Peptide Vaccine Therapy in Colorectal Cancer

Abstract
:1. Introduction
2. Results and Discussion
2.1. The Mechanism of Peptide Vaccine Therapy
2.2. The Mechanism of Immune Destruction of the Tumour
- ●
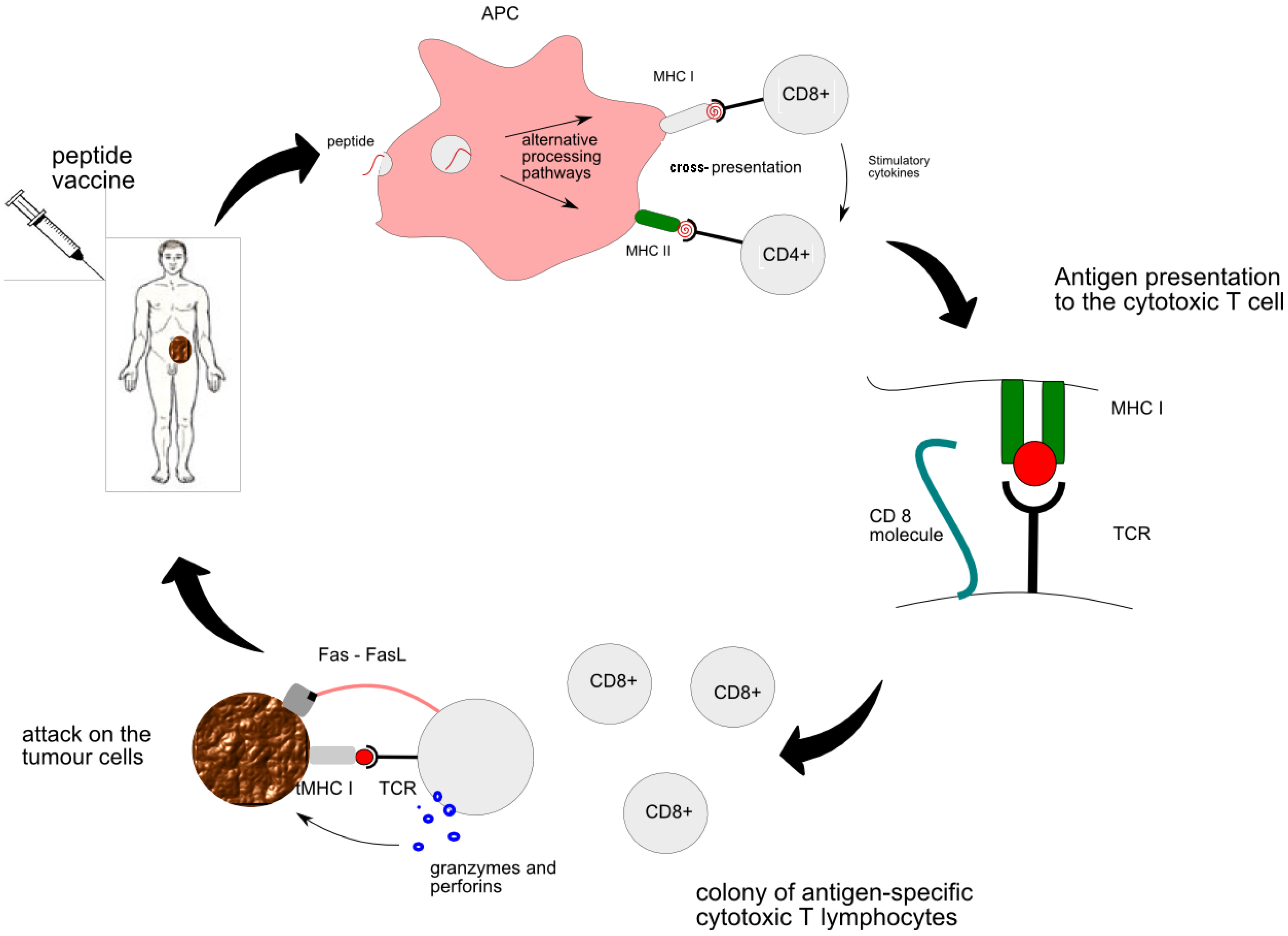
- MHC class I is responsible for presentation of the vaccine-derived peptide between APC and naïve CTLs. Primed CTLs are able to recognize the genuine tumour antigen presented by the MHC I on the surface of the tumour. Thus activated CTL sends out a death signal to the tumour.
- ●
- MHC class II is responsible for “the talk” between APC and CD4+ T cells and subsequent generation of helper T cells. Cytokines secreted by helper T cells, such as IL-2, are essential for activation of strong cytotoxic pathways and potentiation of anti-tumour response [9]. All in all, the CTL encounter with the peptide is at the heart of peptide vaccine therapy. More details of interaction between individual cells can be found in Figure 1.
