
Evolution and Progress of mRNA Vaccines in the Treatment of Melanoma: Future Prospects
Abstract
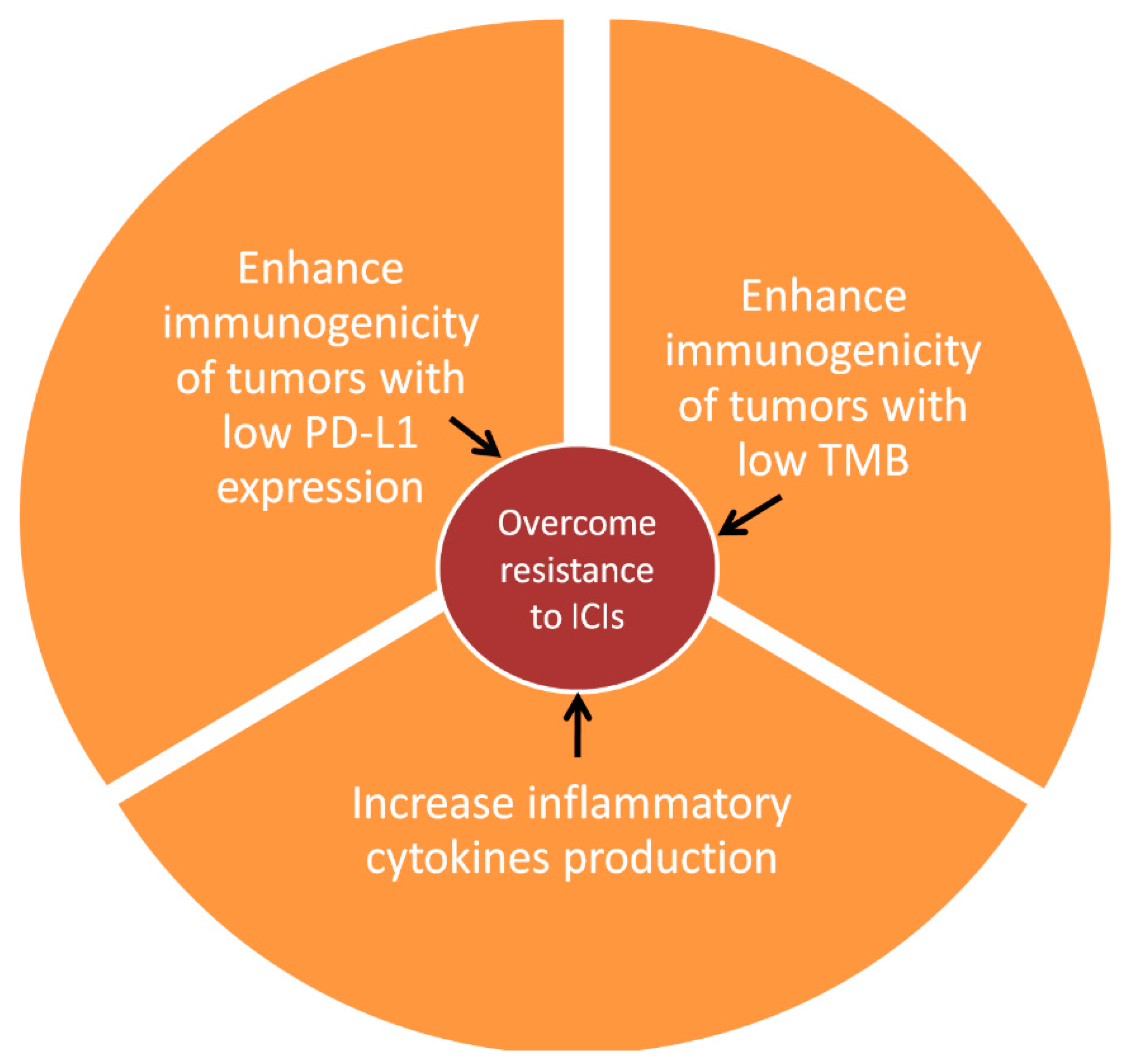
1. Introduction
2. Preclinical Evidence
3. Clinical Evidence
4. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brenner, S.; Jacob, F.; Meselson, M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247 Pt 1, 1465–1468. [Google Scholar] [CrossRef]
- Baklaushev, V.P.; Kilpeläinen, A.; Petkov, S.; Abakumov, M.A.; Grinenko, N.F.; Yusubalieva, G.M.; Latanova, A.A.; Gubskiy, I.L.; Zabozlaev, F.G.; Starodubova, E.S.; et al. Luciferase Expression Allows Bioluminescence Imaging But Imposes Limitations on the Orthotopic Mouse (4T1) Model of Breast Cancer. Sci. Rep. 2017, 7, 7715. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.E.; Hornig, Y.S.; Oei, Y.; Dusich, J.; Purchio, T. Bioluminescent human breast cancer cell lines that permit rapid and sensitive in vivo detection of mammary tumors and multiple metastases in immune deficient mice. Breast Cancer Res. 2005, 7, R444–R454. [Google Scholar] [CrossRef] [PubMed]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Zhou, W.Z.; Hoon, D.S.; Huang, S.K.; Fujii, S.; Hashimoto, K.; Morishita, R.; Kaneda, Y. RNA melanoma vaccine: Induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Hum. Gene Ther. 1999, 10, 2719–2724. [Google Scholar] [CrossRef]
- Boczkowski, D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Shroff, R.T.; Chalasani, P.; Wei, R.; Pennington, D.; Quirk, G.; Schoenle, M.V.; Peyton, K.L.; Uhrlaub, J.L.; Ripperger, T.J.; Jergović, M.; et al. Immune responses to two and three doses of the BNT162b2 mRNA vaccine in adults with solid tumors. Nat. Med. 2021, 27, 2002–2011. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA vaccine against SARS-CoV2dpreliminary report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef]
- Iavarone, C.; O’hagan, D.T.; Yu, D.; Delahaye, N.F.; Ulmer, J.B. Mechanism of action of mRNA-based vaccines. Expert Rev. Vaccines 2017, 16, 871–881. [Google Scholar] [CrossRef]
- Tomba’cz, I.; Weissman, D.; Pardi, N. Vaccination with messenger RNA: A promising alternative to DNA vaccination. Methods Mol. Biol. 2021, 2197, 13–31. [Google Scholar]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Barbier, A.J.; Jiang, A.Y.; Zhang, P.; Wooster, R.; Anderson, D.G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022, 40, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Van