
Multi-View Learning to Unravel the Different Levels Underlying Hepatitis B Vaccine Response
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. RNA-Seq Data Quality Control Steps
2.3. Data Sampling
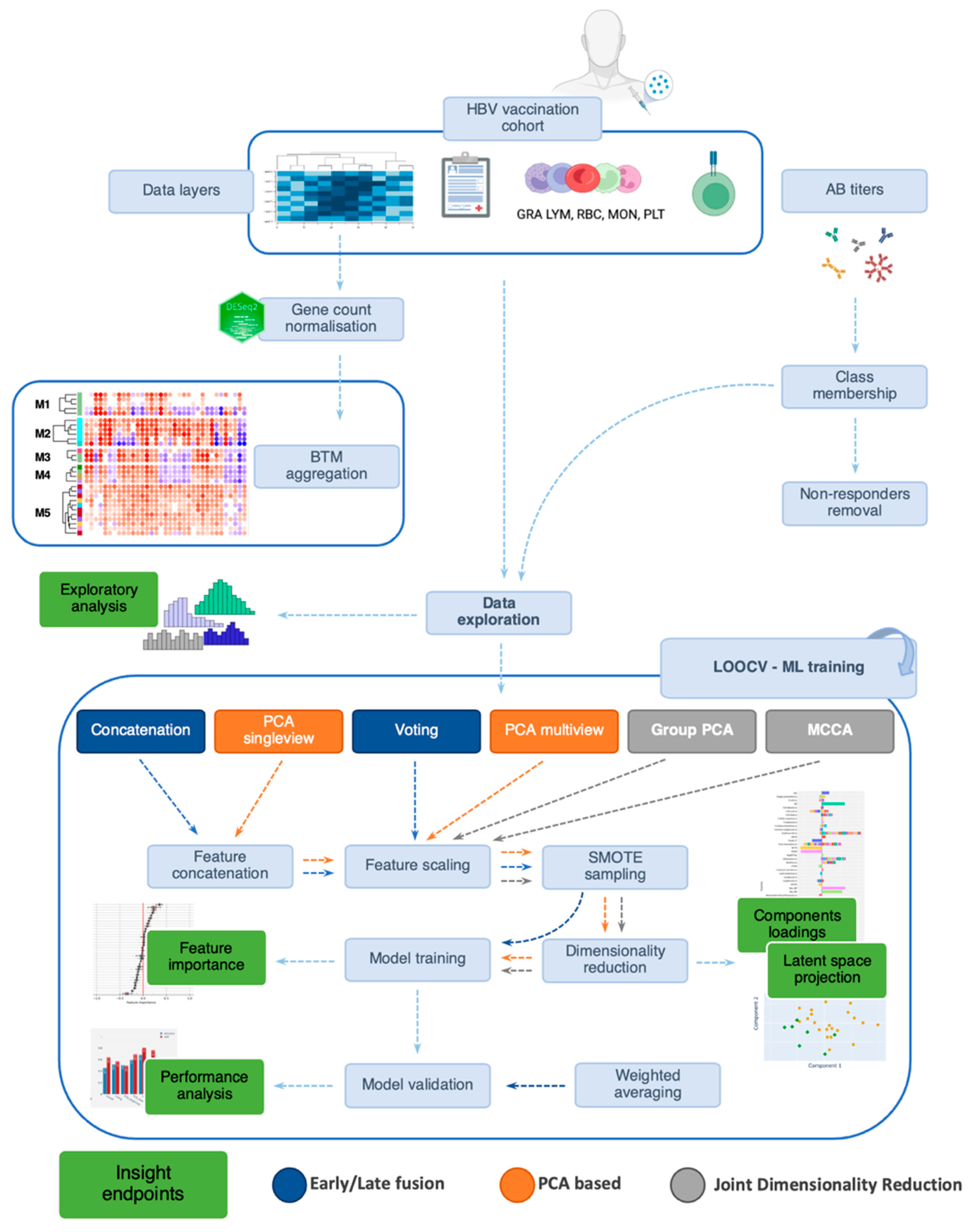
2.4. Integration Methods
3. Results
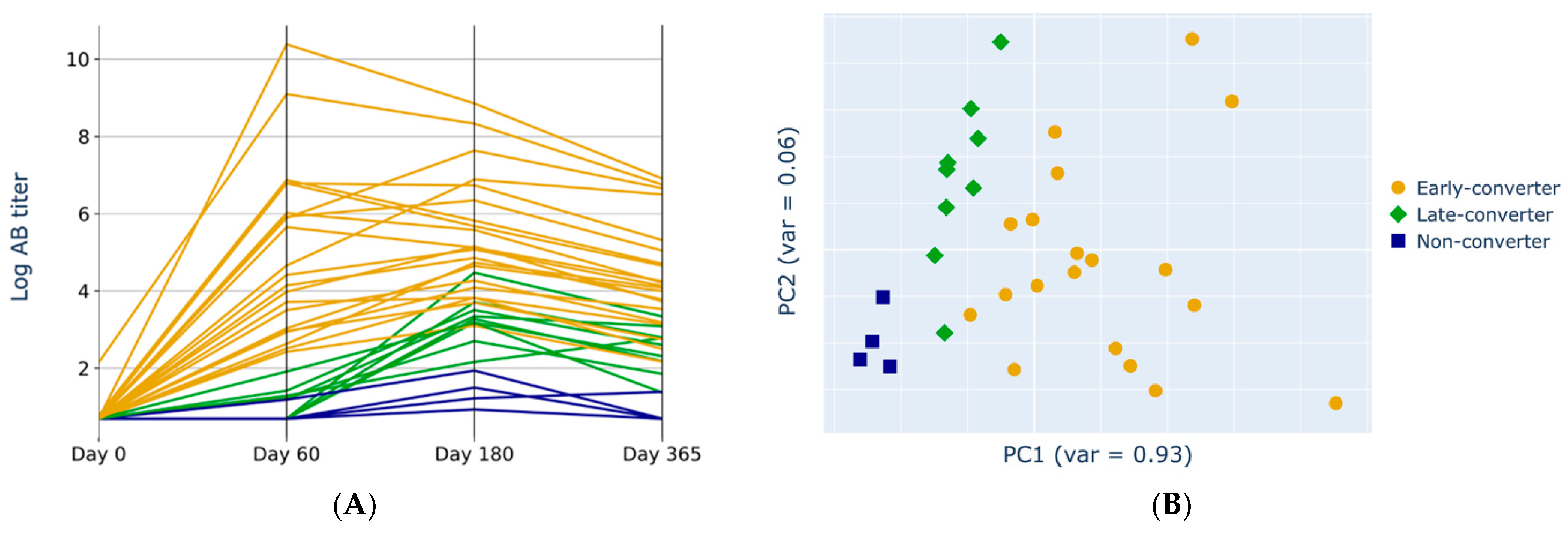
3.1. Anti-HBs Titers Separation
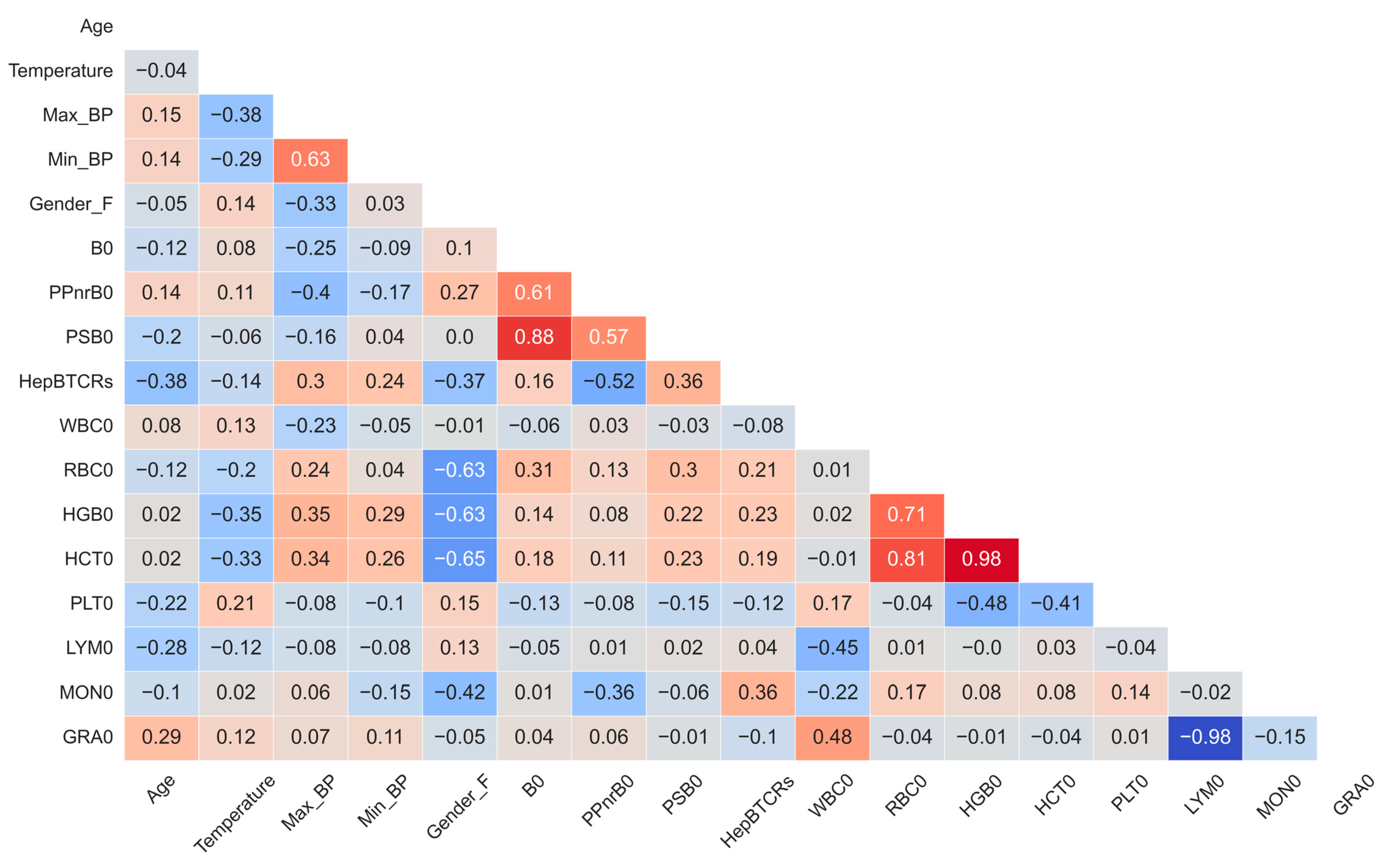
3.2. Independent Data Levels Show Correlated Features
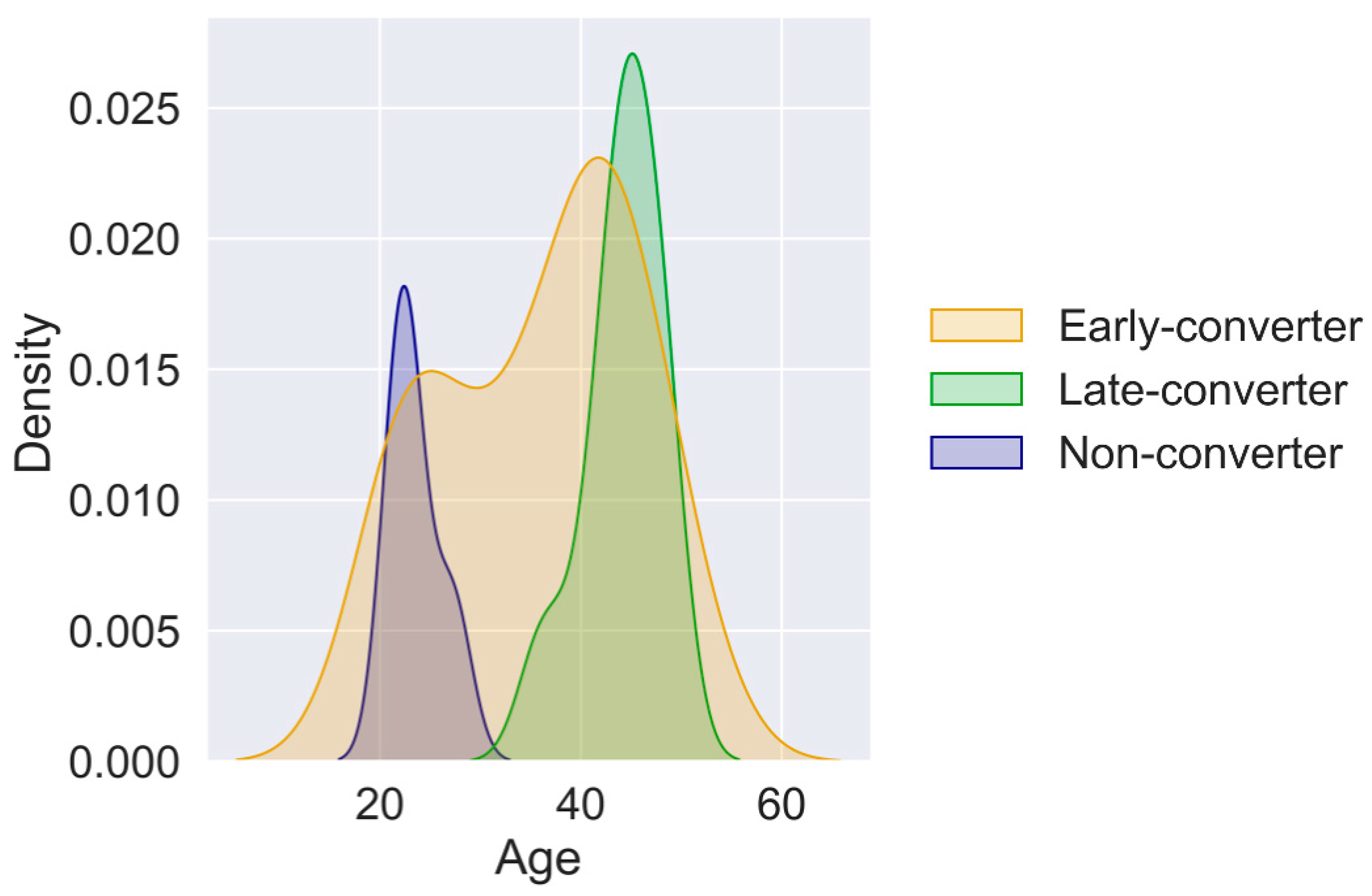
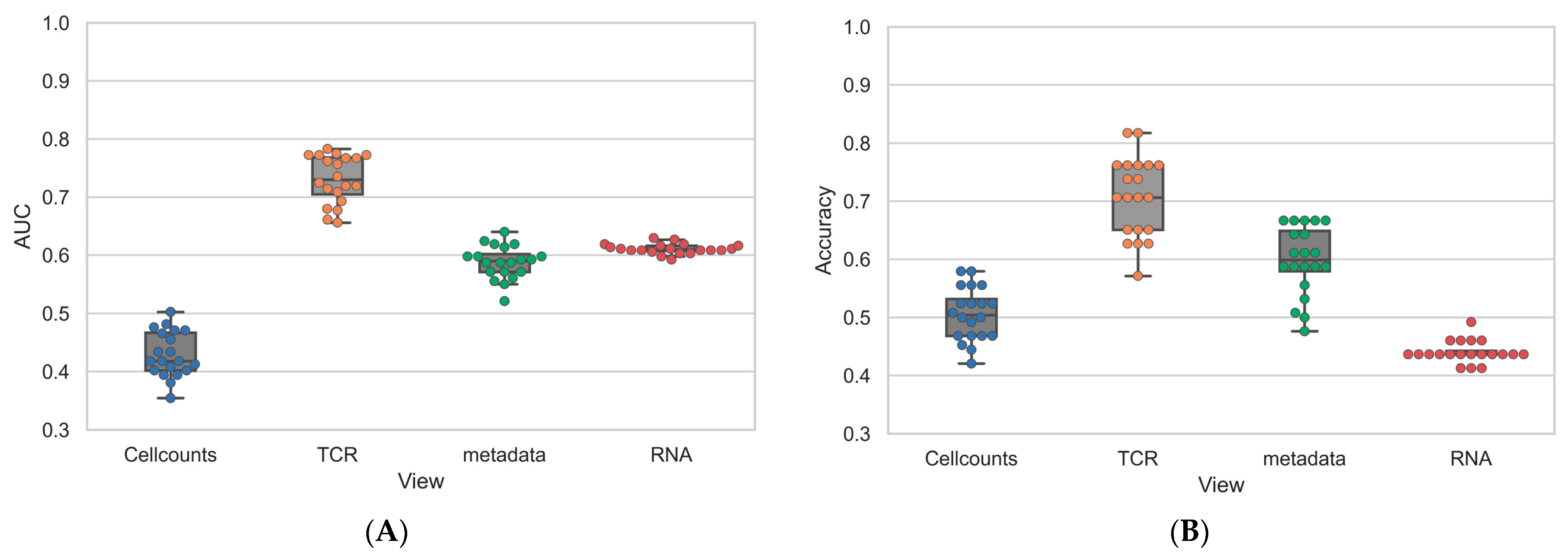
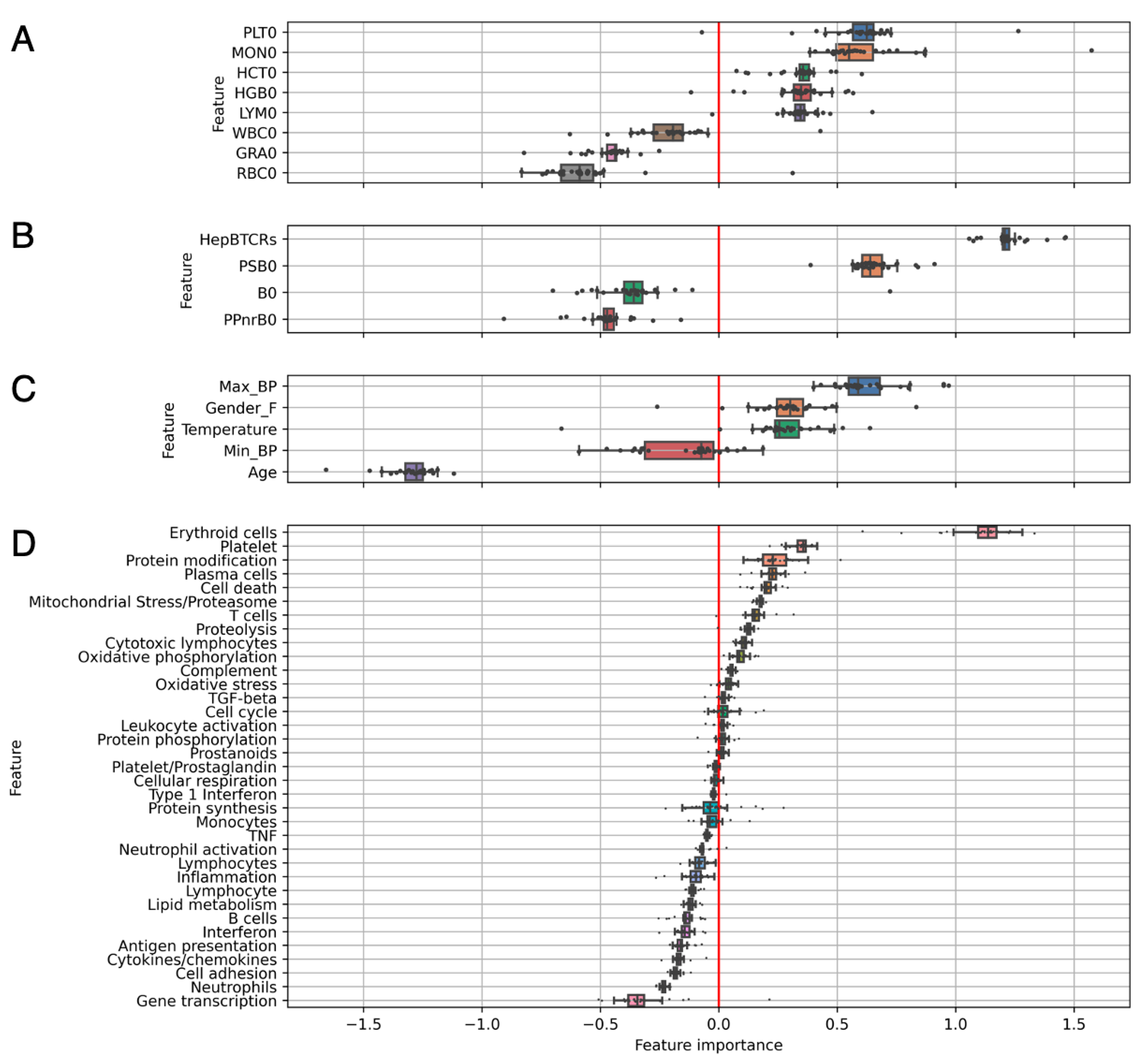
3.3. Individual Levels Contain Elements That Correlate with Response
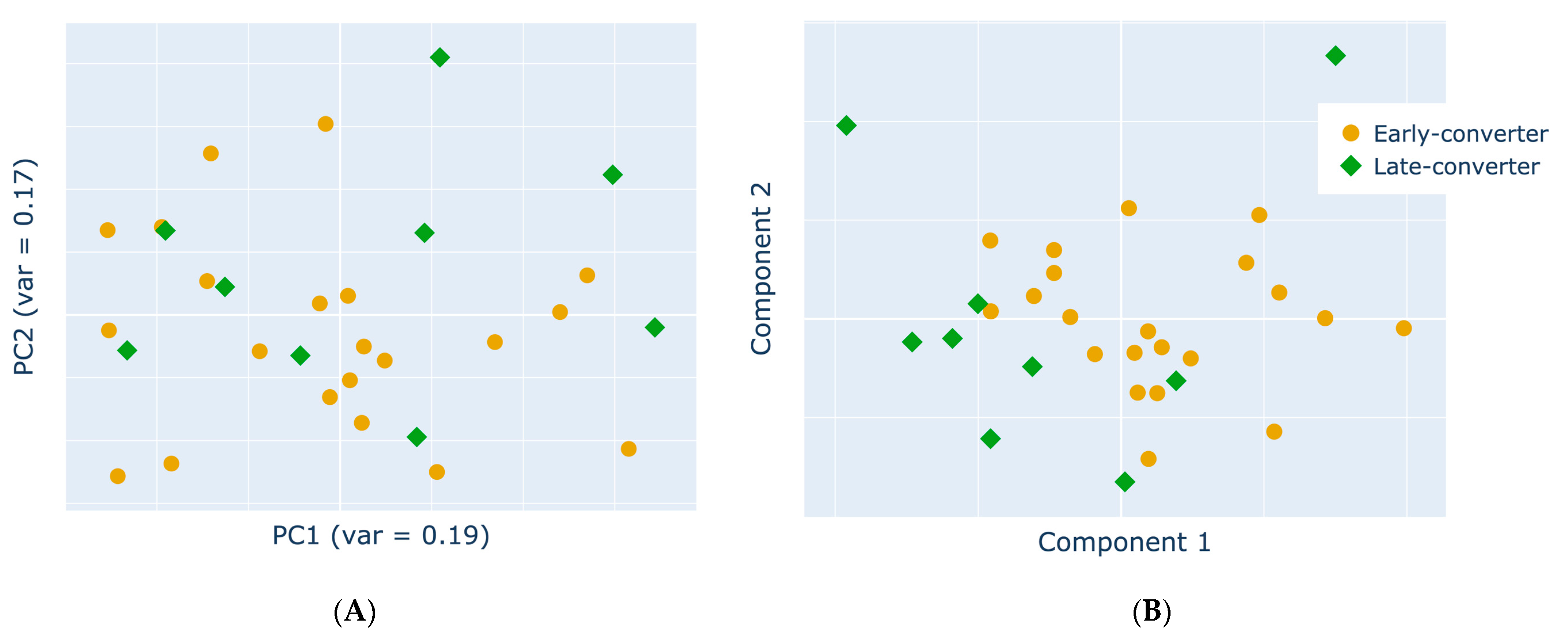
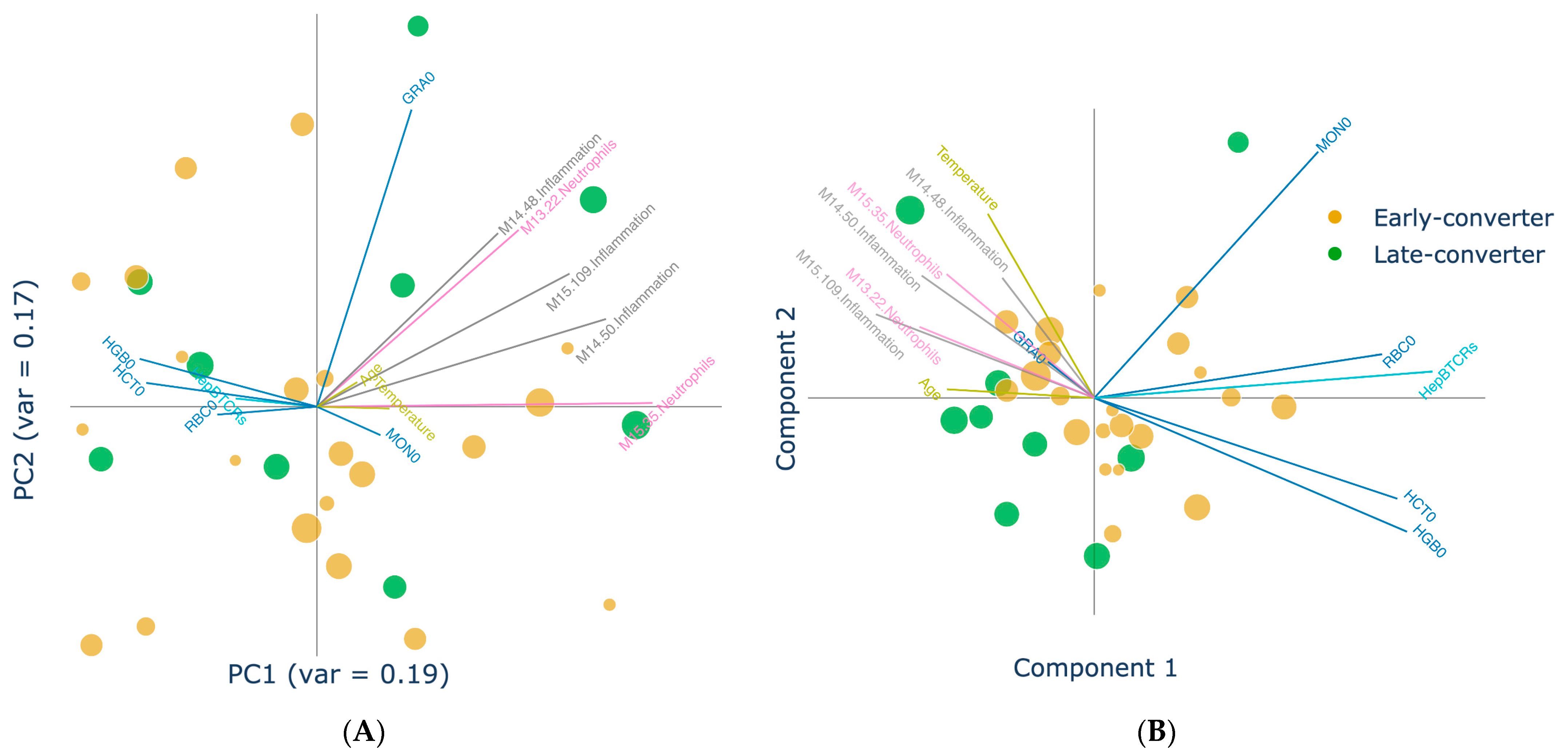
3.4. Projection of Data Views Provides Insights into Response Classes
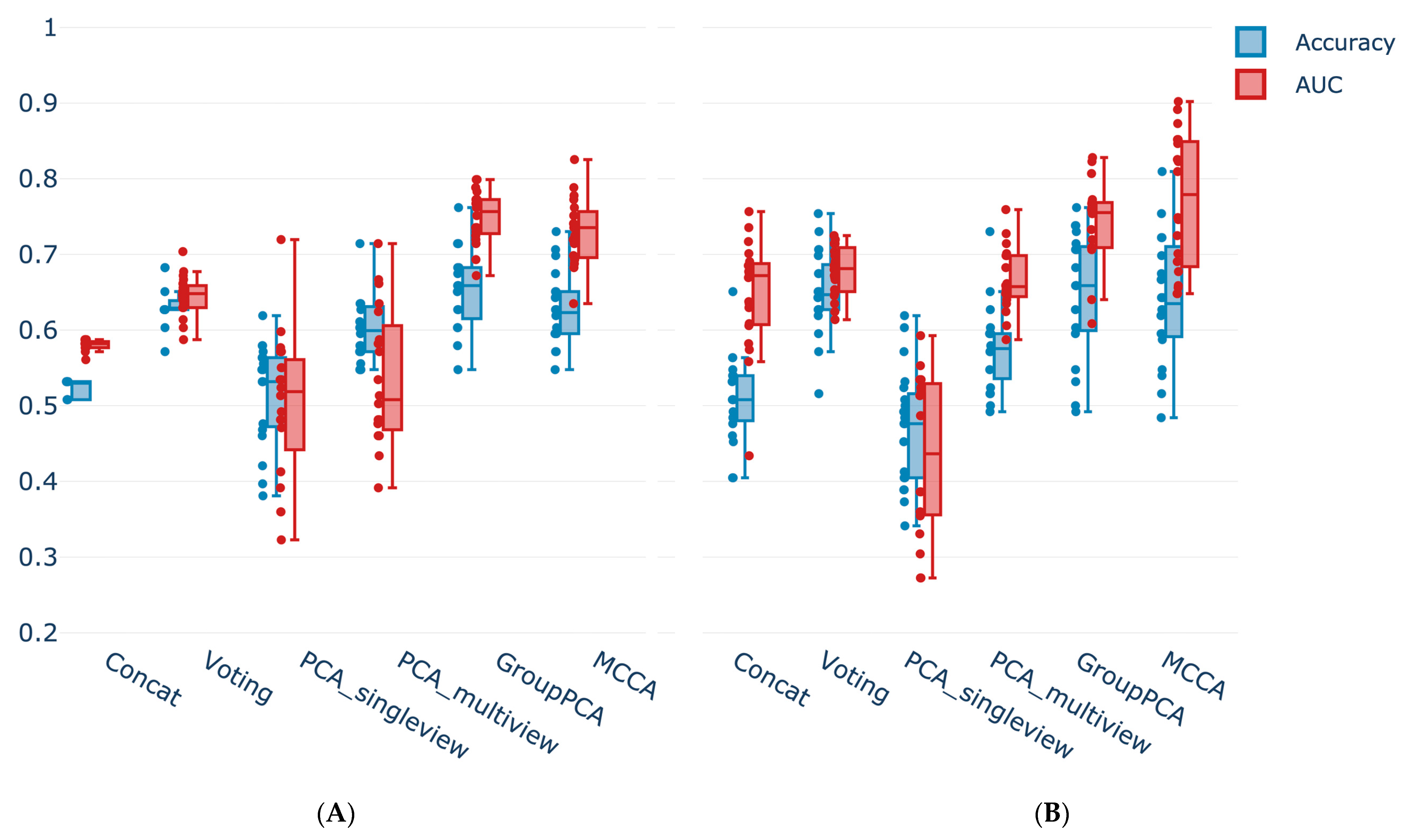
3.5. Multi-View Integration Allows Superior Classification Performance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AB | antibody |
| anti-HBs | antibodies against hepatitis B virus surface antigen |
| AUC | area under the receiver operating characteristic curve |
| BCR | B cell receptor |
| B0 | number of T cell receptors sequenced in the CD4+ memory |
| BTMs | blood transcriptional modules |
| CCA | canonical correlation analysis |
| Gender_F | female gender |
| GRA0 | day 0 granulocytes counts |
| HBV | hepatitis B virus |
| HCT0 | day 0 hematocrit cell counts |
| HepBTCR | normalized ratio of vaccine-specific T-cells |
| HGB0 | day 0 haemoglobin protein counts |
| JDR | joint dimensionality reduction |
| LOOCV | leave-one-out cross validation |
| LR | logistic regression |
| LYM0 | day 0 lymphocytes counts |
| Max_BP | maximum blood pressure |
| MCCA | multi-view canonical correlation analysis |
| Min_BP | minimum blood pressure |
| MON0 | day 0 monocytes counts |
| ML | machine learning |
| MOFA | Multi-Omics Factor Analysis |
| PBMCs | peripheral blood mononuclear cells |
