
FcRn-Driven Nanoengineered Mucosal Vaccine with Multi-Epitope Fusion Induces Robust Dual Immunity and Long-Term Protection Against Brucella


Abstract

1. Introduction
2. Materials and Methods
2.1. Design and Analysis of Bioinformatics
2.1.1. Physicochemical Property Analysis
2.1.2. Homology Assessment
2.1.3. Antigenicity Evaluation
2.1.4. Allergenicity Screening
2.1.5. Transmembrane Topology Prediction
2.1.6. Signal Peptide Identification
2.1.7. Toxicity Profiling
2.1.8. T Cell Epitope Prediction
2.1.9. B Cell Epitope Characterization
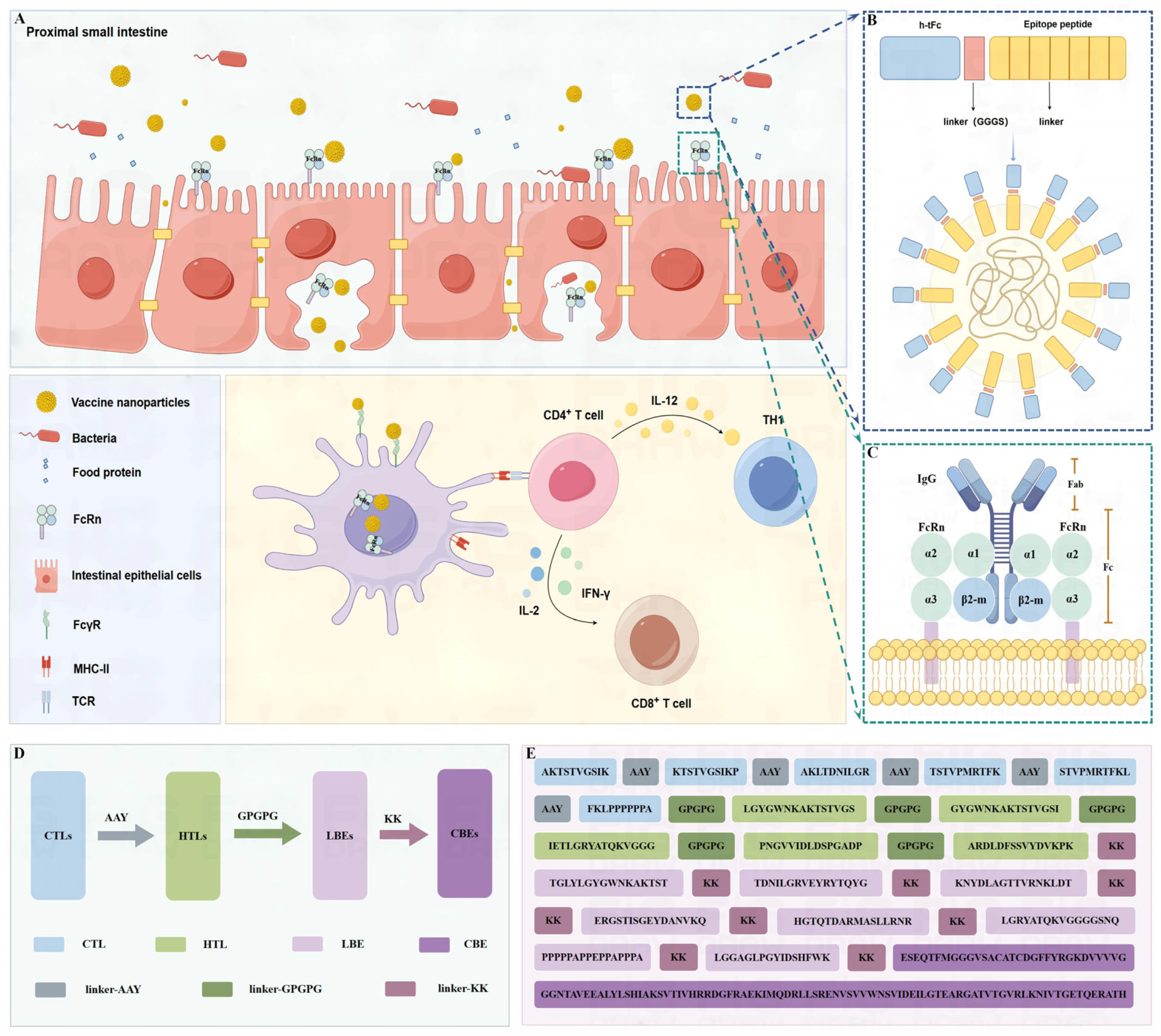
2.1.10. Multi-Epitope Vaccine Construction
2.1.11. Solubility Profiling
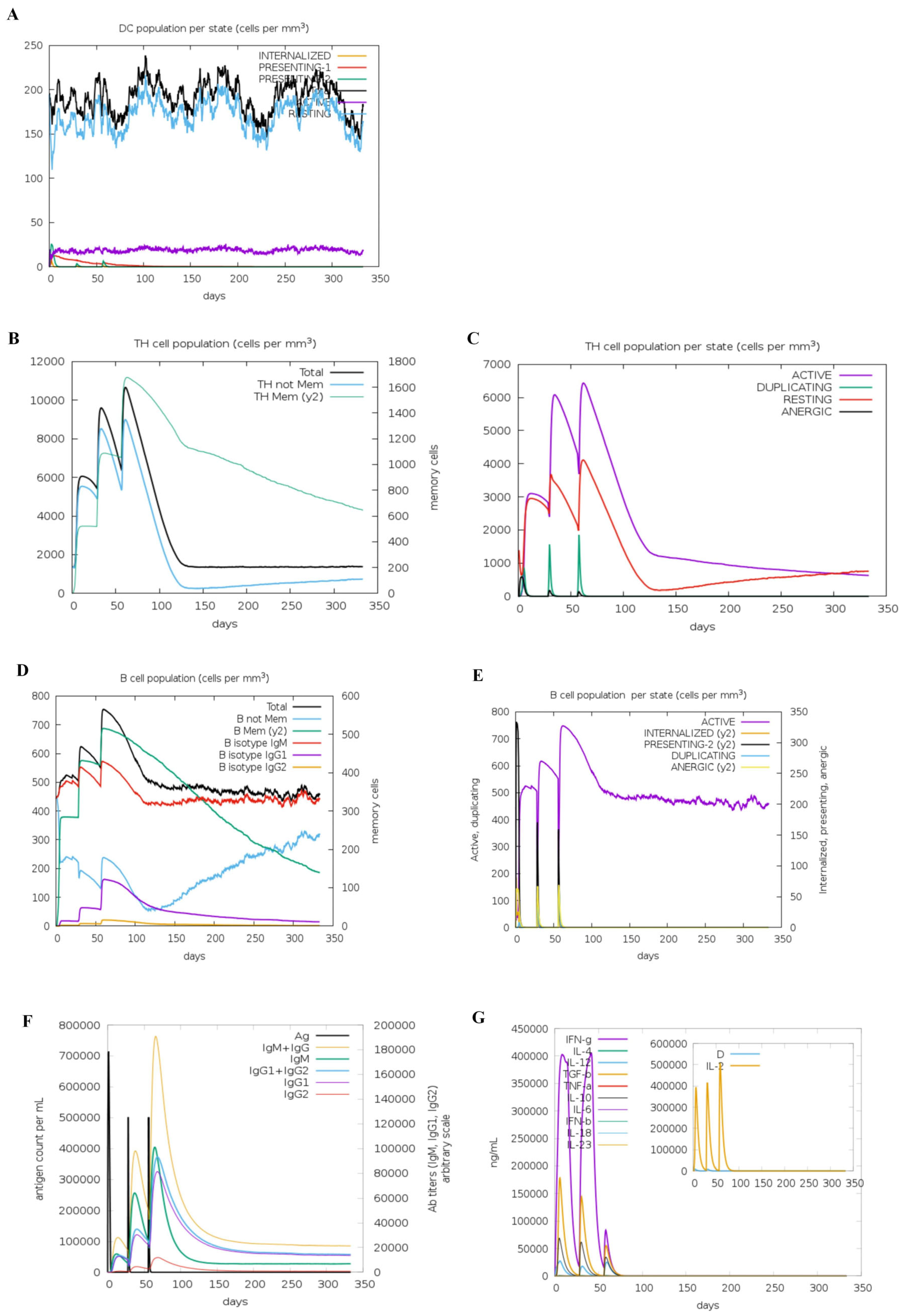
2.1.12. Immunostimulation Simulation
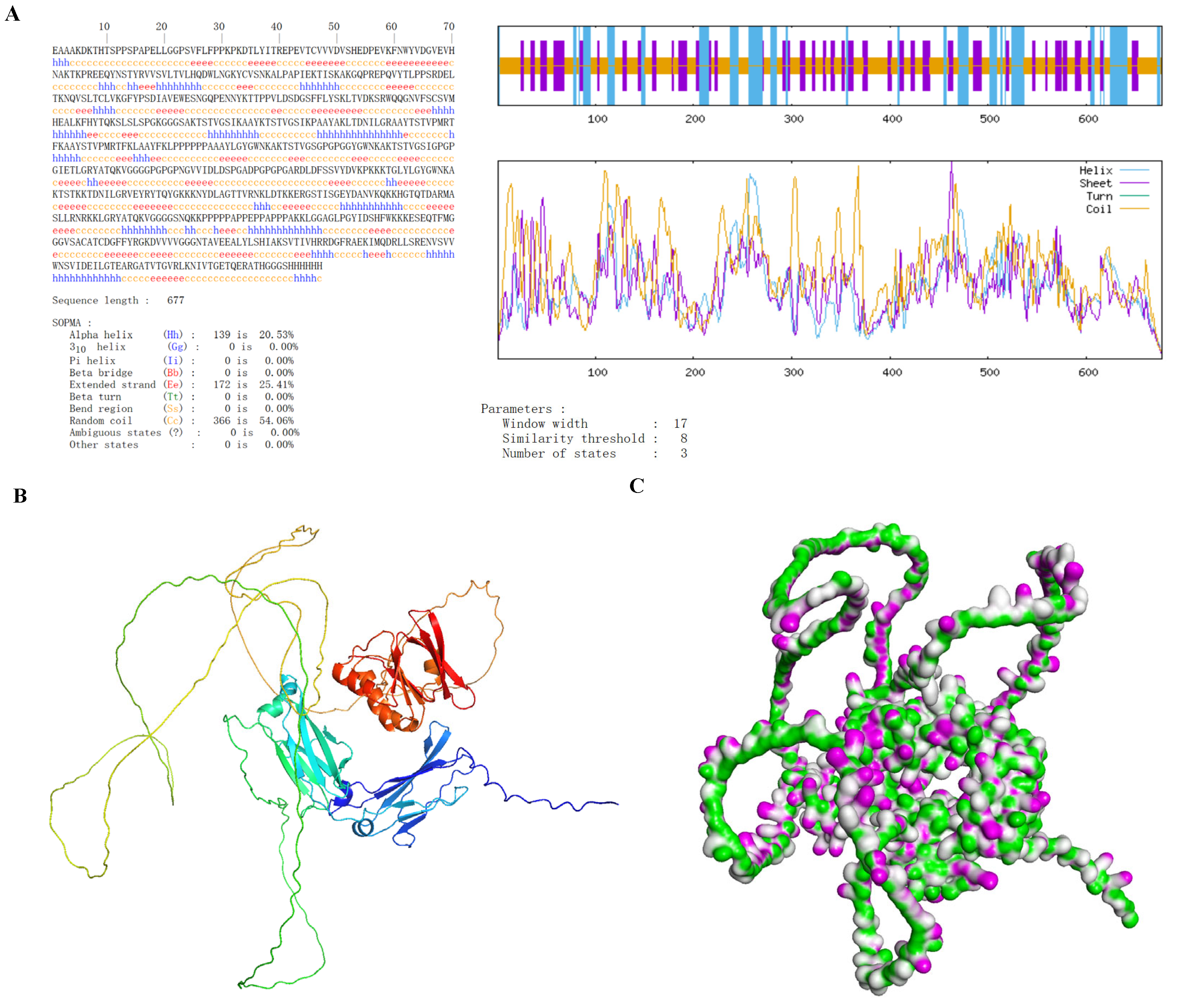
2.1.13. Structural Prediction and Modeling
2.1.14. Structure Refinement and Validation
2.1.15. Molecular Docking and Dynamics Analysis
2.2. In Vivo and In Vitro Experiment
2.2.1. Construction of Recombinant Plasmid
2.2.2. Protein Expression and Purification
2.2.3. Western Blot Analysis
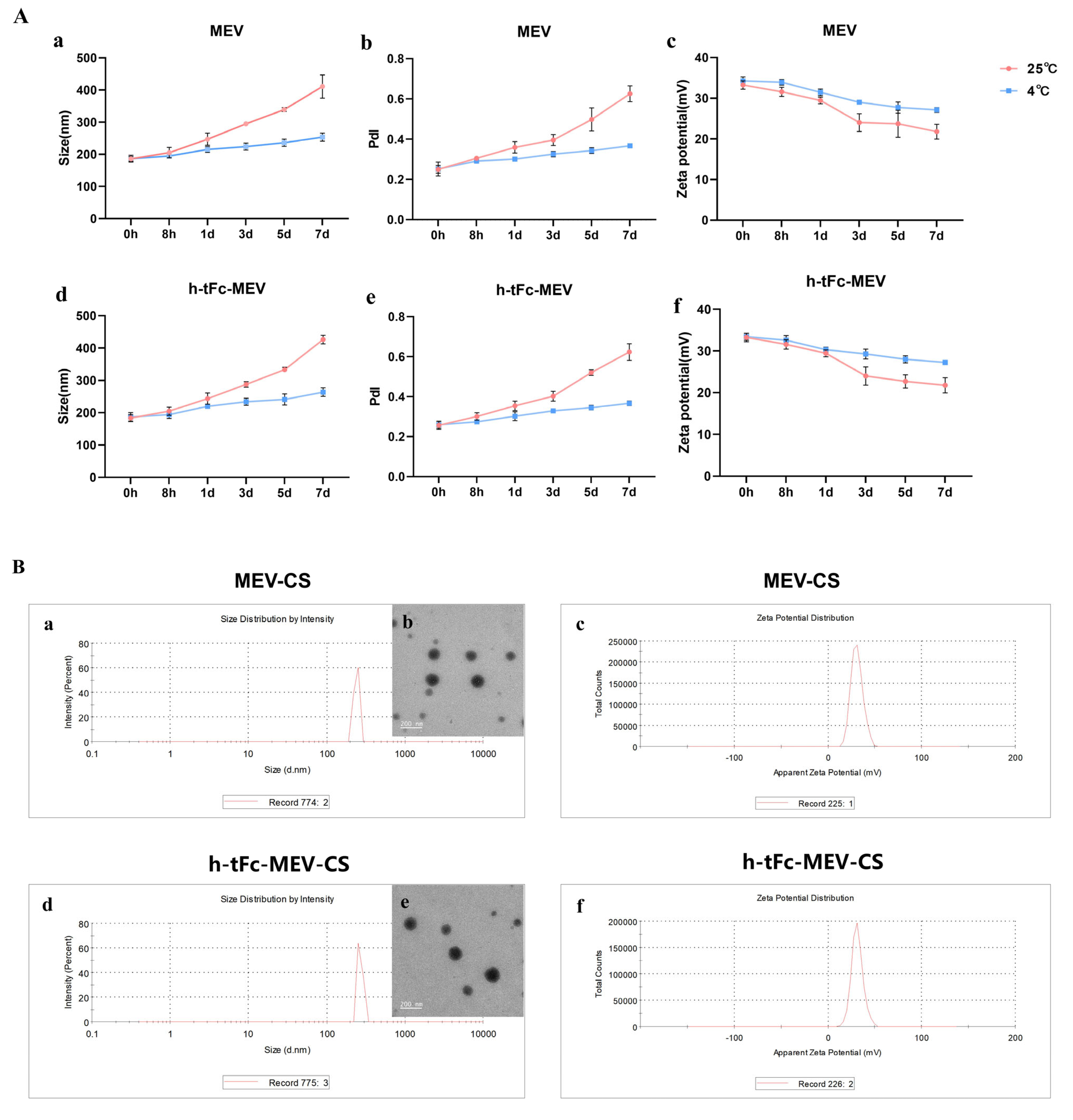
2.2.4. Preparation and Measurement of Chitosan Nanoparticles
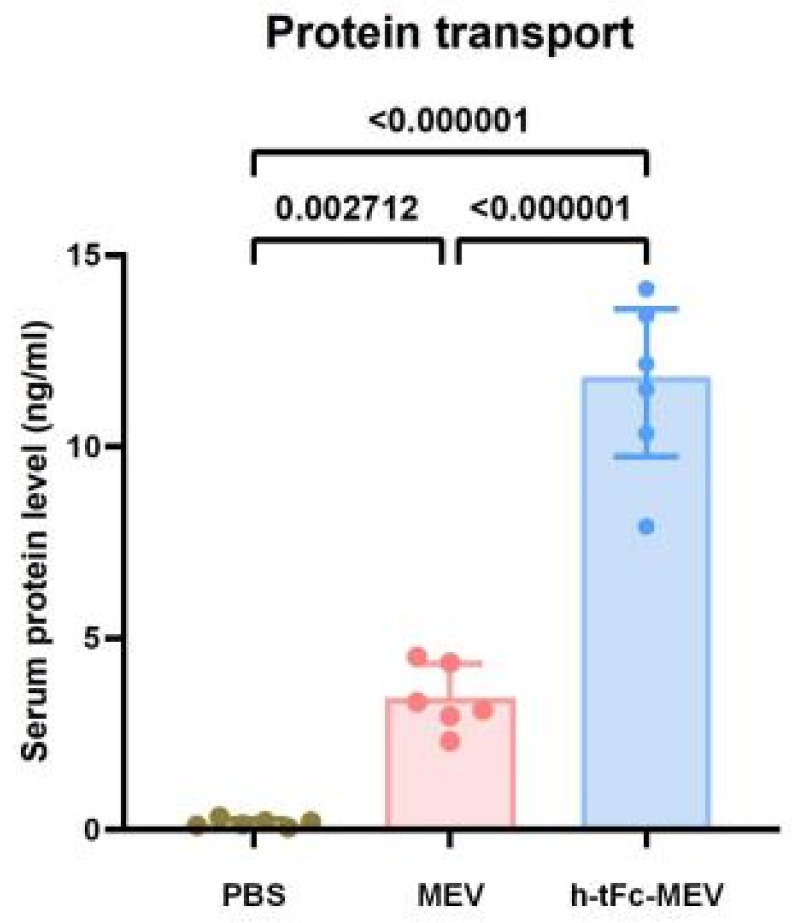
2.2.5. Protein Transport In Vivo
2.2.6. Animal Immunization
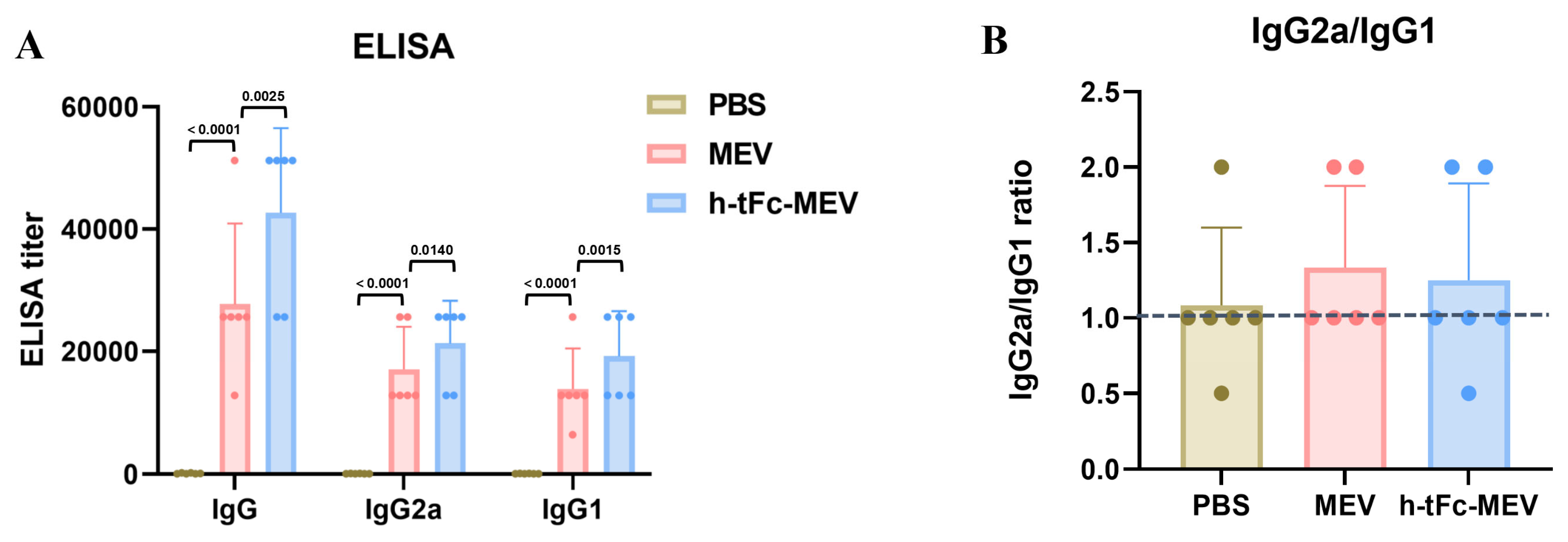
2.2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
Sample Collection and Preparation
Mucosal Lavage Fluids
2.2.8. Flow Cytometry
| The Protocol of Flow Cytometry | |
| IFN-γ | CD45-APC-cy7 (BD Biosciences 557659, Franklin Lakes, NJ, USA); CD3-APC (Biolegend 100236, San Diego, CA, USA); CD4-FITC (Biolegend 100406, San Diego, CA, USA); CD8-APC-Cy7 (BD Biosciences 557654, Franklin Lakes, NJ, USA); IFN-γ-V500 (BD Biosciences 561980, Franklin Lakes, NJ, USA) |
| IL-4 | CD45-APC-cy7 (BD Biosciences 557659, Franklin Lakes, NJ, USA); CD3-APC (Biolegend 100236, San Diego, CA, USA); CD4-FITC (Biolegend 100406, San Diego, CA, USA); CD8-APC-Cy7 (BD Biosciences 557654, Franklin Lakes, NJ, USA); IL-4-V450 (BD Biosciences 560701, Franklin Lakes, NJ, USA) |
| DC | Live/dead-Bv421 (Thermo Fisher Scientific L34955, Waltham, MA, USA); CD45-APC-cy7 (BD Biosciences 557659, Franklin Lakes, NJ, USA); Lin-APC (BioLegend 133306, San Diego, CA, USA); CD11c-PE-cy7 (BD Biosciences 558079, Franklin Lakes, NJ, USA); MHC-II-FITC (Biolegend 116406, San Diego, CA, USA) |
| TCM | CD3-APC (Biolegend 100236, San Diego, CA, USA); CD4-FITC (Biolegend 100406, San Diego, CA, USA); CD8-APC-Cy7 (BD Biosciences 557654, Franklin Lakes, NJ, USA); CD44-PerCP-Cy5.5 (BD Biosciences 1278947, Franklin Lakes, NJ, USA); CD62L-PE-Cy7 (BD Biosciences 1175634, Franklin Lakes, NJ, USA) |
