
Vaccination with an HIV T-Cell Immunogen (HTI) Using DNA Primes Followed by a ChAdOx1-MVA Boost Is Immunogenic in Gut Microbiota-Depleted Mice despite Low IL-22 Serum Levels

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
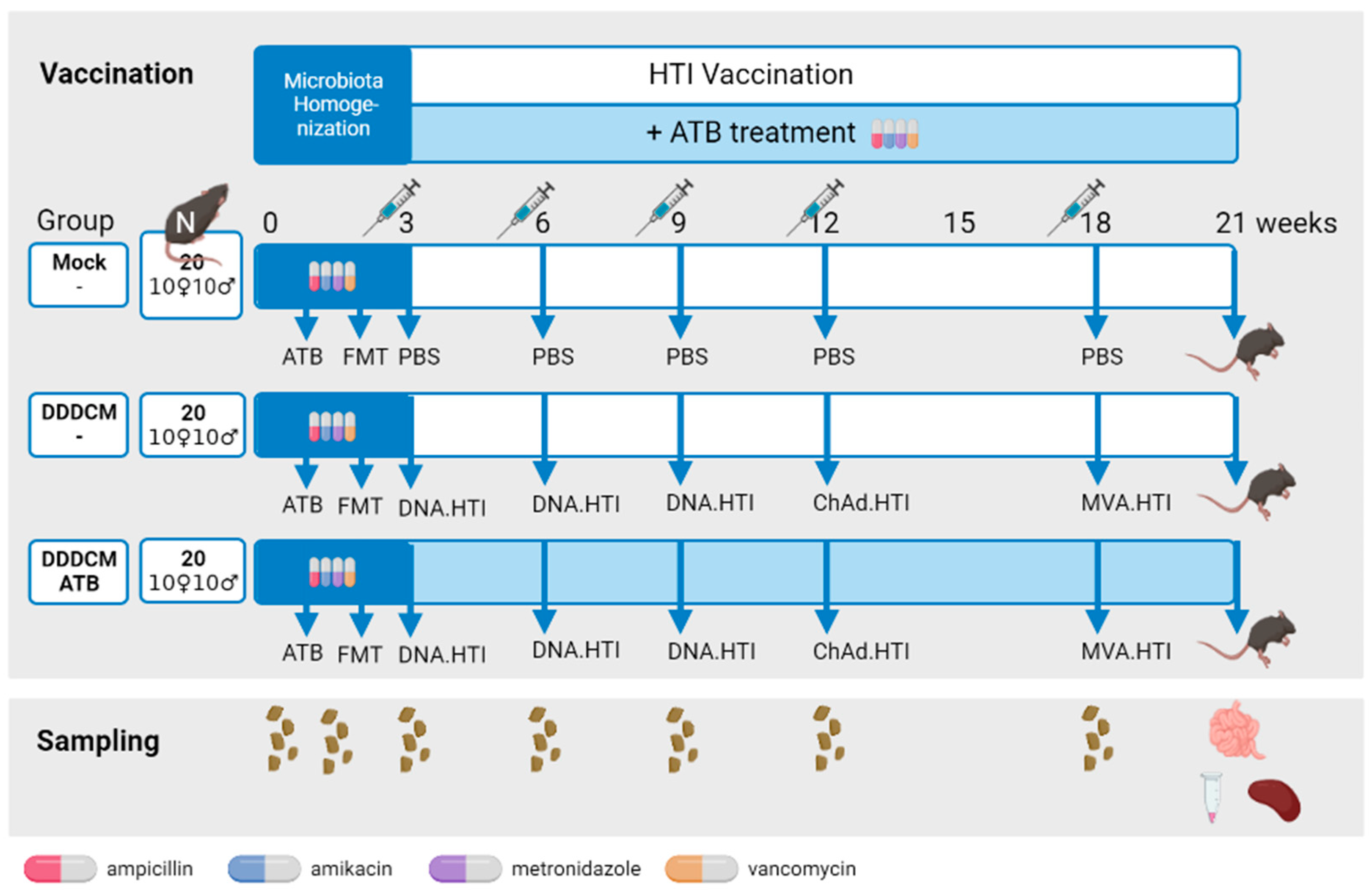
2.1. Experimental Design
2.2. Sample Collection and Processing
2.3. DNA Extraction and 16S rRNA Sequencing
2.4. 16S rRNA Sequence Analysis
2.5. Overlapping Peptides Covering the HTI Sequence
2.6. Mouse IFNγ ELISPOT Assay
2.7. Luminex Assay
2.8. Statistical Analysis of Vaccine Response Data
3. Results
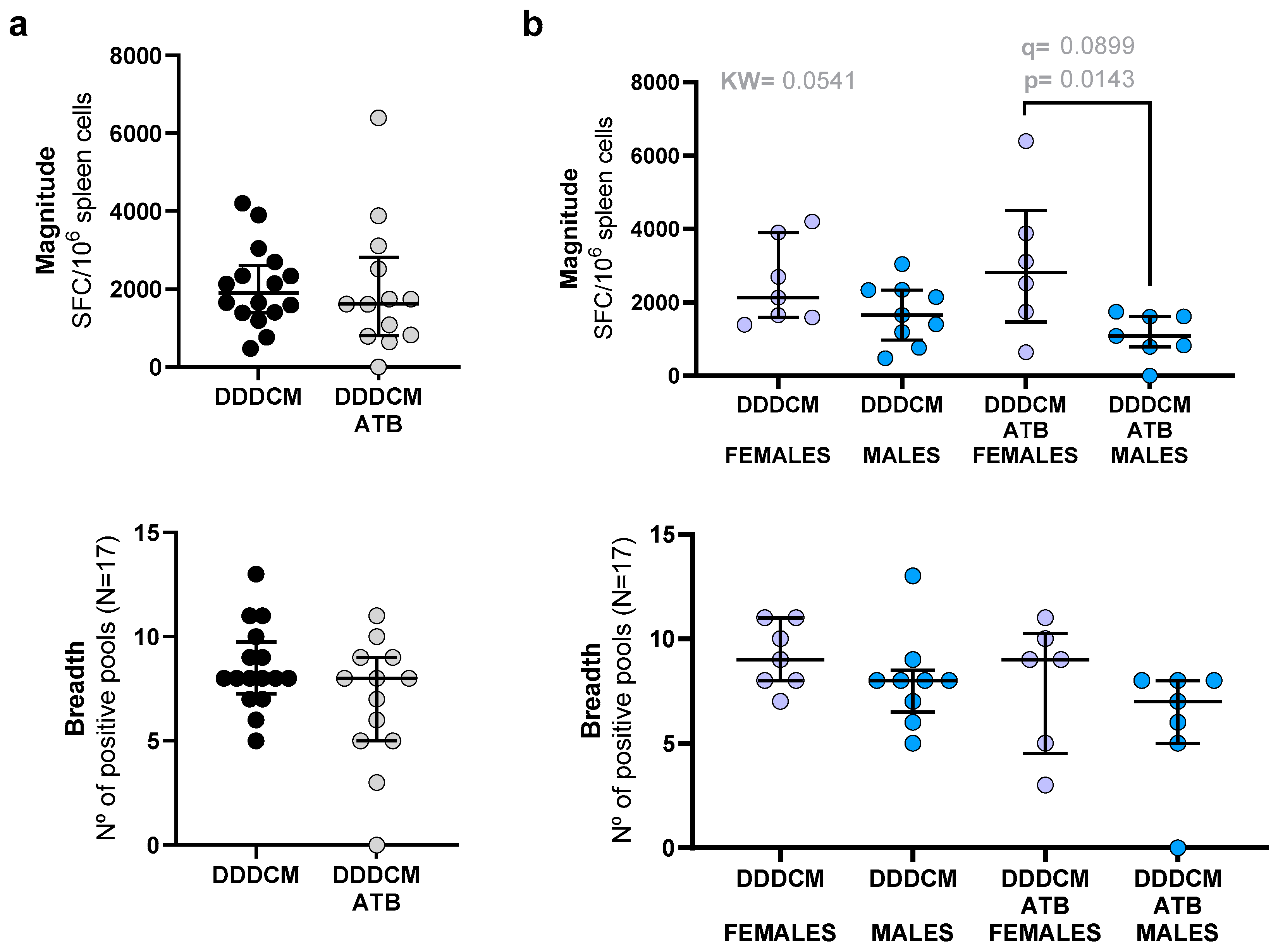
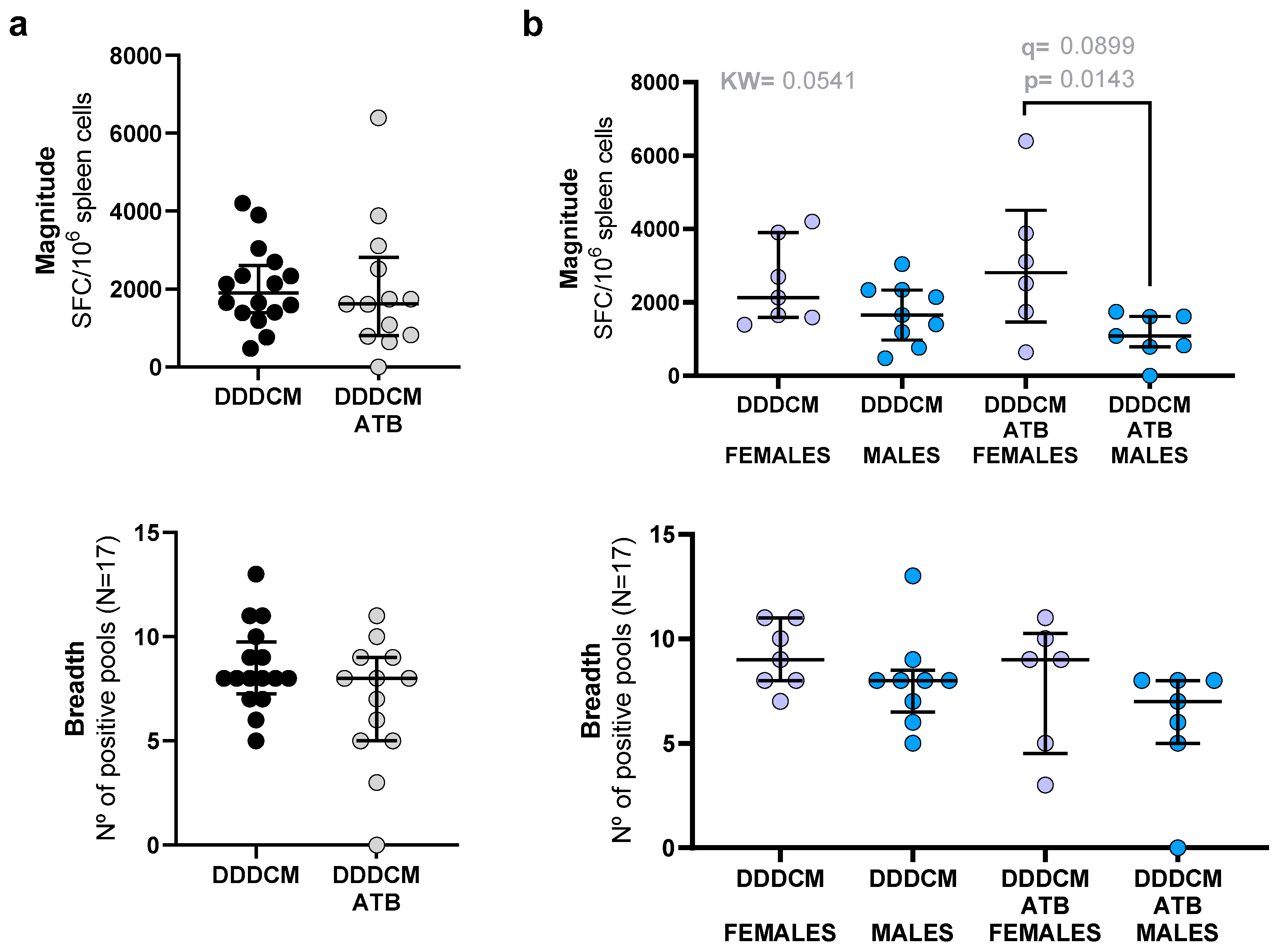
3.1. Vaccination with a Highly Immunogenic Prime-Boost Strategy Triggers Strong HTI-Specific IFNγ Producing T-Cell Responses despite Depleted Microbiota
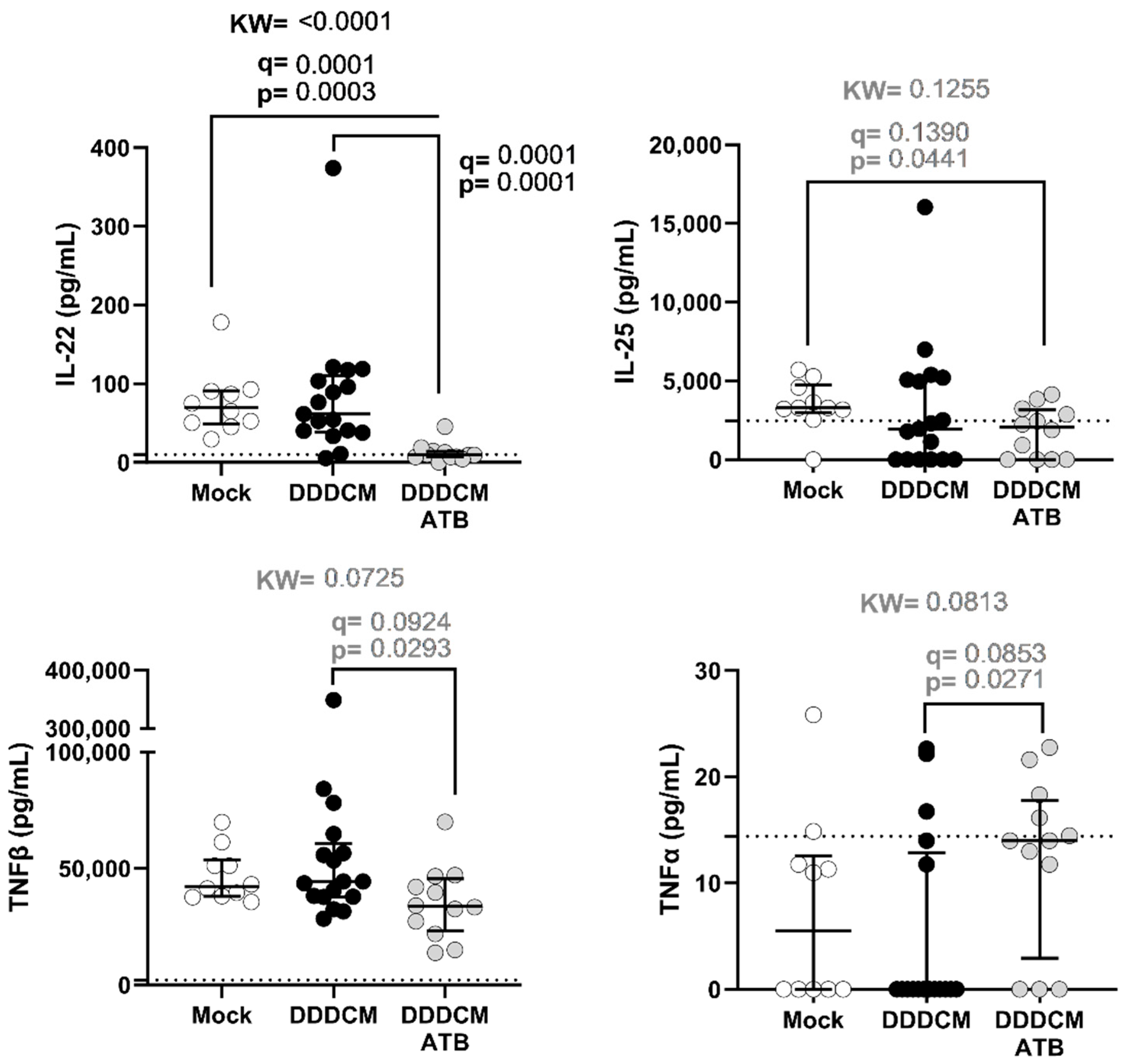
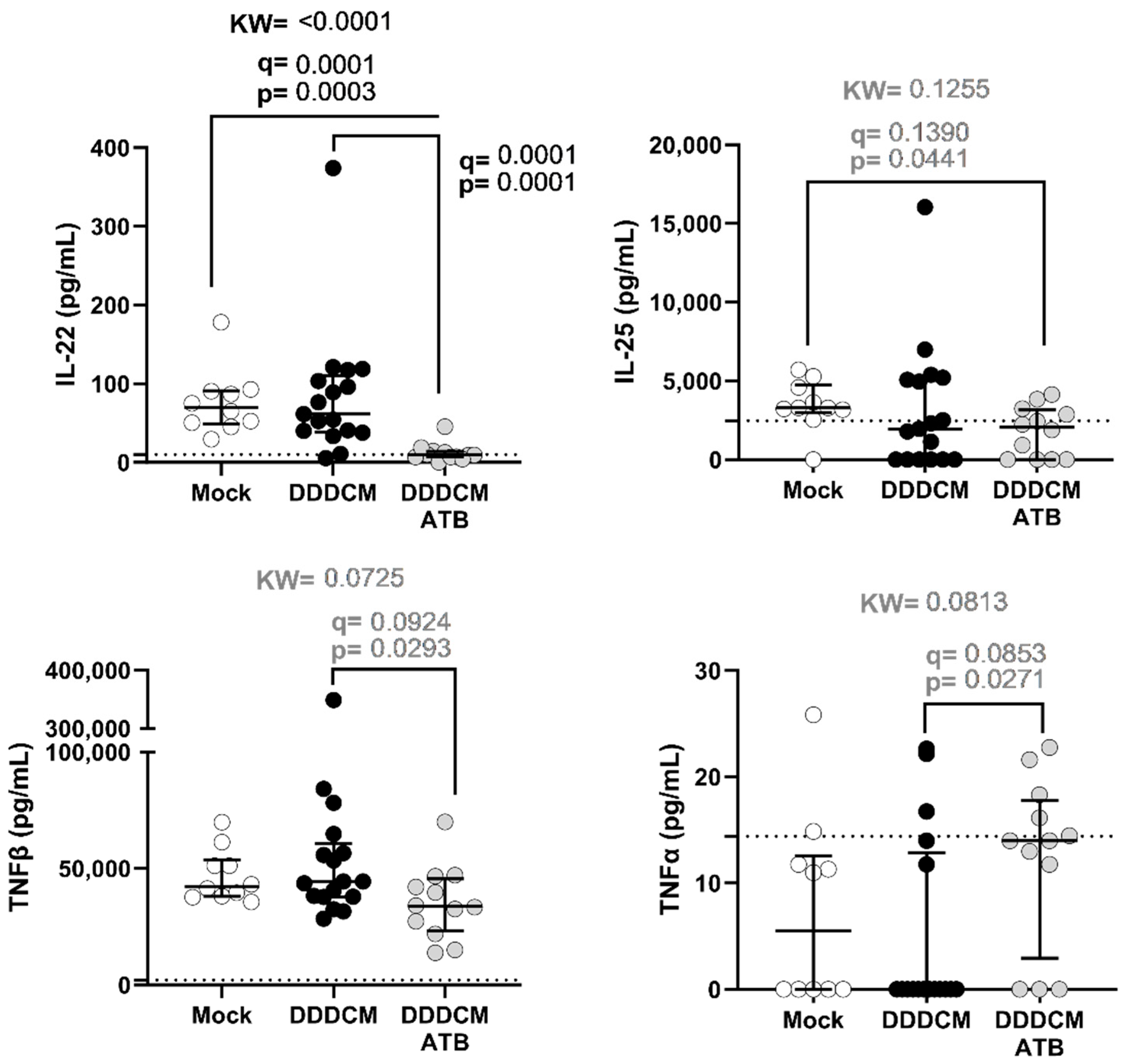
3.2. Gut Microbiota and DDDCM.HTI Vaccination Affect Serum Cytokine Levels
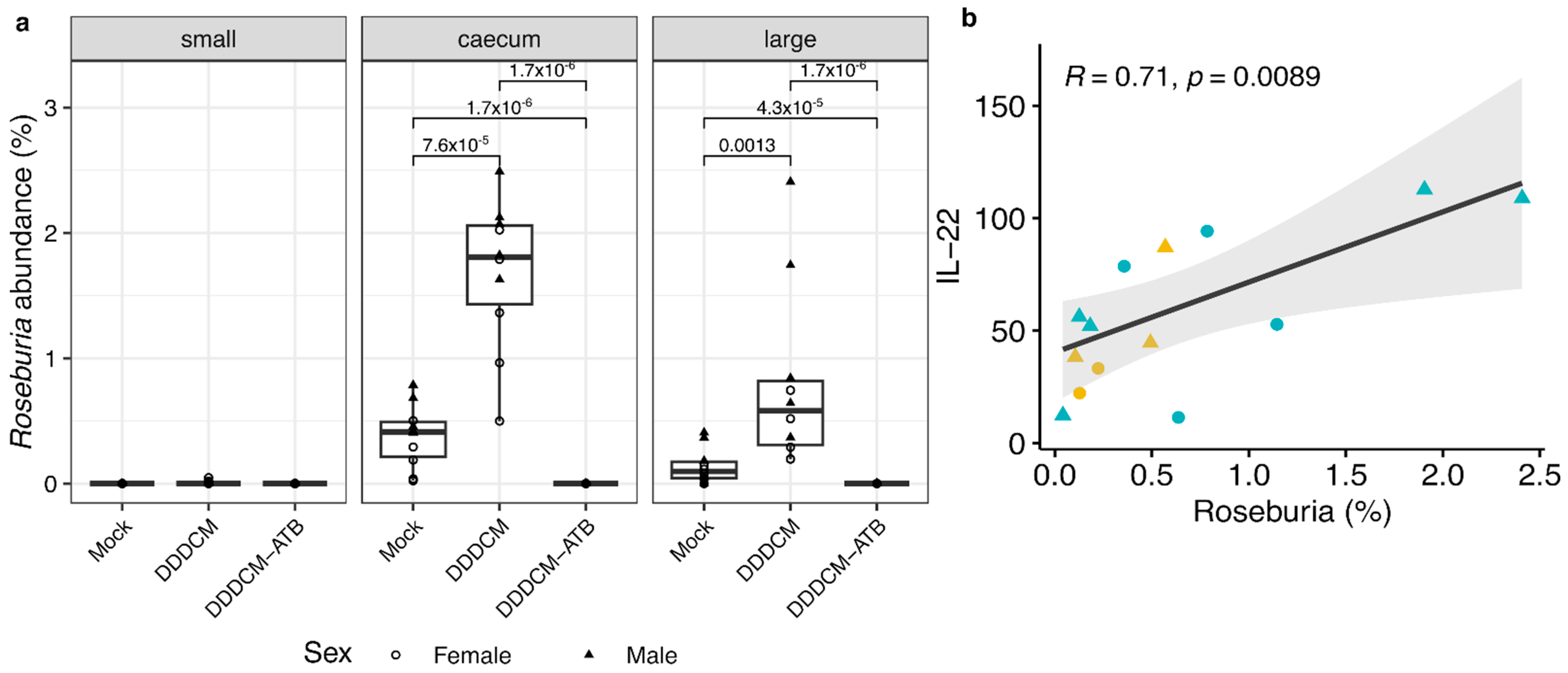
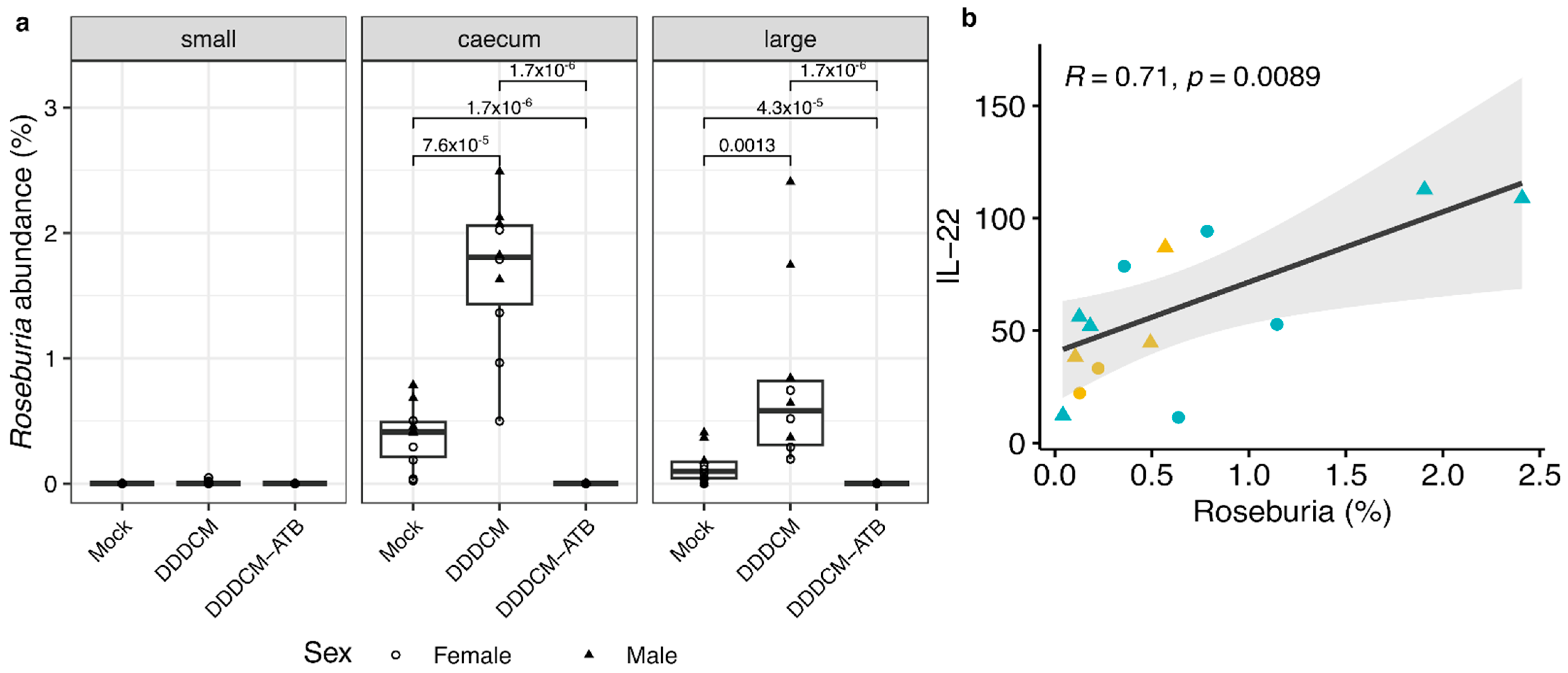
3.3. IL-22 Serum Levels Correlate with the Abundance of Short Chain Fatty Acid (SCFA) Producing Bacteria in Large Intestine
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lynn, D.J.; Benson, S.C.; Lynn, M.A.; Pulendran, B. Modulation of Immune Responses to Vaccination by the Microbiota: Implications and Potential Mechanisms. Nat. Rev. Immunol. 2022, 22, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.J.; Pulendran, B. The Potential of the Microbiota to Influence Vaccine Responses. J. Leukoc. Biol. 2018, 103, 225. [Google Scholar] [CrossRef]
- McCoy, K.D.; Burkhard, R.; Geuking, M.B. The Microbiome and Immune Memory Formation. Immunol. Cell Biol. 2019, 97, 625–635. [Google Scholar] [PubMed]
- De Agüero, M.G.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The Maternal Microbiota Drives Early Postnatal Innate Immune Development. Science 2016, 351, 1296–1302. [Google Scholar] [CrossRef]
- Selma-Royo, M.; Calatayud Arroyo, M.; García-Mantrana, I.; Parra-Llorca, A.; Escuriet, R.; Martínez-Costa, C.; Collado, M.C. Perinatal Environment Shapes Microbiota Colonization and Infant Growth: Impact on Host Response and Intestinal Function. Microbiome 2020, 8, 167. [Google Scholar] [CrossRef]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How Colonization by Microbiota in Early Life Shapes the Immune System. Science 2016, 352, 539–544. [Google Scholar] [CrossRef]
- Ronan, V.; Yeasin, R.; Claud, E.C. Childhood Development and the Microbiome-The Intestinal Microbiota in Maintenance of Health and Development of Disease During Childhood Development. Gastroenterology 2021, 160, 495–506. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Eberl, G. Imprinting of the Immune System by the Microbiota Early in Life. Mucosal Immunol. 2020, 13, 183–189. [Google Scholar] [CrossRef]