| PC | principal component |
| PCA | principal component analysis |
| PPnrB0 | frequency of bystander T cell receptors |
| PSB0 | frequency of vaccine-specific T cell receptors |
| RBC0 | day 0 red blood cells counts |
| RNA-seq | RNA sequencing |
| SMOTE | Synthetic Minority Oversampling |
| SNF | Similarity Network Fusion |
| SUMCOR | sum of pairwise correlations |
| TCR | T cell receptor |
References
- Anaya, J.-M.; Shoenfeld, Y.; Rojas-Villarraga, A.; Levy, R.A.; Cervera, R. (Eds.) Autoimmunity: From Bench to Bedside; El Rosario University Press: Bogota, Colombia, 2013; Available online: http://www.ncbi.nlm.nih.gov/books/NBK459447/ (accessed on 9 January 2023).
- Pulendran, B.; Li, S.; Nakaya, H.I. Systems Vaccinology. Immunity 2010, 33, 516–529. [Google Scholar] [CrossRef]
- Pulendran, B. Systems vaccinology: Probing humanity’s diverse immune systems with vaccines. Proc. Natl. Acad. Sci. USA 2014, 111, 12300–12306. [Google Scholar] [CrossRef]
- Furman, D.; Davis, M.M. New approaches to understanding the immune response to vaccination and infection. Vaccine 2015, 33, 5271–5281. [Google Scholar] [CrossRef]
- Zhao, J.; Xie, X.; Xu, X.; Sun, S. Multi-view learning overview: Recent progress and new challenges. Inf. Fusion 2017, 38, 43–54. [Google Scholar] [CrossRef]
- Hotelling, H. Relations Between Two Sets of Variates. Biometrika 1936, 28, 321. [Google Scholar] [CrossRef]
- Lei, B.; Chen, S.; Ni, D.; Wang, T. Discriminative Learning for Alzheimer’s Disease Diagnosis via Canonical Correlation Analysis and Multimodal Fusion. Front. Aging Neurosci. 2016, 8, 77. [Google Scholar] [CrossRef]
- Singanamalli, A.; Wang, H.; Madabhushi, A.; Weiner, M.; Aisen, P.; Petersen, R.; Jack, C.; Jagust, W.; Trojanowki, J.; Toga, A.; et al. Cascaded Multi-view Canonical Correlation (CaMCCo) for Early Diagnosis of Alzheimer’s Disease via Fusion of Clinical, Imaging and Omic Features. Sci. Rep. 2017, 7, 8137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Adeli, E.; Zhou, T.; Chen, X.; Shen, D. Multi-Layer Multi-View Classification for Alzheimer’s Disease Diagnosis. In Proceedings of the AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018; Volume 2018, pp. 4406–4413. [Google Scholar]