| TFH | CD45-APC-cy7 (BD Biosciences 557659, Franklin Lakes, NJ, USA); CD3-APC (Biolegend 100236, San Diego, CA, USA); CD4-FITC (Biolegend 100406, San Diego, CA, USA); CXCR5-PE (BD Biosciences 1165377, Franklin Lakes, NJ, USA); PD-1-Bv605 (BD Biosciences 748267, Franklin Lakes, NJ, USA) |
| GCB | CD45-APC-cy7 (BD Biosciences 557659, Franklin Lakes, NJ, USA); CD3-APC (Biolegend 100236, San Diego, CA, USA); B220-FITC (Biolegend 103205, San Diego, CA, USA); GL7-PE (Biolegend 144607, San Diego, CA, USA); FAS-Bv711 (BD Biosciences 740716, Franklin Lakes, NJ, USA) |
2.2.9. Immunofluorescence Staining
2.2.10. Evaluation of Protective Immunity
3. Results
3.1. Physicochemical Properties and Immunological Characteristics of Target Proteins
3.2. Screening of Target Protein Sequences
3.3. Vaccine Design and Structural Assembly
3.4. Physicochemical Properties and Immunological Characteristics
3.5. Immune Simulation
3.6. Secondary and Tertiary Structure Prediction of Vaccines
3.7. Molecular Interaction Mechanism of h-tFc-MEV/FcRn Complex
3.8. Western Blot Analysis of MEV and h-tFc-MEV
3.9. Engineering a Chitosan-Based Nanoparticle System for Enhanced Mucosal Vaccine Delivery
3.10. Enhanced Mucosal Penetration of h-tFc-MEV in Oral Delivery Models
3.11. Enhanced Humoral Immunity with Optimized Th1/Th2 Profiling
3.12. FcRn Mediated Subunit Vaccine Strategy Significantly Improve Antigen-Presenting Effect
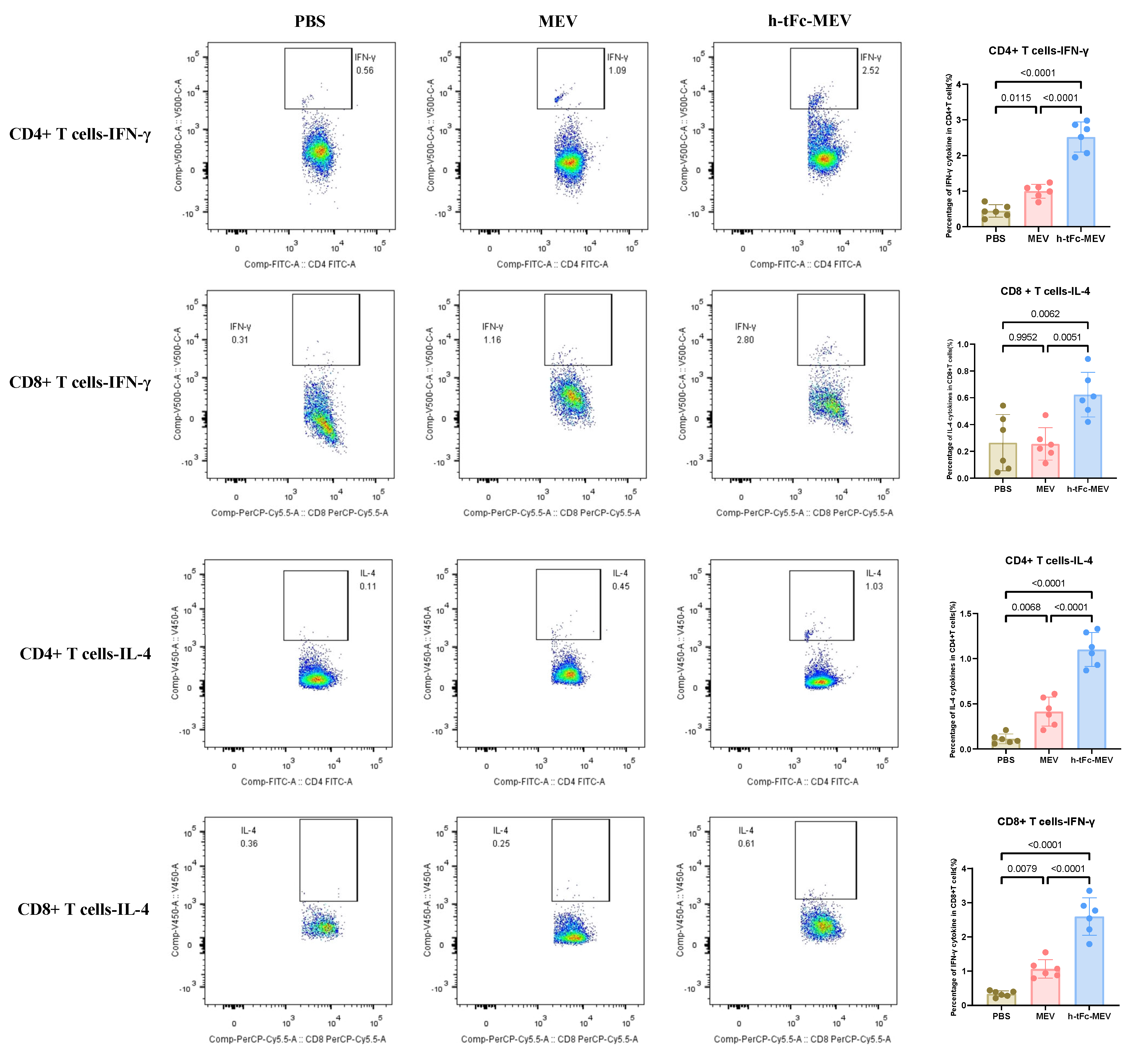
3.13. h-tFc-MEV Elicits Pathogen-Specific T-Cell Immunity
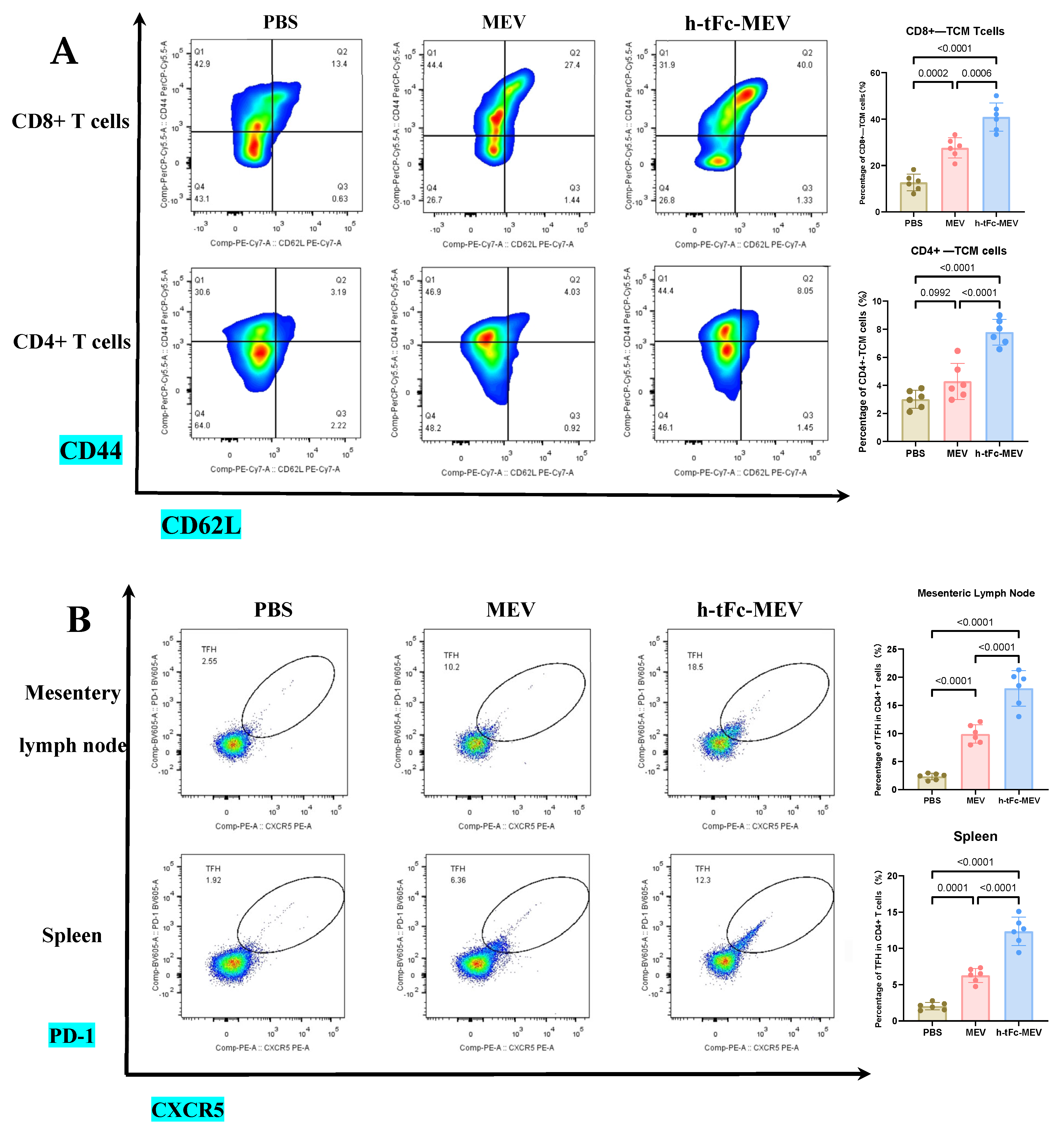
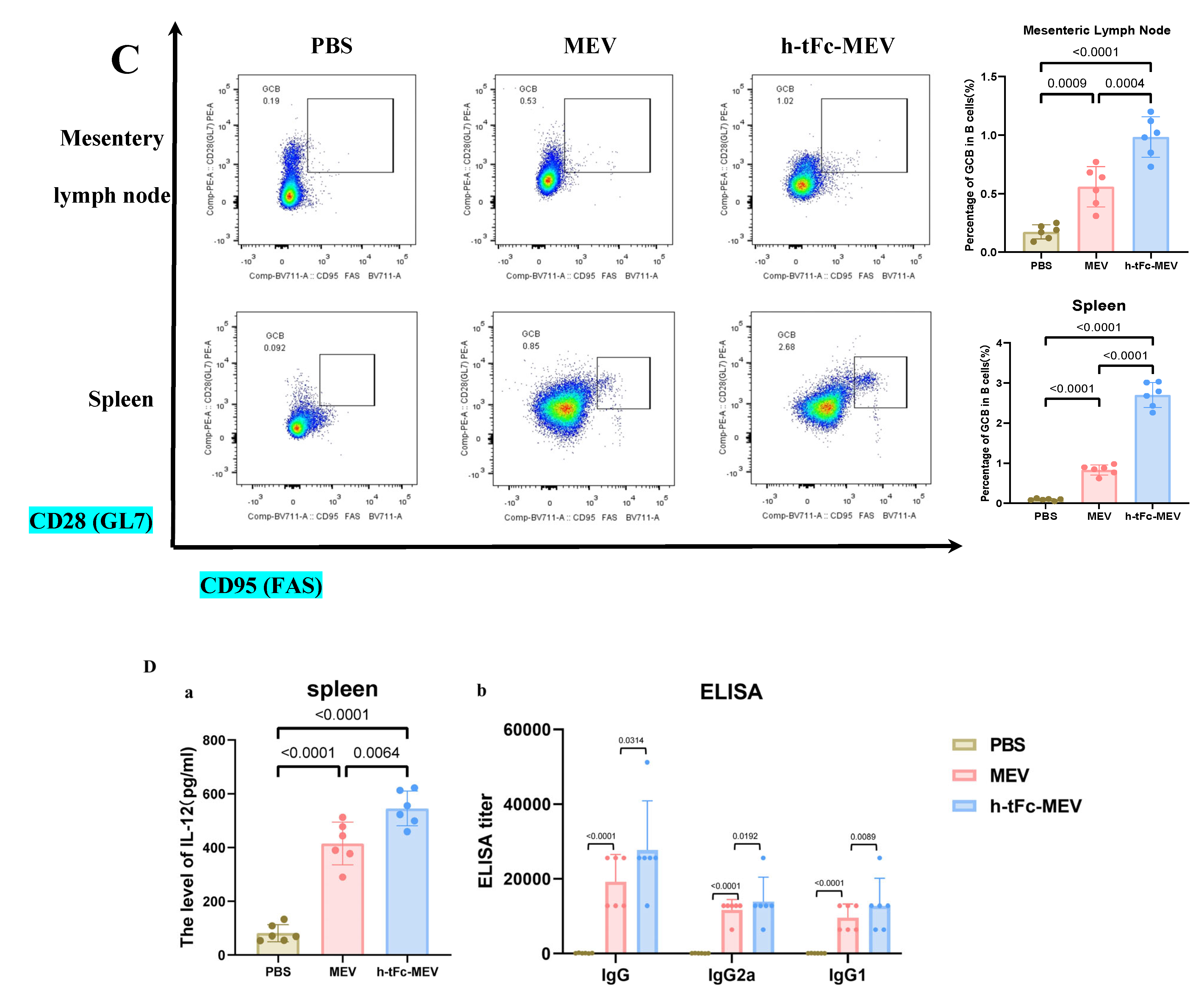
3.14. FcRn-Targeted Strategy Enhances Long-Term Immunological Memory Induced by h-tFc-MEV Vaccine: Pivotal Roles of Central Memory T Cells and Germinal Center Activity
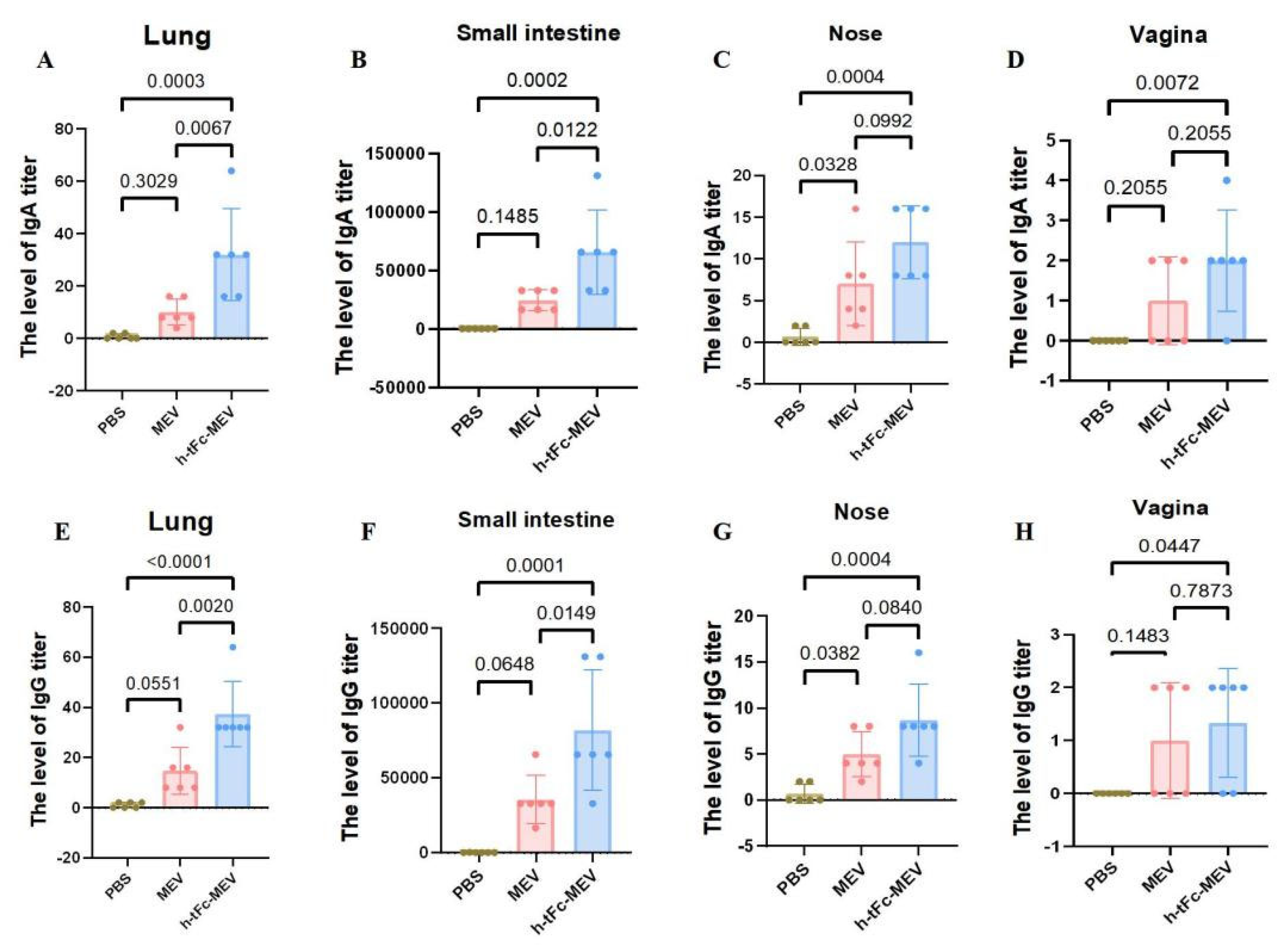
3.15. Mucosal Immune Responses Induced by h-tFc-MEV Vaccine via FcRn-Targeting Strategy
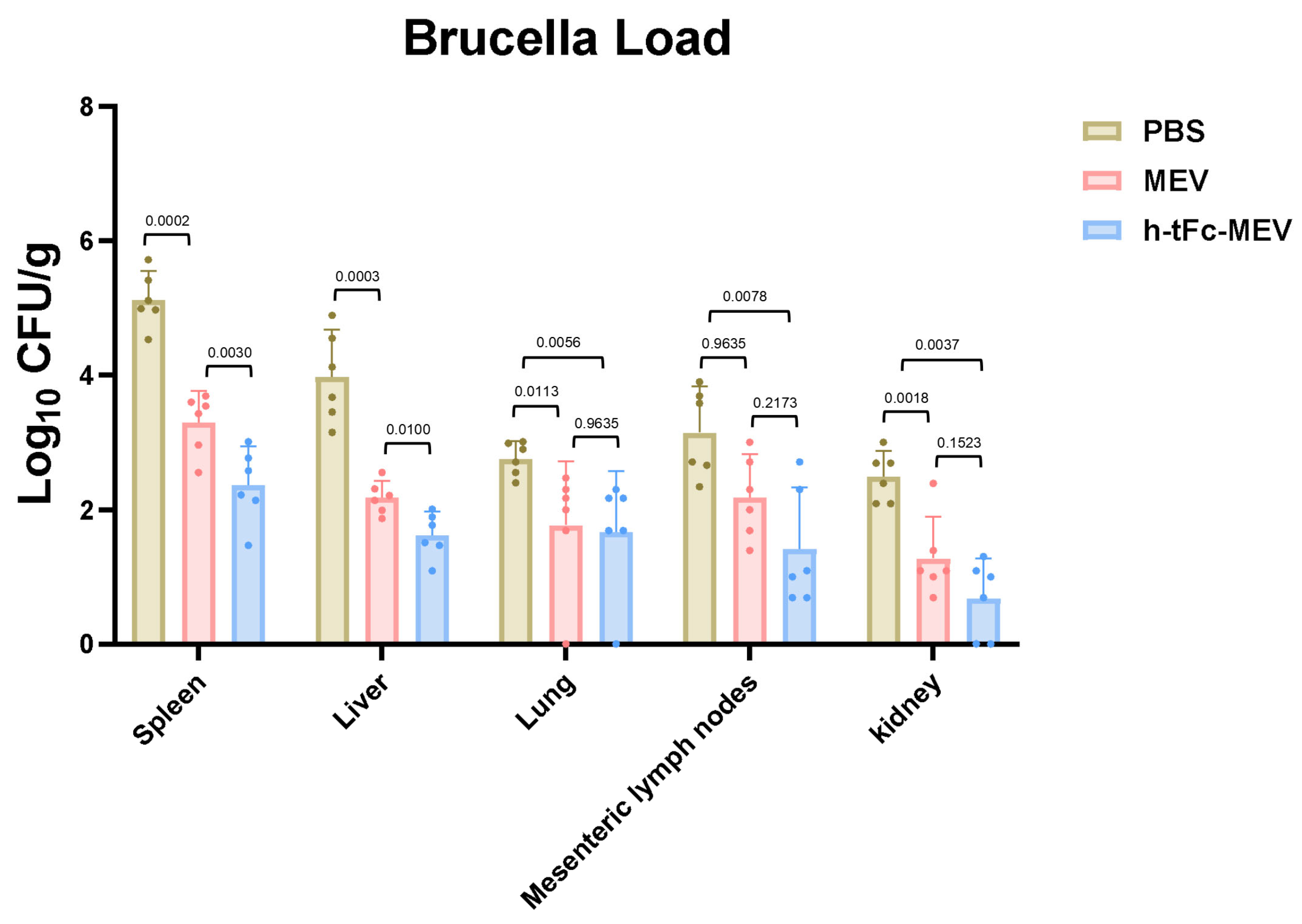
3.16. Protective Efficacy of Orally Administered FcRn-Targeting Vaccine
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suárez-Esquivel, M.; Chaves-Olarte, E.; Moreno, E.; Guzmán-Verri, C. Brucella Genomics: Macro and Micro Evolution. Int. J. Mol. Sci. 2020, 21, 7749. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Zhu, Y.; Shi, J.; Shang, K.; Yin, Z.; Shi, H.; He, Y.; Ding, J.; Zhang, F. The development of a human Brucella mucosal vaccine: What should be considered? Life Sci. 2024, 355, 122986. [Google Scholar] [CrossRef] [PubMed]