Nuffel, A.M.; Wilgenhof, S.; Thielemans, K.; Bonehill, A. Overcoming HLA restriction in clinical trials: Immune monitoring of mRNA-loaded DC therapy. Oncoimmunology 2012, 1, 1392–1394. [Google Scholar] [CrossRef]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA Vaccine Era-Mechanisms, Drug Platform and Clinical Prospection. Int. J. Mol. Sci. 2020, 21, 6582. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target Ther. 2023, 8, 9. [Google Scholar] [CrossRef]
- Liu, C.C.; Yang, H.; Zhang, R.; Zhao, J.J.; Hao, D.J. Tumour-associated antigens and their anti-cancer applications. Eur. J. Cancer Care 2017, 26, e12446. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, M.; Marino, F. Understanding the Pharmacology of COVID-19 mRNA Vaccines: Playing Dice with the Spike? Int. J. Mol. Sci. 2022, 23, 10881. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Ann. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Fundytus, A.; Booth, C.M.; Tannock, I.F. How low can you go? PD-L1 expression as a biomarker in trials of cancer immunotherapy. Ann. Oncol. 2021, 32, 833–836. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Long, H.; Jia, Q.; Wang, L.; Fang, W.; Wang, Z.; Jiang, T.; Zhou, F.; Jin, Z.; Huang, J.; Zhou, L.; et al. Tumor-induced erythroid precursor-differentiated myeloid cells mediate immunosuppression and curtail anti-PD-1/PD-L1 treatment efficacy. Cancer Cell 2022, 40, 674–693.e7. [Google Scholar] [CrossRef]
- Garcia Garcia, C.J.; Huang, Y.; Fuentes, N.R.; Turner, M.C.; Monberg, M.E.; Lin, D.; Nguyen, N.D.; Fujimoto, T.N.; Zhao, J.; Lee, J.J.; et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology 2022, 162, 2018–2031. [Google Scholar] [CrossRef]
- Wu, Y.; Yi, M.; Niu, M.; Mei, Q.; Wu, K. Myeloid-derived suppressor cells: An emerging target for anticancer immunotherapy. Mol. Cancer 2022, 21, 184. [Google Scholar] [CrossRef]
- Bernardo, M.; Tolstykh, T.; Zhang, Y.A.; Bangari, D.S.; Cao, H.; Heyl, K.A.; Lee, J.S.; Malkova, N.V.; Malley, K.; Marquez, E.; et al. An experimental model of anti-PD-1 resistance exhibits activation of TGFß and Notch pathways and is sensitive to local mRNA immunotherapy. Oncoimmunology 2021, 10, 1881268. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019, 9, 1148. [Google Scholar] [CrossRef] [PubMed]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Xu, Z.; Miao, L.; Huang, L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol. Ther. 2018, 26, 420–434. [Google Scholar] [CrossRef]