- Zhuang, X.; Yang, Z.; Cordes, D. A technical review of canonical correlation analysis for neuroscience applications. Hum. Brain Mapp. 2020, 41, 3807–3833. [Google Scholar] [CrossRef]
- Yang, J.; Wang, L.; Qin, J.; Du, J.; Ding, M.; Niu, T.; Li, R. Multi-view learning for lymph node metastasis prediction using tumor and nodal radiomics in gastric cancer. Phys. Med. Biol. 2022, 67, 055007. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K. On lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901, 2, 559–572. [Google Scholar] [CrossRef]
- Jolliffe, I.T. Principal Component Analysis; Springer: New York, NY, USA, 1986. [Google Scholar] [CrossRef]
- Kettenring, J.R. Canonical analysis of several sets of variables. Biometrika 1971, 58, 433–451. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis—A framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Rasa, S.M.M.; Annunziata, F.; Krepelova, A.; Nunna, S.; Omrani, O.; Gebert, N.; Adam, L.; Käppel, S.; Höhn, S.; Donati, G.; et al. Inflammaging is driven by upregulation of innate immune receptors and systemic interferon signaling and is ameliorated by dietary restriction. Cell Rep. 2022, 39, 111017. [Google Scholar] [CrossRef]
- Bartholomeus, E.; De Neuter, N.; Meysman, P.; Suls, A.; Keersmaekers, N.; Elias, G.; Jansens, H.; Hens, N.; Smits, E.; Van Tendeloo, V.; et al. Transcriptome profiling in blood before and after hepatitis B vaccination shows significant differences in gene expression between responders and non-responders. Vaccine 2018, 36, 6282–6289. [Google Scholar] [CrossRef]
- Querec, T.D.; Akondy, R.S.; Lee, E.K.; Cao, W.; Nakaya, H.I.; Teuwen, D.; Pirani, A.; Gernert, K.; Deng, J.; Marzolf, B.; et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 2009, 10, 116–125. [Google Scholar] [CrossRef]