- Hensel, M.E.; Stranahan, L.W.; Edwards, J.F.; Arenas-Gamboa, A.M. Intratracheal inoculation results in Brucella-associated reproductive disease in male mouse and guinea pig models of infection. Front. Microbiol. 2022, 13, 1029199. [Google Scholar] [CrossRef]
- Ke, Y.; Wang, Y.; Li, W.; Chen, Z. Type IV secretion system of Brucella spp. and its effectors. Front. Cell Infect. Microbiol. 2015, 5, 72. [Google Scholar] [CrossRef]
- Wang, H.; Hoffman, C.; Yang, X.; Clapp, B.; Pascual, D.W. Targeting resident memory T cell immunity culminates in pulmonary and systemic protection against Brucella infection. PLoS Pathog. 2020, 16, e1008176. [Google Scholar] [CrossRef]
- Bao, Y.; Tian, M.; Li, P.; Liu, J.; Ding, C.; Yu, S. Characterization of Brucella abortus mutant strain Δ22915, a potential vaccine candidate. Vet. Res. 2017, 48, 17. [Google Scholar] [CrossRef]
- Dorneles, E.M.; Sriranganathan, N.; Lage, A.P. Recent advances in Brucella abortus vaccines. Vet. Res. 2015, 46, 76. [Google Scholar] [CrossRef]
- O’Callaghan, D. Human brucellosis: Recent advances and future challenges. Infect. Dis. Poverty 2020, 9, 101. [Google Scholar] [CrossRef]
- Ko, J.; Splitter, G.A. Molecular host-pathogen interaction in brucellosis: Current understanding and future approaches to vaccine development for mice and humans. Clin. Microbiol. Rev. 2003, 16, 65–78. [Google Scholar] [CrossRef]
- Perkins, S.D.; Smither, S.J.; Atkins, H.S. Towards a Brucella vaccine for humans. FEMS Microbiol. Rev. 2010, 34, 379–394. [Google Scholar] [CrossRef]
- Xie, J.; Wang, J.; Li, Z.; Wang, W.; Pang, Y.; He, Y. Ontology-Based Meta-Analysis of Animal and Human Adverse Events Associated with Licensed Brucellosis Vaccines. Front. Pharmacol. 2018, 9, 503. [Google Scholar] [CrossRef] [PubMed]
- Vives-Soto, M.; Puerta-García, A.; Rodríguez-Sánchez, E.; Pereira, J.L.; Solera, J. What risk do Brucella vaccines pose to humans? A systematic review of the scientific literature on occupational exposure. PLoS Negl. Trop. Dis. 2024, 18, e0011889. [Google Scholar] [CrossRef] [PubMed]
- Pasquevich, K.A.; Ibañez, A.E.; Coria, L.M.; García Samartino, C.; Estein, S.M.; Zwerdling, A.; Barrionuevo, P.; Oliveira, F.S.; Seither, C.; Warzecha, H.; et al. An oral vaccine based on U-Omp19 induces protection against B. abortus mucosal challenge by inducing an adaptive IL-17 immune response in mice. PLoS ONE 2011, 6, e16203. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, Z.I.; Yang, X.; Hoffman, C.; Pascual, D.W. Live mucosal vaccination stimulates potent protection via varied CD4(+) and CD8(+) T cell subsets against wild-type Brucella melitensis 16M challenge. Front. Immunol. 2022, 13, 995327. [Google Scholar] [CrossRef]