- Kim, J.E.; Li, B.; Fei, L.; Horne, R.; Lee, D.; Loe, A.K.; Miyake, H.; Ayar, E.; Kim, D.K.; Surette, M.G.; et al. Gut Microbiota Promotes Stem Cell Differentiation through Macrophage and Mesenchymal Niches in Early Postnatal Development. Immunity 2022, 55, 2300–2317.e6. [Google Scholar] [CrossRef]
- Köhler, A.; Delbauve, S.; Smout, J.; Torres, D.; Flamand, V. Very Early-Life Exposure to Microbiota-Induced TNF Drives the Maturation of Neonatal Pre-CDC1. Gut 2021, 70, 511–521. [Google Scholar] [CrossRef]
- Zegarra-Ruiz, D.F.; Kim, D.V.; Norwood, K.; Kim, M.; Wu, W.J.H.; Saldana-Morales, F.B.; Hill, A.A.; Majumdar, S.; Orozco, S.; Bell, R.; et al. Thymic Development of Gut-Microbiota-Specific T Cells. Nature 2021, 594, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Lai, G.C.; Yao, L.J.; Aung, T.T.; Shental, N.; Rotter-Maskowitz, A.; Shepherdson, E.; Singh, G.S.N.; Pai, R.; Shanti, A.; et al. Microbial Exposure during Early Human Development Primes Fetal Immune Cells. Cell 2021, 184, 3394–3409.e20. [Google Scholar] [CrossRef]
- de Jong, S.E.; Olin, A.; Pulendran, B. The Impact of the Microbiome on Immunity to Vaccination in Humans. Cell Host Microbe 2020, 28, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Ruff, W.E.; Greiling, T.M.; Kriegel, M.A. Host-Microbiota Interactions in Immune-Mediated Diseases. Nat. Rev. Microbiol. 2020, 18, 521–538. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Shoenfeld, Y. The Microbiome in Autoimmune Diseases. Clin. Exp. Immunol. 2019, 195, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yao, M.; Lv, L.; Ling, Z.; Li, L. The Human Microbiota in Health and Disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- Niewiem, M.; Grzybowska-Chlebowczyk, U. Intestinal Barrier Permeability in Allergic Diseases. Nutrients 2022, 14, 1893. [Google Scholar] [CrossRef]
- Fujimura, K.E.; Sitarik, A.R.; Havstad, S.; Lin, D.L.; Levan, S.; Fadrosh, D.; Panzer, A.R.; Lamere, B.; Rackaityte, E.; Lukacs, N.W.; et al. Neonatal Gut Microbiota Associates with Childhood Multisensitized Atopy and T Cell Differentiation. Nat. Med. 2016, 22, 1187–1191. [Google Scholar] [CrossRef]
- Hedin, C.R.; McCarthy, N.E.; Louis, P.; Farquharson, F.M.; McCartney, S.; Taylor, K.; Prescott, N.J.; Murrells, T.; Stagg, A.J.; Whelan, K.; et al. Altered Intestinal Microbiota and Blood T Cell Phenotype Are Shared by Patients with Crohn’s Disease and Their Unaffected Siblings. Gut 2014, 63, 1578–1586. [Google Scholar] [CrossRef]
- Zhang, Q.; Cheng, L.; Wang, J.; Hao, M.; Che, H. Antibiotic-Induced Gut Microbiota Dysbiosis Damages the Intestinal Barrier, Increasing Food Allergy in Adult Mice. Nutrients 2021, 13, 3315. [Google Scholar] [CrossRef]