- Tsang, J.S.; Schwartzberg, P.L.; Kotliarov, Y.; Biancotto, A.; Xie, Z.; Germain, R.N.; Wang, E.; Olnes, M.J.; Narayanan, M.; Golding, H.; et al. Global Analyses of Human Immune Variation Reveal Baseline Predictors of Postvaccination Responses. Cell 2014, 157, 499–513. [Google Scholar] [CrossRef]
- Tsang, J.S.; Dobaño, C.; VanDamme, P.; Moncunill, G.; Marchant, A.; Ben Othman, R.; Sadarangani, M.; Koff, W.C.; Kollmann, T.R. Improving Vaccine-Induced Immunity: Can Baseline Predict Outcome? Trends Immunol. 2020, 41, 457–465. [Google Scholar] [CrossRef]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 2015, 5, srep10775. [Google Scholar] [CrossRef]
- Elias, G.; Meysman, P.; Bartholomeus, E.; De Neuter, N.; Keersmaekers, N.; Suls, A.; Jansens, H.; Souquette, A.; De Reu, H.; Emonds, M.-P.; et al. Preexisting memory CD4 T cells in naïve individuals confer robust immunity upon hepatitis B vaccination. elife 2022, 11, e68388. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Bailey, S. Principal component analysis with noisy and/or missing data. Publ. Astron. Soc. Pac. 2012, 124, 1015–1023. [Google Scholar] [CrossRef]
- Altman, M.C.; Rinchai, D.; Baldwin, N.; Toufiq, M.; Whalen, E.; Garand, M.; Kabeer, B.S.A.; Alfaki, M.; Presnell, S.R.; Khaenam, P.; et al. Development of a fixed module repertoire for the analysis and interpretation of blood transcriptome data. Nat. Commun. 2021, 12, 4385. [Google Scholar] [CrossRef]
- Blagus, R.; Lusa, L. SMOTE for high-dimensional class-imbalanced data. BMC Bioinform. 2013, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-A. An Introduction to Logistic Regression: From Basic Concepts to Interpretation with Particular Attention to Nursing Domain. J. Korean Acad. Nurs. 2013, 43, 154–164. [Google Scholar] [CrossRef]