- Kazemi-Roudsari, M.; Doosti, A.; Jami, M.S. Design of an oral vaccine using Lactococcus lactis against brucellosis: An in vitro and in vivo study. AMB Express 2024, 14, 2. [Google Scholar] [CrossRef]
- Nazifi, N.; Tahmoorespur, M.; Sekhavati, M.H.; Haghparast, A.; Behroozikhah, A.M. In vivo immunogenicity assessment and vaccine efficacy evaluation of a chimeric tandem repeat of epitopic region of OMP31 antigen fused to interleukin 2 (IL-2) against Brucella melitensis in BALB/c mice. BMC Vet. Res. 2019, 15, 402. [Google Scholar] [CrossRef]
- Xiong, X.; Li, B.; Zhou, Z.; Gu, G.; Li, M.; Liu, J.; Jiao, H. The VirB System Plays a Crucial Role in Brucella Intracellular Infection. Int. J. Mol. Sci. 2021, 22, 13637. [Google Scholar] [CrossRef]
- Yin, Z.; Li, M.; Niu, C.; Yu, M.; Xie, X.; Haimiti, G.; Guo, W.; Shi, J.; He, Y.; Ding, J.; et al. Design of multi-epitope vaccine candidate against Brucella type IV secretion system (T4SS). PLoS ONE 2023, 18, e0286358. [Google Scholar] [CrossRef]
- Edmonds, M.D.; Cloeckaert, A.; Elzer, P.H. Brucella species lacking the major outer membrane protein Omp25 are attenuated in mice and protect against Brucella melitensis and Brucella ovis. Vet. Microbiol. 2002, 88, 205–221. [Google Scholar] [CrossRef]
- Cloeckaert, A.; Vizcaíno, N.; Paquet, J.Y.; Bowden, R.A.; Elzer, P.H. Major outer membrane proteins of Brucella spp.: Past, present and future. Vet. Microbiol. 2002, 90, 229–247. [Google Scholar] [CrossRef]
- Li, Z.; Wang, S.; Wei, S.; Yang, G.; Zhang, C.; Xi, L.; Zhang, J.; Cui, Y.; Hao, J.; Zhang, H.; et al. Immunization with a combination of recombinant Brucella abortus proteins induces T helper immune response and confers protection against wild-type challenge in BALB/c mice. Microb. Biotechnol. 2022, 15, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Martín-Martín, A.I.; Caro-Hernández, P.; Sancho, P.; Tejedor, C.; Cloeckaert, A.; Fernández-Lago, L.; Vizcaíno, N. Analysis of the occurrence and distribution of the Omp25/Omp31 family of surface proteins in the six classical Brucella species. Vet. Microbiol. 2009, 137, 74–82. [Google Scholar] [CrossRef]