- Blázquez, A.B.; Berin, M.C. Microbiome and Food Allergy. Transl. Res. 2017, 179, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.H.; Baek, M.G.; Choi, S.M.; Bae, B.; Kim, R.Y.; Kim, Y.C.; Kim, H.Y.; Yi, H.; Kang, H.R. Alteration of Lung and Gut Microbiota in IL-13-Transgenic Mice Simulating Chronic Asthma. J. Microbiol. Biotechnol. 2020, 30, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The Microbiome and Innate Immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal Microbiota-Derived Short-Chain Fatty Acids Regulation of Immune Cell IL-22 Production and Gut Immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef]
- Xing, C.; Wang, M.; Ajibade, A.A.; Tan, P.; Fu, C.; Chen, L.; Zhu, M.; Hao, Z.Z.; Chu, J.; Yu, X.; et al. Microbiota Regulate Innate Immune Signaling and Protective Immunity against Cancer. Cell Host Microbe 2021, 29, 959–974.e7. [Google Scholar] [CrossRef]
- Hu, C.; Xu, B.; Wang, X.; Wan, W.H.; Lu, J.; Kong, D.; Jin, Y.; You, W.; Sun, H.; Mu, X.; et al. Gut Microbiota-Derived Short-Chain Fatty Acids Regulate Group 3 Innate Lymphoid Cells in HCC. Hepatology 2023, 77, 48–64. [Google Scholar] [CrossRef]
- Singh, R.; Chandrashekharappa, S.; Bodduluri, S.R.; Baby, B.V.; Hegde, B.; Kotla, N.G.; Hiwale, A.A.; Saiyed, T.; Patel, P.; Vijay-Kumar, M.; et al. Enhancement of the Gut Barrier Integrity by a Microbial Metabolite through the Nrf2 Pathway. Nat. Commun. 2019, 10, 89. [Google Scholar] [CrossRef]
- Liang, L.; Liu, L.; Zhou, W.; Yang, C.; Mai, G.; Li, H.; Chen, Y. Gut Microbiota-Derived Butyrate Regulates Gut Mucus Barrier Repair by Activating the Macrophage/WNT/ERK Signaling Pathway. Clin. Sci. 2022, 136, 291–307. [Google Scholar] [CrossRef]
- Jiao, Y.; Wu, L.; Huntington, N.D.; Zhang, X. Crosstalk Between Gut Microbiota and Innate Immunity and Its Implication in Autoimmune Diseases. Front. Immunol. 2020, 11, 282. [Google Scholar]
- Li, G.; Lin, J.; Zhang, C.; Gao, H.; Lu, H.; Gao, X.; Zhu, R.; Li, Z.; Li, M.; Liu, Z. Microbiota Metabolite Butyrate Constrains Neutrophil Functions and Ameliorates Mucosal Inflammation in Inflammatory Bowel Disease. Gut Microbes 2021, 13, e1968257. [Google Scholar] [CrossRef]
- Silva, L.M.; Doyle, A.D.; Greenwell-Wild, T.; Dutzan, N.; Tran, C.L.; Abusleme, L.; Juang, L.J.; Leung, J.; Chun, E.M.; Lum, A.G.; et al. Fibrin Is a Critical Regulator of Neutrophil Effector Function at the Oral Mucosal Barrier. Science 2021, 374, eabl5450. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 9th ed.; Elsevier: Philadelphia, PA, USA, 2018; ISBN 978-0-323-47978-3. [Google Scholar]