- Zhang, H.; You, X.; Wang, X.; Cui, L.; Wang, Z.; Xu, F.; Li, M.; Yang, Z.; Liu, J.; Huang, P.; et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through Toll-like receptor 4 signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2005191118. [Google Scholar] [CrossRef]
- Li, Q.; Ren, J.; Liu, W.; Jiang, G.; Hu, R. CpG Oligodeoxynucleotide Developed to Activate Primate Immune Responses Promotes Antitumoral Effects in Combination with a Neoantigen-Based mRNA Cancer Vaccine. Drug Des. Dev. Ther. 2021, 15, 3953–3963. [Google Scholar] [CrossRef]
- Chen, J.; Ye, Z.; Huang, C.; Qiu, M.; Song, D.; Li, Y.; Xu, Q. Lipid nanoparticle-mediated lymph node-targeting delivery of mRNA cancer vaccine elicits robust CD8+ T cell response. Proc. Natl. Acad. Sci. USA 2022, 119, e2207841119. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; Pak, B.J.; Bani, M.R.; Kapoor, M.; Lu, S.J.; Tamir, A.; Kerbel, R.S.; Ben-David, Y. Tyrosinase-related protein 2 as a mediator of melanoma specific resistance to cis-diamminedichloroplatinum(II): Therapeutic implications. Oncogene 2000, 19, 395–402. [Google Scholar] [CrossRef]
- He, M.; Huang, L.; Hou, X.; Zhong, C.; Bachir, Z.A.; Lan, M.; Chen, R.; Gao, F. Efficient ovalbumin delivery using a novel multifunctional micellar platform for targeted melanoma immunotherapy. Int. J. Pharm. 2019, 560, 1–10. [Google Scholar] [CrossRef]
- Kyte, J.A.; Mu, L.; Aamdal, S.; Kvalheim, G.; Dueland, S.; Hauser, M.; Gullestad, H.P.; Ryder, T.; Lislerud, K.; Hammerstad, H.; et al. Phase I/II trial of melanoma therapy with dendritic cells transfected with autologous tumor-mRNA. Cancer Gene Ther. 2006, 13, 905–918. [Google Scholar] [CrossRef]
- Kyte, J.A.; Kvalheim, G.; Lislerud, K.; Thor Straten, P.; Dueland, S.; Aamdal, S.; Gaudernack, G. T cell responses in melanoma patients after vaccination with tumor-mRNA transfected dendritic cells. Cancer Immunol. Immunother. 2007, 56, 659–675. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.G.; Garbe, C. Direct injection of protamine-protected mRNA: Results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Van Nuffel, A.M.; Corthals, J.; Heirman, C.; Tuyaerts, S.; Benteyn, D.; De Coninck, A.; Van Riet, I.; Verfaillie, G.; Vandeloo, J.; et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J. Immunother. 