- Markello, R.D.; Shafiei, G.; Tremblay, C.; Postuma, R.B.; Dagher, A.; Misic, B. Multimodal phenotypic axes of Parkinson’s disease. npj Park. Dis. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Kittler, J.; Hatef, M.; Duin, R.; Matas, J. On combining classifiers. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 226–239. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Trendafilov, N.T.; Uddin, M. A Modified Principal Component Technique Based on the LASSO. J. Comput. Graph. Stat. 2003, 12, 531–547. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T.; Tibshirani, R. Sparse Principal Component Analysis. J. Comput. Graph. Stat. 2006, 15, 265–286. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Perry, R.; Mischler, G.; Guo, R.; Lee, T.; Chang, A.; Koul, A.; Franz, C.; Richard, H.; Carmichael, I.; Ablin, P.; et al. mvlearn: Multiview Machine Learning in Python. J. Mach. Learn. Res. 2021, 22, 4938–4944. [Google Scholar]
- Tenenhaus, A.; Tenenhaus, M. Regularized Generalized Canonical Correlation Analysis. Psychometrika 2011, 76, 257–284. [Google Scholar] [CrossRef]
- Lee, G.; Singanamalli, A.; Wang, H.; Feldman, M.D.; Master, S.R.; Shih, N.N.C.; Spangler, E.; Rebbeck, T.; Tomaszewski, J.E.; Madabhushi, A. Supervised Multi-View Canonical Correlation Analysis (sMVCCA): Integrating Histologic and Proteomic Features for Predicting Recurrent Prostate Cancer. IEEE Trans. Med. Imaging 2015, 34, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Witten, D.M.; Tibshirani, R.J. Extensions of Sparse Canonical Correlation Analysis with Applications to Genomic Data. Stat. Appl. Genet. Mol. Biol. 2009, 8, 1–27. [Google Scholar] [CrossRef]
- Murphy, W.G. The sex difference in haemoglobin levels in adults—Mechanisms, causes, and consequences. Blood Rev. 2014, 28, 41–47. [Google Scholar] [CrossRef]
- Tian, Y.; Hua, W.; Wu, Y.; Zhang, T.; Wang, W.; Wu, H.; Guo, C.; Huang, X. Immune Response to Hepatitis B Virus Vaccine Among People Living With HIV: A Meta-Analysis. Front. Immunol. 2021, 12, 745541. [Google Scholar] [CrossRef]