- Mishima, Y.; Oka, A.; Liu, B.; Herzog, J.W.; Eun, C.S.; Fan, T.J.; Bulik-Sullivan, E.; Carroll, I.M.; Hansen, J.J.; Chen, L.; et al. Microbiota Maintain Colonic Homeostasis by Activating TLR2/MyD88/PI3K Signaling in IL-10-Producing Regulatory B Cells. J. Clin. Investig. 2019, 129, 3702–3716. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Z.; Ravindran, R.; Chassaing, B.; Carvalho, F.A.; Maddur, M.S.; Bower, M.; Hakimpour, P.; Gill, K.P.; Nakaya, H.I.; Yarovinsky, F.; et al. TLR5-Mediated Sensing of Gut Microbiota Is Necessary for Antibody Responses to Seasonal Influenza Vaccination. Immunity 2014, 41, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M.; Longman, R.S.; Iliev, I.D.; Sonnenberg, G.F.; Artis, D. Regulation of Inflammation by Microbiota Interactions with the Host. Nat. Immunol. 2017, 18, 851–860. [Google Scholar] [CrossRef]
- Sanchez, H.N.; Moroney, J.B.; Gan, H.; Shen, T.; Im, J.L.; Li, T.; Taylor, J.R.; Zan, H.; Casali, P. B Cell-Intrinsic Epigenetic Modulation of Antibody Responses by Dietary Fiber-Derived Short-Chain Fatty Acids. Nat. Commun. 2020, 11, 60. [Google Scholar] [CrossRef]
- Abdalkareem Jasim, S.; Jade Catalan Opulencia, M.; Alexis Ramírez-Coronel, A.; Kamal Abdelbasset, W.; Hasan Abed, M.; Markov, A.; Raheem Lateef Al-Awsi, G.; Azamatovich Shamsiev, J.; Thaeer Hammid, A.; Nader Shalaby, M.; et al. The Emerging Role of Microbiota-Derived Short-Chain Fatty Acids in Immunometabolism. Int. Immunopharmacol. 2022, 110, 108983. [Google Scholar] [CrossRef]
- Singh, N.; Thangaraju, M.; Prasad, P.D.; Martin, P.M.; Lambert, N.A.; Boettger, T.; Offermanns, S.; Ganapathy, V. Blockade of Dendritic Cell Development by Bacterial Fermentation Products Butyrate and Propionate through a Transporter (Slc5a8)-Dependent Inhibition of Histone Deacetylases. J. Biol. Chem. 2010, 285, 27601–27608. [Google Scholar] [CrossRef]
- Kim, S.H.; Cho, B.H.; Kiyono, H.; Jang, Y.S. Microbiota-Derived Butyrate Suppresses Group 3 Innate Lymphoid Cells in Terminal Ileal Peyer’s Patches. Sci. Rep. 2017, 7, 3980. [Google Scholar] [CrossRef]
- Tang, B.; Tang, L.; He, W.; Jiang, X.; Hu, C.; Li, Y.; Zhang, Y.; Pang, K.; Lei, Y.; Li, S.; et al. Correlation of Gut Microbiota and Metabolic Functions with the Antibody Response to the BBIBP-CorV Vaccine. Cell Rep. Med. 2022, 3, 100752. [Google Scholar] [CrossRef]
- Fix, J.; Chandrashekhar, K.; Perez, J.; Bucardo, F.; Hudgens, M.G.; Yuan, L.; Twitchell, E.; Azcarate-Peril, M.A.; Vilchez, S.; Becker-Dreps, S. Association between Gut Microbiome Composition and Rotavirus Vaccine Response among Nicaraguan Infants. Am. J. Trop. Med. Hyg. 2020, 102, 213–219. [Google Scholar] [CrossRef]
- Che, Y.; Fu, S.; Wang, H.; Suo, J.; Chen, C.; Pu, D.; Li, C.; Yang, Y. Correlation of the Gut Microbiota and Antitumor Immune Responses Induced by a Human Papillomavirus Therapeutic Vaccine. ACS Infect. Dis. 2022, 8, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, J.; Fu, Y.; Ye, H.; Liu, X.; Li, G.; Yang, X.; Yang, J. Influence of Gut Microbiota on Mucosal IgA Antibody Response to the Polio Vaccine. npj Vaccines 2020, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.; Ali, A.; Fuentes, S.; Korpela, K.; Kazi, M.; Tate, J.; Parashar, U.; Wiersinga, W.J.; Giaquinto, C.; de Weerth, C.; et al. Rotavirus Vaccine Response Correlates with the Infant Gut Microbiota Composition in Pakistan. Gut Microbes 2018, 9, 93–101. [Google Scholar] [CrossRef]