2011, 34, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Benteyn, D.; Van Nuffel, A.M.; Wilgenhof, S.; Corthals, J.; Heirman, C.; Neyns, B.; Thielemans, K.; Bonehill, A. Characterization of CD8+ T-cell responses in the peripheral blood and skin injection sites of melanoma patients treated with mRNA electroporated autologous dendritic cells (TriMixDC-MEL). Biomed Res Int. 2013, 2013, 976383. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Van Nuffel, A.M.; Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Neyns, B. Long-term clinical outcome of melanoma patients treated with messenger RNA-electroporated dendritic cell therapy following complete resection of metastases. Cancer Immunol. Immunother. 2015, 64, 381–388. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef]
- Arance Fernandez, A.M.; Baurain, J.-F.; Vulsteke, C.; Rutten, A.; Soria, A.; Carrasco, J.; Neyns, B.; De Keersmaecker, B.; Van Assche, T.; Lindmark, B. A phase I study (E011-MEL) of a TriMix-based mRNA immunotherapy (ECI-006) in resected melanoma patients: Analysis of safety and immunogenicity. J. Clin. Oncol. 2019, 37, 2641. [Google Scholar] [CrossRef]
- Ping, H.; Yu, W.; Gong, X.; Tong, X.; Lin, C.; Chen, Z.; Cai, C.; Guo, K.; Ke, H. Analysis of melanoma tumor antigens and immune subtypes for the development of mRNA vaccine. Investig. New Drugs 2022, 40, 1173–1184. [Google Scholar] [CrossRef]
- Lorentzen, C.L.; Haanen, J.B.; Met, Ö.; Svane, I.M. Clinical advances and ongoing trials on mRNA vaccines for cancer treatment. Lancet Oncol. 2022, 23, e450–e458. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ (accessed on 10 January 2023).
- Moderna and Merck Announce mRNA-4157/V940, an Investigational Personalized mRNA Cancer Vaccine, in Combination with KEYTRUDA® (Pembrolizumab), Met Primary Efficacy Endpoint in Phase 2b KEYNOTE-942 Trial. Available online: https://www.merck.com/news/moderna-and-merck-announce-mrna-4157-v940-an-investigational-personalized-mrna-cancer-vaccine-in-combination-with-keytruda-pembrolizumab-met-primary-efficacy-endpoint-in-phase-2b-keynote-94/ (accessed on 13 December 2022).
- Breakthrough in mRNA Vaccines for Melanoma. Available online: https://www1.racgp.org.au/newsgp/clinical/breakthrough-in-mrna-vaccines-for-melanoma (accessed on 10 January 2023).
- mRNA Vaccine Plus KEYTRUDA® Improve Melanoma Survival. Available online: https://www.europeanpharmaceuticalreview.com/news/177505/mrna-vaccine-plus-keytruda-improve-melanoma-survival/ (accessed on 10 January 2023).


