Yepez S, Gonzalez JD, Sheik N, Beraki S, Kathirvel E, Rodriguez-Frandsen A, Chen P-C, Sargsyan T, Mahammad S, Dybul MR, et al. Therapeutic Efficacy of CD34-Derived Allogeneic Dendritic Cells Engineered to Express CD93, CD40L, and CXCL13 in Humanized Mouse Models of Pancreatic Cancer. Vaccines. 2025; 13(7):749. https://doi.org/10.3390/vaccines13070749
Chicago/Turabian StyleHuerta-Yepez, Sara, Jose D. Gonzalez, Neha Sheik, Senay Beraki, Elango Kathirvel, Ariel Rodriguez-Frandsen, Po-Chun Chen, Tiran Sargsyan, Saleemulla Mahammad, Mark R. Dybul, and et al. 2025. "Therapeutic Efficacy of CD34-Derived Allogeneic Dendritic Cells Engineered to Express CD93, CD40L, and CXCL13 in Humanized Mouse Models of Pancreatic Cancer" Vaccines 13, no. 7: 749. https://doi.org/10.3390/vaccines13070749
APA StyleHuerta-Yepez, S., Gonzalez, J. D., Sheik, N., Beraki, S., Kathirvel, E., Rodriguez-Frandsen, A., Chen, P.-C., Sargsyan, T., Mahammad, S., Dybul, M. R., Chen, L., Binette, F., & Jewett, A. (2025). Therapeutic Efficacy of CD34-Derived Allogeneic Dendritic Cells Engineered to Express CD93, CD40L, and CXCL13 in Humanized Mouse Models of Pancreatic Cancer. Vaccines, 13(7), 749. https://doi.org/10.3390/vaccines13070749