- Chac, D.; Bhuiyan, T.R.; Saha, A.; Alam, M.M.; Salma, U.; Jahan, N.; Chowdhury, F.; Khan, A.I.; Ryan, E.T.; LaRocque, R.; et al. Gut Microbiota and Development of Vibrio Cholerae-Specific Long-Term Memory b Cells in Adults after Whole-Cell Killed Oral Cholera Vaccine. Infect. Immun. 2021, 89, e0021721. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, Y.; Gui, J.; Nadeau, K.C.; Morrison, H.G.; Madan, J.; Karagas, M.R. A Prospective Study of the Infant Gut Microbiome in Relation to Vaccine Response. Pediatr. Res. 2023, 93, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.L.; Mullish, B.H.; Danckert, N.P.; Liu, Z.; Olbei, M.L.; Saifuddin, A.; Torkizadeh, M.; Ibraheim, H.; Blanco, J.M.; Roberts, L.A.; et al. The Gut Microbiota and Metabolome Are Associated with Diminished COVID-19 Vaccine-Induced Antibody Responses in Immunosuppressed Inflammatory Bowel Disease Patients. eBioMedicine 2023, 88, 104430. [Google Scholar] [CrossRef] [PubMed]
- Macbeth, J.C.; Liu, R.; Alavi, S.; Hsiao, A. A Dysbiotic Gut Microbiome Suppresses Antibody Mediated-Protection against Vibrio Cholerae. iScience 2021, 24, 103443. [Google Scholar] [CrossRef]
- Hagan, T.; Cortese, M.; Rouphael, N.; Boudreau, C.; Linde, C.; Maddur, M.S.; Das, J.; Wang, H.; Guthmiller, J.; Zheng, N.Y.; et al. Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 2019, 178, 1313–1328.e13. [Google Scholar] [CrossRef]
- Ng, S.C.; Peng, Y.; Zhang, L.; Mok, C.K.P.; Zhao, S.; Li, A.; Ching, J.Y.L.; Liu, Y.; Yan, S.; Chan, D.L.S.; et al. Gut Microbiota Composition Is Associated with SARS-CoV-2 Vaccine Immunogenicity and Adverse Events. Gut 2022, 71, 1106–1116. [Google Scholar] [CrossRef]
- Harris, V.C.; Armah, G.; Fuentes, S.; Korpela, K.E.; Parashar, U.; Victor, J.C.; Tate, J.; De Weerth, C.; Giaquinto, C.; Wiersinga, W.J.; et al. Significant Correlation Between the Infant Gut Microbiome and Rotavirus Vaccine Response in Rural Ghana. J. Infect. Dis. 2017, 215, 34–41. [Google Scholar] [CrossRef]
- Stražar, M.; Mourits, V.P.; Koeken, V.A.C.M.; de Bree, L.C.J.; Moorlag, S.J.C.F.M.; Joosten, L.A.B.; van Crevel, R.; Vlamakis, H.; Netea, M.G.; Xavier, R.J. The Influence of the Gut Microbiome on BCG-Induced Trained Immunity. Genome Biol. 2021, 22, 275. [Google Scholar] [CrossRef] [PubMed]
- Huda, M.N.; Lewis, Z.; Kalanetra, K.M.; Rashid, M.; Ahmad, S.M.; Raqib, R.; Qadri, F.; Underwood, M.A.; Mills, D.A.; Stephensen, C.B. Stool Microbiota and Vaccine Responses of Infants. Pediatrics 2014, 134, e362–e372. [Google Scholar] [CrossRef] [PubMed]