- Basireddy, P.; Avileli, S.; Beldono, N.; Gundela, S.L. Evaluation of Immune Response to Hepatitis B Vaccine in Healthcare Workers at a Tertiary Care Hospital. Indian J. Med. Microbiol. 2018, 36, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- De Maeyer, R.P.; Chambers, E.S. The impact of ageing on monocytes and macrophages. Immunol. Lett. 2021, 230, 1–10. [Google Scholar] [CrossRef]
- Cao, W. IFN-Aging: Coupling Aging With Interferon Response. Front. Aging 2022, 3, 870489. [Google Scholar] [CrossRef]
- Smith, D.L. Anemia in the elderly. Am. Fam. Physician 2000, 62, 1565–1572. [Google Scholar]
- Song, M.; Graubard, B.I.; Rabkin, C.S.; Engels, E.A. Neutrophil-to-lymphocyte ratio and mortality in the United States general population. Sci. Rep. 2021, 11, 464. [Google Scholar] [CrossRef]
- Le, P.-H.; Liang, K.-H.; Chang, M.-L.; Hsu, C.-W.; Chen, Y.-C.; Lin, C.-L.; Lin, W.-R.; Lai, M.-W.; Yeh, C.-T. Clinical Predictors for Neutrophil–to–Lymphocyte Ratio Changes in Patients with Chronic Hepatitis B Receiving Peginterferon Treatment. In Vivo 2017, 31, 723–729. [Google Scholar] [CrossRef]
- Liu, K.; Wang, F.-S.; Xu, R.; Liu, K.; Wang, F.-S.; Xu, R. Neutrophils in liver diseases: Pathogenesis and therapeutic targets. Cell. Mol. Immunol. 2021, 18, 38–44. [Google Scholar] [CrossRef]
- Evans, S.S.; Repasky, E.A.; Fisher, D.T. Fever and the thermal regulation of immunity: The immune system feels the heat. Nat. Rev. Immunol. 2015, 15, 335–349. [Google Scholar] [CrossRef]
- Milan-Mattos, J.; Anibal, F.; Perseguini, N.; Minatel, V.; Rehder-Santos, P.; Castro, C.; Vasilceac, F.; Mattiello, S.; Faccioli, L.; Catai, A. Effects of natural aging and gender on pro-inflammatory markers. Braz. J. Med Biol. Res. 2019, 52, e8392. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, A.; Frasson, C.; De Nuzzo, D.; Nicolli, A.; Scapellato, M.L. Significance of anti-HB levels below 10 IU/L after vaccination against hepatitis B in infancy or adolescence: An update in relation to sex. Hum. Vaccines Immunother. 2020, 16, 460–464. [Google Scholar] [CrossRef]