- Edmonds, M.D.; Cloeckaert, A.; Hagius, S.D.; Samartino, L.E.; Fulton, W.T.; Walker, J.V.; Enright, F.M.; Booth, N.J.; Elzer, P.H. Pathogenicity and protective activity in pregnant goats of a Brucella melitensis Deltaomp25 deletion mutant. Res. Vet. Sci. 2002, 72, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Liu, W.; Wang, X.; Chen, Y.; Du, Q.; Zhao, X.; Zhang, H.; Liu, S.L.; Tong, D.; Huang, Y. Brucella Omp25 Upregulates miR-155, miR-21-5p, and miR-23b to Inhibit Interleukin-12 Production via Modulation of Programmed Death-1 Signaling in Human Monocyte/Macrophages. Front. Immunol. 2017, 8, 708. [Google Scholar] [CrossRef]
- Zhi, F.; Zhou, D.; Bai, F.; Li, J.; Xiang, C.; Zhang, G.; Jin, Y.; Wang, A. VceC Mediated IRE1 Pathway and Inhibited CHOP-induced Apoptosis to Support Brucella Replication in Goat Trophoblast Cells. Int. J. Mol. Sci. 2019, 20, 4104. [Google Scholar] [CrossRef]
- Lei, S.; Zhong, Z.; Ke, Y.; Yang, M.; Xu, X.; Ren, H.; An, C.; Yuan, J.; Yu, J.; Xu, J.; et al. Deletion of the Small RNA Chaperone Protein Hfq down Regulates Genes Related to Virulence and Confers Protection against Wild-Type Brucella Challenge in Mice. Front. Microbiol. 2015, 6, 1570. [Google Scholar] [CrossRef]
- Fieux, M.; Le Quellec, S.; Bartier, S.; Coste, A.; Louis, B.; Giroudon, C.; Nourredine, M.; Bequignon, E. FcRn as a Transporter for Nasal Delivery of Biologics: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 6475. [Google Scholar] [CrossRef]
- Li, W.; Wang, T.; Rajendrakumar, A.M.; Acharya, G.; Miao, Z.; Varghese, B.P.; Yu, H.; Dhakal, B.; LeRoith, T.; Karunakaran, A.; et al. An FcRn-targeted mucosal vaccine against SARS-CoV-2 infection and transmission. Nat. Commun. 2023, 14, 7114. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- He, Y.; Li, J.; Mao, W.; Zhang, D.; Liu, M.; Shan, X.; Zhang, B.; Zhu, C.; Shen, J.; Deng, Z.; et al. HLA common and well-documented alleles in China. Hla 2018, 92, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Tarrahimofrad, H.; Rahimnahal, S.; Zamani, J.; Jahangirian, E.; Aminzadeh, S. Designing a multi-epitope vaccine to provoke the robust immune response against influenza A H7N9. Sci. Rep. 2021, 11, 24485. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Ye, L.; Zeng, R.; Bai, Y.; Roopenian, D.C.; Zhu, X. Efficient mucosal vaccination mediated by the neonatal Fc receptor. Nat. Biotechnol. 2011, 29, 158–163. [Google Scholar] [CrossRef]
- Dasari, V.; McNeil, L.K.; Beckett, K.; Solomon, M.; Ambalathingal, G.; Thuy, T.L.; Panikkar, A.; Smith, C.; Steinbuck, M.P.; Jakubowski, A.; et al. Lymph node targeted multi-epitope subunit vaccine promotes effective immunity to EBV in HLA-expressing mice. Nat. Commun. 2023, 14, 4371. [Google Scholar] [CrossRef]