- Williams, W.B.; Han, Q.; Haynes, B.F. Cross-Reactivity of HIV Vaccine Responses and the Microbiome. Curr. Opin. HIV AIDS 2018, 13, 9–14. [Google Scholar] [CrossRef]
- Cram, J.A.; Fiore-Gartland, A.J.; Srinivasan, S.; Karuna, S.; Pantaleo, G.; Tomaras, G.D.; Fredricks, D.N.; Kublin, J.G. Human Gut Microbiota Is Associated with HIV-Reactive Immunoglobulin at Baseline and Following HIV Vaccination. PLoS ONE 2019, 14, e0225622. [Google Scholar] [CrossRef]
- Gonçalves, E.; Guillén, Y.; Lama, J.R.; Sanchez, J.; Brander, C.; Paredes, R.; Combadière, B. Host Transcriptome and Microbiota Signatures Prior to Immunization Profile Vaccine Humoral Responsiveness. Front. Immunol. 2021, 12, 657162. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, S.; Maurya, S.K.; Das, D.K.; Khan, N.; Agrewala, J.N. Gut Dysbiosis Thwarts the Efficacy of Vaccine Against Mycobacterium Tuberculosis. Front. Immunol. 2020, 11, 726. [Google Scholar] [CrossRef]
- Hirota, M.; Tamai, M.; Yukawa, S.; Taira, N.; Matthews, M.M.; Toma, T.; Seto, Y.; Yoshida, M.; Toguchi, S.; Miyagi, M.; et al. Human Immune and Gut Microbial Parameters Associated with Inter-Individual Variations in COVID-19 MRNA Vaccine-Induced Immunity. Commun. Biol. 2023, 6, 368. [Google Scholar] [CrossRef]
- Borgognone, A.; Noguera-Julian, M.; Oriol, B.; Noël-Romas, L.; Ruiz-Riol, M.; Guillén, Y.; Parera, M.; Casadellà, M.; Duran, C.; Puertas, M.C.; et al. Gut Microbiome Signatures Linked to HIV-1 Reservoir Size and Viremia Control. Microbiome 2022, 10, 59. [Google Scholar] [CrossRef]
- Pastor-Ibáñez, R.; Díez-Fuertes, F.; Sánchez-Palomino, S.; Alcamí, J.; Plana, M.; Torrents, D.; Leal, L.; García, F. Impact of Transcriptome and Gut Microbiome on the Response of Hiv-1 Infected Individuals to a Dendritic Cell-Based Hiv Therapeutic Vaccine. Vaccines 2021, 9, 694. [Google Scholar] [CrossRef]
- Mothe, B.; Llano, A.; Ibarrondo, J.; Daniels, M.; Miranda, C.; Zamarreno, J.; Bach, V.; Zuniga, R.; Perez-Alvarez, S.; Berger, C.T.; et al. Definition of the Viral Targets of Protective HIV-1-Specific T Cell Responses. J. Transl. Med. 2011, 9, 208. [Google Scholar] [CrossRef]
- Mothe, B.; Hu, X.; Llano, A.; Rosati, M.; Olvera, A.; Kulkarni, V.; Valentin, A.; Alicea, C.; Pilkington, G.R.; Sardesai, N.Y.; et al. A Human Immune Data-Informed Vaccine Concept Elicits Strong and Broad T-Cell Specificities Associated with HIV-1 Control in Mice and Macaques. J. Transl. Med. 2015, 13, 60. [Google Scholar] [CrossRef]
- Mothe, B.; Llano, A.; Ibarrondo, J.; Zamarreno, J.; Schiaulini, M.; Miranda, C.; Ruiz-Riol, M.; Berger, C.T.; Herrero, M.J.; Palou, E.; et al. CTL Responses of High Functional Avidity and Broad Variant Cross-Reactivity Are Associated with HIV Control. PLoS ONE 2012, 7, e29717. [Google Scholar] [CrossRef] [PubMed]