- Yager, E.J.; Ahmed, M.; Lanzer, K.; Randall, T.D.; Woodland, D.L.; Blackman, M.A. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J. Exp. Med. 2008, 205, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.M.; Ansari, A.M.; Frater, J.; Klenerman, P.; Dunachie, S.; Barnes, E.; Ogbe, A. The impact of pre-existing cross-reactive immunity on SARS-CoV-2 infection and vaccine responses. Nat. Rev. Immunol. 2022, 23, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.I. Platelet function and ageing. Mamm. Genome 2016, 27, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ge, S.; Cheng, Y.; Wang, X. Multi-View Spectral Clustering Based on Multi-Smooth Representation Fusion for Cancer Subtype Prediction. Front. Genet. 2021, 12, 718915. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Chierici, M.; Bussola, N.; Marcolini, A.; Francescatto, M.; Zandonà, A.; Trastulla, L.; Agostinelli, C.; Jurman, G.; Furlanello, C. Integrative Network Fusion: A Multi-Omics Approach in Molecular Profiling. Front. Oncol. 2020, 10, 1065. [Google Scholar] [CrossRef]
- Pai, S.; Hui, S.; Isserlin, R.; Shah, M.A.; Kaka, H.; Bader, G.D. netDx: Interpretable patient classification using integrated patient similarity networks. Mol. Syst. Biol. 2019, 15, e8497. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Arora, R.; Livescu, K.; Bilmes, J. On Deep Multi-View Representation Learning: Objectives and Optimization. arXiv 2016, arXiv:1602.01024. [Google Scholar] [CrossRef]
- Simpson, E.H. The Interpretation of Interaction in Contingency Tables. J. R. Stat. Soc. Ser. B Methodol. 1951, 13, 238–241. [Google Scholar] [CrossRef]
- Tu, Y.-K.; Gunnell, D.; Gilthorpe, M.S. Simpson’s Paradox, Lord’s Paradox, and Suppression Effects are the same phenomenon—The reversal paradox. Emerg. Themes Epidemiol. 2008, 5, 2. [Google Scholar] [CrossRef]