{kind=link}
| Experiment Subject | Vaccine Composition | Vaccine Transport | Results | Reference |
|---|---|---|---|---|
| Aggressive B16F10 murine melanoma models | Lipid nanoparticles-mRNA encoding gp100, TRP-2 | Direct vaccine administration | Tumor shrinkage Prolonged overall survival of the treated mice | Oberli et al., 2017 [33] |
| Immune-competent murine B16F10 melanoma model | LCP-based vaccine mRNA encoding TRP-2 siRNA targeting PD-L1 | Transfected DCs transported to mice | Efficient mRNA delivery to DCs in lymph nodes T cell specific reaction to TRP-2 Reduced tumor growth Enhanced CD8+ T cell proliferation | Wang et al., 2018 [34] |
| Murine melanoma models | Nanovaccine with C1 lipid nanoparticle mRNA encoding TRP-2 | Vaccine enters APCs via phagocytosis | TLR4 activation-Robust T cell activation Inflammatory cytokines inductionReduced tumor growth | Zhang et al., 2021 [35] |
| Syngeneic murine models | Tumor neoantigen mRNA, encapsulated in lipid nanoparticles | Intratumoral vaccine administration | Melanoma growth inhibition Immunogenically ”cold” tumors turn into “hot” | Li et al., 2021 [36] |
| B16F10 melanoma murine models | Lymph node-targeting lipid nanoparticle with mRNA encoding for ovalbumin, TRP-2 | Targeted delivery of mRNA to lymph nodes | Increased CD8+ T cell response Long term immune memory | Chen et al., 2022 [37] |
| Patient Population | Vaccine-Encoded Antigens | Outcomes | Reference |
|---|---|---|---|
| 22 patients with advanced malignant melanoma | Autologous tumor mRNA | Vaccine-specific immune response in 9/19 patients evaluable by T cell assays and in 8/18 patients evaluable by delayed-type hypersensitivity reaction | Kyte et al. 2006 [40] |
| 21 metastatic melanoma patients | Melan-A, Tyrosinase, gp100, MAGE-A1, MAGE-A3, Survivin | Safe, tolerable Antigen-specific T cell reaction in 2/4 patients CR in 1/7 patients | Weide et al. 2009 [42] |
| 35 advanced melanoma patients | Tyrosinase, gp100, MAGE-A3, MAGE-C2 | In patients treated by autologous DCs electroporated with mRNA vaccine plus IFN-α-2b: PR:1/17 SD: 5/17 | Wilgenhof et al. 2011 [43] |
| 14 recurrent melanoma patients | CD40L, TLR4, CD70 plus tyrosinase or MAGE-A3 or MAGE-C2 or gp100 | T cell-specific reaction in 11/14 patients (peripheral blood) and in 12/14 patients (tissue) CR: 2/14 PR: 1/14 SD: 4/14 | Benteyn et al. 2013 [44] |
| 30 patients with resected melanoma | Autologous mRNA | mRFS: 22 months (95% CI 12–32 months) 4yr OS 70% | Wilgenhof et al. 2015 [45] |
| 39 advanced melanoma patients | Tyrosinase, gp100, MAGE-A3, MAGE-C2 | 6mo DCR 51% CR: 20.5% PR: 17.9% T cell stimulation in 12/15 evaluable patients T cell response related to objective response | De Keersmaecker et al. 2020 [46] |
| 157 patients with resected melanoma | 20 tumor neoantigens | Decreased risk of relapse/death by 44% compared to pembrolizumab monotherapy | KEYNOTE-942, press release 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bafaloukos, D.; Gazouli, I.; Koutserimpas, C.; Samonis, G. Evolution and Progress of mRNA Vaccines in the Treatment of Melanoma: Future Prospects. Vaccines 2023, 11, 636. https://doi.org/10.3390/vaccines11030636
Bafaloukos D, Gazouli I, Koutserimpas C, Samonis G. Evolution and Progress of mRNA Vaccines in the Treatment of Melanoma: Future Prospects. Vaccines. 2023; 11(3):636. https://doi.org/10.3390/vaccines11030636
Chicago/Turabian StyleBafaloukos, Dimitrios, Ioanna Gazouli, Christos Koutserimpas, and George Samonis. 2023. "Evolution and Progress of mRNA Vaccines in the Treatment of Melanoma: Future Prospects" Vaccines 11, no. 3: 636. https://doi.org/10.3390/vaccines11030636
APA StyleBafaloukos, D., Gazouli, I., Koutserimpas, C., & Samonis, G. (2023). Evolution and Progress of mRNA Vaccines in the Treatment of Melanoma: Future Prospects. Vaccines, 11(3), 636. https://doi.org/10.3390/vaccines11030636