- Saubi, N.; Kilpeläinen, A.; Eto, Y.; Chen, C.-W.; Olvera, À.; Hanke, T.; Brander, C.; Joseph-Munné, J. Priming with Recombinant BCG Expressing HTI Enhances the Magnitude and Breadth of the T-Cell Immune Responses Elicited by MVA.HTI in BALB/c Mice. Vaccines 2020, 8, 678. [Google Scholar] [CrossRef] [PubMed]
- Kilpeläinen, A.; Saubi, N.; Guitart, N.; Olvera, A.; Hanke, T.; Brander, C.; Joseph, J. Recombinant BCG Expressing HTI Prime and Recombinant ChAdOx1 Boost Is Safe and Elicits HIV-1-Specific T-Cell Responses in BALB/c Mice. Vaccines 2019, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Gallinaro, A.; Borghi, M.; Pirillo, M.F.; Cecchetti, S.; Bona, R.; Canitano, A.; Michelini, Z.; Di Virgilio, A.; Olvera, A.; Brander, C.; et al. Development and Preclinical Evaluation of an Integrase Defective Lentiviral Vector Vaccine Expressing the HIVACAT T Cell Immunogen in Mice. Mol. Ther.-Methods Clin. Dev. 2020, 17, 418–428. [Google Scholar] [CrossRef]
- Guardo, A.C.; Joe, P.T.; Miralles, L.; Bargalló, M.E.; Mothe, B.; Krasniqi, A.; Heirman, C.; García, F.; Thielemans, K.; Brander, C.; et al. Preclinical Evaluation of an MRNA HIV Vaccine Combining Rationally Selected Antigenic Sequences and Adjuvant Signals (HTI-TriMix). AIDS 2017, 31, 321–332. [Google Scholar] [CrossRef]
- Borgognone, A.; Elizalde-Torrent, A.; Casadellà, M.; Romero, L.; Escribà, T.; Parera, M.; Català-Moll, F.; Noguera-Julian, M.; Brander, C.; Olvera, A.; et al. Vaccination with an HIV T-Cell Immunogen Induces Alterations in the Mouse Gut Microbiota. npj Biofilms Microbiomes 2022, 8, 104. [Google Scholar] [CrossRef]
- Bailón, L.; Llano, A.; Cedeño, S.; Escribà, T.; Rosás-Umbert, M.; Parera, M.; Casadellà, M.; Lopez, M.; Pérez, F.; Oriol-Tordera, B.; et al. Safety, Immunogenicity and Effect on Viral Rebound of HTI Vaccines in Early Treated HIV-1 Infection: A Randomized, Placebo-Controlled Phase 1 Trial. Nat. Med. 2022, 28, 2611–2621. [Google Scholar] [CrossRef]
- Bailon, L.; Llano, A.; Cedeño, S.; Lopez, M.B.; Alarcon, Y.; Coll, P.; Rivero, À.; Leselbaum, A.R.; McGowan, I.; SenGupta, D.; et al. A Placebo-Controlled Ati Trial of HTI Vaccines in Early Treated HIV Infection. Top. Antivir. Med. 2021, 29, 48–49. [Google Scholar]
- Le Roy, T.; Debédat, J.; Marquet, F.; Da-Cunha, C.; Ichou, F.; Guerre-Millo, M.; Kapel, N.; Aron-Wisnewsky, J.; Clément, K. Comparative Evaluation of Microbiota Engraftment Following Fecal Microbiota Transfer in Mice Models: Age, Kinetic and Microbial Status Matter. Front. Microbiol. 2019, 10, 3289. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of Commensal Microflora by Toll-like Receptors Is Required for Intestinal Homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef]
- 16S Metagenomic Sequencing Library Preparation. Available online: https://support.illumina.com/downloads/16s_metagenomic_sequencing_library_preparation.html (accessed on 23 December 2021).