- Sidorczuk, K.; Mackiewicz, P.; Pietluch, F.; Gagat, P. Characterization of signal and transit peptides based on motif composition and taxon-specific patterns. Sci. Rep. 2023, 13, 15751. [Google Scholar] [CrossRef]
- Bajrovic, I.; Croyle, M.A. Challenges in vaccine transport: Can we deliver without the cold chain? Expert. Rev. Vaccines 2023, 22, 933–936. [Google Scholar] [CrossRef]
- Uddin, M.N.; Roni, M.A. Challenges of Storage and Stability of mRNA-Based COVID-19 Vaccines. Vaccines 2021, 9, 1033. [Google Scholar] [CrossRef]
- Zhao, T.; Cai, Y.; Jiang, Y.; He, X.; Wei, Y.; Yu, Y.; Tian, X. Vaccine adjuvants: Mechanisms and platforms. Signal Transduct. Target. Ther. 2023, 8, 283. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.; Byun, J.; Wu, Y.; Park, J.; Oh, Y.K. In vivo fate and intracellular trafficking of vaccine delivery systems. Adv. Drug Deliv. Rev. 2022, 186, 114325. [Google Scholar] [CrossRef]
- Sampath, V.; Rabinowitz, G.; Shah, M.; Jain, S.; Diamant, Z.; Jesenak, M.; Rabin, R.; Vieths, S.; Agache, I.; Akdis, M.; et al. Vaccines and allergic reactions: The past, the current COVID-19 pandemic, and future perspectives. Allergy 2021, 76, 1640–1660. [Google Scholar] [CrossRef] [PubMed]
- Stöppler, D.; Macpherson, A.; Smith-Penzel, S.; Basse, N.; Lecomte, F.; Deboves, H.; Taylor, R.D.; Norman, T.; Porter, J.; Waters, L.C.; et al. Insight into small molecule binding to the neonatal Fc receptor by X-ray crystallography and 100 kHz magic-angle-spinning NMR. PLoS Biol. 2018, 16, e2006192. [Google Scholar] [CrossRef] [PubMed]
- Schlothauer, T.; Rueger, P.; Stracke, J.O.; Hertenberger, H.; Fingas, F.; Kling, L.; Emrich, T.; Drabner, G.; Seeber, S.; Auer, J.; et al. Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. MAbs 2013, 5, 576–586. [Google Scholar] [CrossRef]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J. Clin. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef]
- Heo, Y.A. Efgartigimod: First Approval. Drugs 2022, 82, 341–348. [Google Scholar] [CrossRef]
- Milillo, M.A.; Trotta, A.; Serafino, A.; Marin Franco, J.L.; Marinho, F.V.; Alcain, J.; Genoula, M.; Balboa, L.; Oliveira, S.C.; Giambartolomei, G.H.; et al. Bacterial RNA Contributes to the Down-Modulation of MHC-II Expression on Monocytes/Macrophages Diminishing CD4(+) T Cell Responses. Front. Immunol. 2019, 10, 2181. [Google Scholar] [CrossRef]