- Gilbert, S.C. T-cell-inducing vaccines—What’s the future. Immunology 2012, 135, 19–26. [Google Scholar] [CrossRef]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Collier, A.-R.Y.; Yu, J.; McMahan, K.; Liu, J.; Chandrashekar, A.; Maron, J.S.; Atyeo, C.; Martinez, D.R.; Ansel, J.L.; Aguayo, R.; et al. Differential Kinetics of Immune Responses Elicited by COVID-19 Vaccines. N. Engl. J. Med. 2021, 385, 2010–2012. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Outcome | AB Titer | Time | # in Group |
|---|---|---|---|
| non-converters | <10 IU/L | - | 4 * |
| early converters | >10 IU/L | 60 days | 21 |
| late-converters | >10 IU/L | 180 days | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Affaticati, F.; Bartholomeus, E.; Mullan, K.; Damme, P.V.; Beutels, P.; Ogunjimi, B.; Laukens, K.; Meysman, P. Multi-View Learning to Unravel the Different Levels Underlying Hepatitis B Vaccine Response. Vaccines 2023, 11, 1236. https://doi.org/10.3390/vaccines11071236
Affaticati F, Bartholomeus E, Mullan K, Damme PV, Beutels P, Ogunjimi B, Laukens K, Meysman P. Multi-View Learning to Unravel the Different Levels Underlying Hepatitis B Vaccine Response. Vaccines. 2023; 11(7):1236. https://doi.org/10.3390/vaccines11071236
Chicago/Turabian StyleAffaticati, Fabio, Esther Bartholomeus, Kerry Mullan, Pierre Van Damme, Philippe Beutels, Benson Ogunjimi, Kris Laukens, and Pieter Meysman. 2023. "Multi-View Learning to Unravel the Different Levels Underlying Hepatitis B Vaccine Response" Vaccines 11, no. 7: 1236. https://doi.org/10.3390/vaccines11071236
APA StyleAffaticati, F., Bartholomeus, E., Mullan, K., Damme, P. V., Beutels, P., Ogunjimi, B., Laukens, K., & Meysman, P. (2023). Multi-View Learning to Unravel the Different Levels Underlying Hepatitis B Vaccine Response. Vaccines, 11(7), 1236. https://doi.org/10.3390/vaccines11071236