- LaMar, D. FastQC. Available online: https://qubeshub.org/resources/fastqc (accessed on 23 April 2022).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naıve Bayesian Classifier for Rapid Assignment of RRNA Sequences.Pdf. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Volume 2, ISBN 3-900051-07-0.
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-2. CRAN R 2019, 1, 2. [Google Scholar]
- Dray, S.; Dufour, A.-B. The Ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2015, 22, 1–20. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2; Springer: New York, NY, USA, 2009; ISBN 9780387981406. [Google Scholar]
- Los Alamos HIV Databases PeptGen Peptide Generator. Available online: https://www.hiv.lanl.gov/content/sequence/PEPTGEN/peptgen.html (accessed on 23 December 2021).
- Martínez-López, M.; Iborra, S.; Conde-Garrosa, R.; Mastrangelo, A.; Danne, C.; Mann, E.R.; Reid, D.M.; Gaboriau-Routhiau, V.; Chaparro, M.; Lorenzo, M.P.; et al. Microbiota Sensing by Mincle-Syk Axis in Dendritic Cells Regulates Interleukin-17 and -22 Production and Promotes Intestinal Barrier Integrity. Immunity 2019, 50, 446–461.e9. [Google Scholar] [CrossRef]
- van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-Chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of Propionate and Butyrate by the Human Colonic Microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Kilpeläinen, A.; Saubi, N.; Guitart, N.; Moyo, N.; Wee, E.G.; Ravi, K.; Hanke, T.; Joseph, J. Priming with Recombinant BCG Expressing Novel HIV-1 Conserved Mosaic Immunogens and Boosting with Recombinant CHADOX1 Is Safe, Stable, and Elicits HIV-1specific T-Cell Responses in BALB/c Mice. Front. Immunol. 2019, 10, 923. [Google Scholar] [CrossRef]
- Kennedy, E.A.; King, K.Y.; Baldridge, M.T. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front. Physiol. 2018, 9, 1534. [Google Scholar] [CrossRef]
- Fiebiger, U.; Bereswill, S.; Heimesaat, M.M. Dissecting the Interplay between Intestinal Microbiota and Host Immunity in Health and Disease: Lessons Learned from Germfree and Gnotobiotic Animal Models. Eur. J. Microbiol. Immunol. 2016, 6, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Frahm, N.; Korber, B.T.; Adams, C.M.; Szinger, J.J.; Draenert, R.; Addo, M.M.; Feeney, M.E.; Yusim, K.; Sango, K.; Brown, N.V.; et al. Consistent Cytotoxic-T-Lymphocyte Targeting of Immunodominant Regions in Human Immunodeficiency Virus across Multiple Ethnicities. J. Virol. 2004, 78, 2187–2200. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, S.; Flanders, M.; Cutler, S.; Soghoian, D.Z.; Ghebremichael, M.; Davis, I.; Lindqvist, M.; Pereyra, F.; Walker, B.D.; Heckerman, D.; et al. HIV-Specific CD4 T Cell Responses to Different Viral Proteins Have Discordant Associations with Viral Load and Clinical Outcome. J. Virol. 2012, 86, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Pennell, L.M.; Galligan, C.L.; Fish, E.N. Sex Affects Immunity. J. Autoimmun. 2012, 38, J282–J291. [Google Scholar]
- Klein, S.L.; Flanagan, K.L. Sex Differences in Immune Responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Dupraz, L.; Magniez, A.; Rolhion, N.; Richard, M.L.; Da Costa, G.; Touch, S.; Mayeur, C.; Planchais, J.; Agus, A.; Danne, C.; et al. Gut Microbiota-Derived Short-Chain Fatty Acids Regulate IL-17 Production by Mouse and Human Intestinal Γδ T Cells. Cell Rep. 2021, 36, 109332. [Google Scholar] [CrossRef]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The Gut Microbiota and Inflammation: An Overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef]
- Tamanai-Shacoori, Z.; Smida, I.; Bousarghin, L.; Loreal, O.; Meuric, V.; Fong, S.B.; Bonnaure-Mallet, M.; Jolivet-Gougeon, A. Roseburia spp.: A Marker of Health? Future Microbiol. 2017, 12, 157–170. [Google Scholar] [CrossRef]
- Nie, K.; Ma, K.; Luo, W.; Shen, Z.; Yang, Z.; Xiao, M.; Tong, T.; Yang, Y.; Wang, X. Roseburia Intestinalis: A Beneficial Gut Organism From the Discoveries in Genus and Species. Front. Cell. Infect. Microbiol. 2021, 11, 757718. [Google Scholar] [CrossRef]
- Lindahl, H.; Olsson, T. Interleukin-22 Influences the Th1/Th17 Axis. Front. Immunol. 2021, 12, 618110. [Google Scholar]
- Rutz, S.; Eidenschenk, C.; Ouyang, W. IL-22, Not Simply a Th17 Cytokine. Immunol. Rev. 2013, 252, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Gubernatorova, E.O.; Tumanov, A.V. Tumor Necrosis Factor and Lymphotoxin in Regulation of Intestinal Inflammation. Biochemistry 2016, 81, 1309–1325. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elizalde-Torrent, A.; Borgognone, A.; Casadellà, M.; Romero-Martin, L.; Escribà, T.; Parera, M.; Rosales-Salgado, Y.; Díaz-Pedroza, J.; Català-Moll, F.; Noguera-Julian, M.; et al. Vaccination with an HIV T-Cell Immunogen (HTI) Using DNA Primes Followed by a ChAdOx1-MVA Boost Is Immunogenic in Gut Microbiota-Depleted Mice despite Low IL-22 Serum Levels. Vaccines 2023, 11, 1663. https://doi.org/10.3390/vaccines11111663
Elizalde-Torrent A, Borgognone A, Casadellà M, Romero-Martin L, Escribà T, Parera M, Rosales-Salgado Y, Díaz-Pedroza J, Català-Moll F, Noguera-Julian M, et al. Vaccination with an HIV T-Cell Immunogen (HTI) Using DNA Primes Followed by a ChAdOx1-MVA Boost Is Immunogenic in Gut Microbiota-Depleted Mice despite Low IL-22 Serum Levels. Vaccines. 2023; 11(11):1663. https://doi.org/10.3390/vaccines11111663
Chicago/Turabian StyleElizalde-Torrent, Aleix, Alessandra Borgognone, Maria Casadellà, Luis Romero-Martin, Tuixent Escribà, Mariona Parera, Yaiza Rosales-Salgado, Jorge Díaz-Pedroza, Francesc Català-Moll, Marc Noguera-Julian, and et al. 2023. "Vaccination with an HIV T-Cell Immunogen (HTI) Using DNA Primes Followed by a ChAdOx1-MVA Boost Is Immunogenic in Gut Microbiota-Depleted Mice despite Low IL-22 Serum Levels" Vaccines 11, no. 11: 1663. https://doi.org/10.3390/vaccines11111663
APA StyleElizalde-Torrent, A., Borgognone, A., Casadellà, M., Romero-Martin, L., Escribà, T., Parera, M., Rosales-Salgado, Y., Díaz-Pedroza, J., Català-Moll, F., Noguera-Julian, M., Brander, C., Paredes, R., & Olvera, A. (2023). Vaccination with an HIV T-Cell Immunogen (HTI) Using DNA Primes Followed by a ChAdOx1-MVA Boost Is Immunogenic in Gut Microbiota-Depleted Mice despite Low IL-22 Serum Levels. Vaccines, 11(11), 1663. https://doi.org/10.3390/vaccines11111663