- Charles, N.; Watford, W.T.; Ramos, H.L.; Hellman, L.; Oettgen, H.C.; Gomez, G.; Ryan, J.J.; O’Shea, J.J.; Rivera, J. Lyn kinase controls basophil GATA-3 transcription factor expression and induction of Th2 cell differentiation. Immunity 2009, 30, 533–543. [Google Scholar] [CrossRef]
- Xu, M.; Qi, Y.; Liu, G.; Song, Y.; Jiang, X.; Du, B. Size-Dependent In Vivo Transport of Nanoparticles: Implications for Delivery, Targeting, and Clearance. ACS Nano 2023, 17, 20825–20849. [Google Scholar] [CrossRef]
- Zhu, C.D.; Zheng, Q.; Wang, L.X.; Xu, H.F.; Tong, J.L.; Zhang, Q.A.; Wan, Y.; Wu, J.Q. Synthesis of novel galactose functionalized gold nanoparticles and its radiosensitizing mechanism. J. Nanobiotechnol. 2015, 13, 67. [Google Scholar] [CrossRef]
- Macri, C.; Paxman, M.; Jenika, D.; Lin, X.P.; Elahi, Z.; Gleeson, P.A.; Caminschi, I.; Lahoud, M.H.; Villadangos, J.A.; Mintern, J.D. FcRn regulates antigen presentation in dendritic cells downstream of DEC205-targeted vaccines. NPJ Vaccines 2024, 9, 76. [Google Scholar] [CrossRef]
- Gray, J.I.; Westerhof, L.M.; MacLeod, M.K.L. The roles of resident, central and effector memory CD4 T-cells in protective immunity following infection or vaccination. Immunology 2018, 154, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef] [PubMed]
- Peter, H.H.; Ochs, H.D.; Cunningham-Rundles, C.; Vinh, D.C.; Kiessling, P.; Greve, B.; Jolles, S. Targeting FcRn for immunomodulation: Benefits, risks, and practical considerations. J. Allergy Clin. Immunol. 2020, 146, 479–491.e475. [Google Scholar] [CrossRef]
- Rashki, S.; Asgarpour, K.; Tarrahimofrad, H.; Hashemipour, M.; Ebrahimi, M.S.; Fathizadeh, H.; Khorshidi, A.; Khan, H.; Marzhoseyni, Z.; Salavati-Niasari, M.; et al. Chitosan-based nanoparticles against bacterial infections. Carbohydr. Polym. 2021, 251, 117108. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W., Jr. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef]
- Ehrenberg, M.S.; Friedman, A.E.; Finkelstein, J.N.; Oberdörster, G.; McGrath, J.L. The influence of protein adsorption on nanoparticle association with cultured endothelial cells. Biomaterials 2009, 30, 603–610. [Google Scholar] [CrossRef]
- Pridgen, E.M.; Alexis, F.; Kuo, T.T.; Levy-Nissenbaum, E.; Karnik, R.; Blumberg, R.S.; Langer, R.; Farokhzad, O.C. Transepithelial transport of Fc-targeted nanoparticles by the neonatal fc receptor for oral delivery. Sci. Transl. Med. 2013, 5, 213ra167. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug Deliv. Rev. 2015, 91, 109–124. [Google Scholar] [CrossRef]
- Baker, K.; Rath, T.; Pyzik, M.; Blumberg, R.S. The Role of FcRn in Antigen Presentation. Front. Immunol. 2014, 5, 408. [Google Scholar] [CrossRef]
- Baker, K.; Qiao, S.W.; Kuo, T.T.; Aveson, V.G.; Platzer, B.; Andersen, J.T.; Sandlie, I.; Chen, Z.; de Haar, C.; Lencer, W.I.; et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9927–9932. [Google Scholar] [CrossRef]
- Wen, J.; Li, Z.; Lv, Y.; Ding, S.; Zhu, Y.; Yang, J.; Tang, J.; Zhu, M.; Zhao, Y.; Zhao, W. A subunit vaccine based on Brucella rBP26 induces Th1 immune responses and M1 macrophage activation. Acta Biochim. Biophys. Sin. 2024, 56, 879–891. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Omp25 | VirB10 |
| Accession number | Q45321 | Q8YDZ0 |
| Amino acid number | 213aa | 380aa |
| Subcellular localization | Cell outer membrane | Cell outer membrane |
| Antigenicity | 0.8164 | 0.6906 |
| Allergenicity | non-allergen | non-allergen |
| Stability | 23.00 | 36.52 |
| Hydrophilicity | −0.317 | −0.155 |
| Protein | h-tFc-MEV |
| Molecular weight | 71.54 kDa |
| Formula | C3198H5027N883O957S12 |
| Theoretical pI | 9.59 |
| Antigenicity | 0.6878 |
| Allergenicity | non-allergen |
| Stability | stable |
| Hydrophilicity | −0.577 |
| Solubility | Good (Supplementary Material S4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, T.; Zhu, Y.; Shang, K.; Shi, H.; Xu, R.; Li, M.; Pu, F.; Kuang, J.; Ding, J.; Zhang, F. FcRn-Driven Nanoengineered Mucosal Vaccine with Multi-Epitope Fusion Induces Robust Dual Immunity and Long-Term Protection Against Brucella. Vaccines 2025, 13, 567. https://doi.org/10.3390/vaccines13060567
Tian T, Zhu Y, Shang K, Shi H, Xu R, Li M, Pu F, Kuang J, Ding J, Zhang F. FcRn-Driven Nanoengineered Mucosal Vaccine with Multi-Epitope Fusion Induces Robust Dual Immunity and Long-Term Protection Against Brucella. Vaccines. 2025; 13(6):567. https://doi.org/10.3390/vaccines13060567
Chicago/Turabian StyleTian, Tingting, Yuejie Zhu, Kaiyu Shang, Huidong Shi, Ruixue Xu, Mingzhe Li, Fuling Pu, Junyu Kuang, Jianbing Ding, and Fengbo Zhang. 2025. "FcRn-Driven Nanoengineered Mucosal Vaccine with Multi-Epitope Fusion Induces Robust Dual Immunity and Long-Term Protection Against Brucella" Vaccines 13, no. 6: 567. https://doi.org/10.3390/vaccines13060567
APA StyleTian, T., Zhu, Y., Shang, K., Shi, H., Xu, R., Li, M., Pu, F., Kuang, J., Ding, J., & Zhang, F. (2025). FcRn-Driven Nanoengineered Mucosal Vaccine with Multi-Epitope Fusion Induces Robust Dual Immunity and Long-Term Protection Against Brucella. Vaccines, 13(6), 567. https://doi.org/10.3390/vaccines13060567