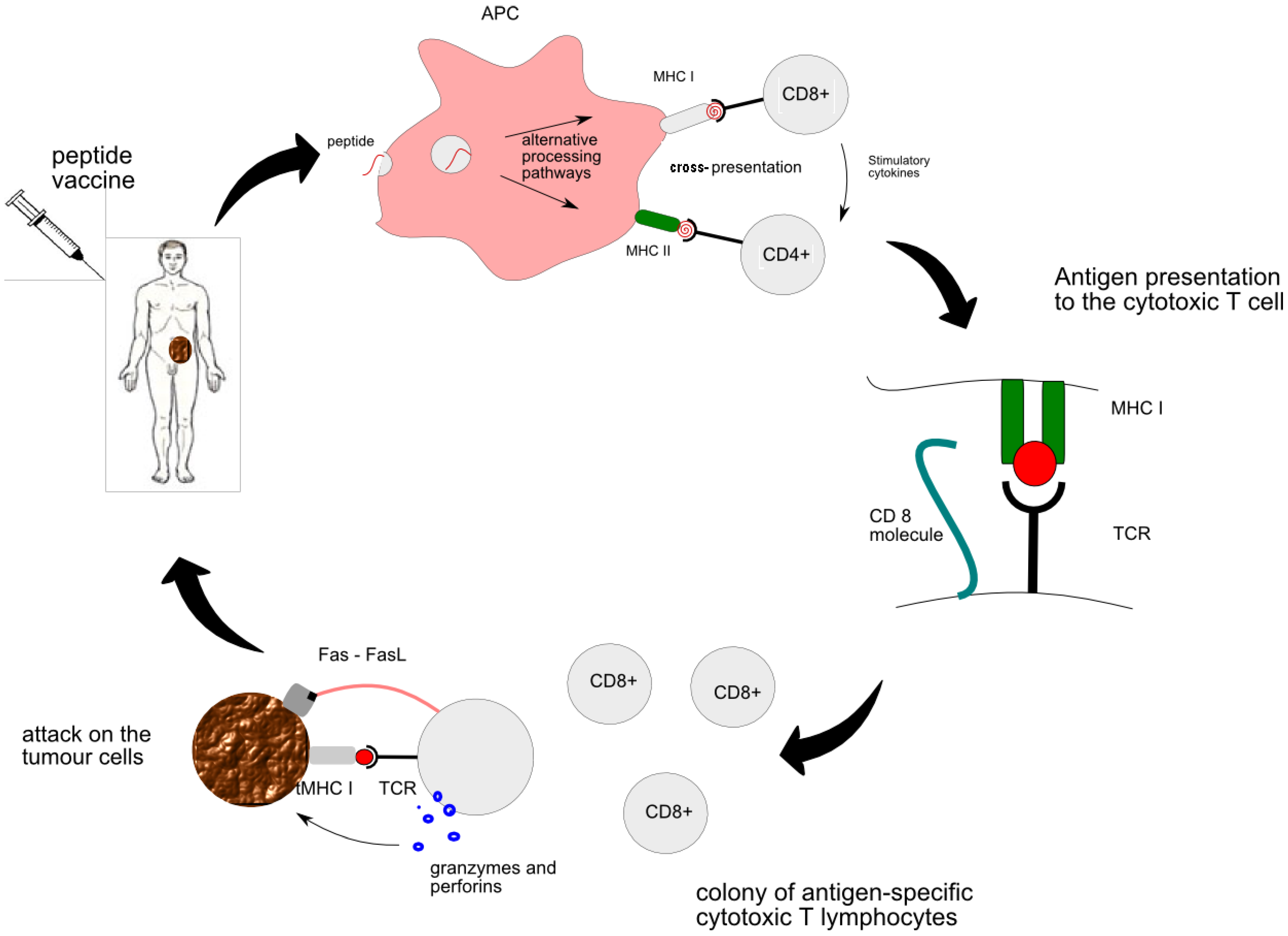
2.3. The Mechanisms whereby Tumour Evades Immune System
- ●
- One such mechanism is expression of FasL, which is a ligand for Fas receptor present on CTL. Fas functions as a death receptor which, upon binding of its ligand expressed on the tumour surface, induces CTL to undergo apoptosis. Additionally, expression of FasL provides a degree of resistance to Fas-induced apoptosis in tumour cells themselves [13].
- ●
- Another mechanism involves expression of altered peptide ligands for T cell receptors (TCR) by the tumour. “Altered Peptide Ligand” (APL) are analogs of immunogenic peptides in which the TCR contact sites have been manipulated [14]. Recognition of a peptide ligand by CTL leads to lysis of the tumour cell. However, ligands that are slightly altered retain their ability to bind the TCR but the outcome of this interaction is different. The altered peptides may be related to the agonist ligand on the basis of their structural homology. Thus, partially activating APL is a subset of the antagonists and thereby modulates the activity of CTL [15].
- ●
- Oncogenic signalling pathways within tumour cells and immunologic checkpoints in the tumour microenvironment also play a crucial role in promoting immunologic tolerance. For example, tumour releases factors that induce inhibition of both innate and adaptive antitumor immunity. Stat3 activation in tumours, as well as Braf activation, can induce release of factors such as IL10 that induce Stat3 signalling in NK cells, granulocytes, inhibiting their tumouricidal activity. Stat3 is also activated within conventional dendritic cells (CDC) in the tumour, converting them to toleragenic DC, which can induce T cell anergy and possibly regulatory T cells (Treg). Plasmacytoid DC (PDC) or PDC-related cells in the tumour microenvironment upregulate indoleamine 2,3-dioxygenase (IDO), an enzyme that metabolizes tryptophan. T cells are very sensitive to tryptophan depletion. Tumours can express co-inhibitory B7 family members, such as B7-H1 and B7-H4, which downregulate T cell activation and/or cytolytic activity. They can also induce B7-H1 and B7-H4 expression on tumour associated macrophages (TAM). Related immature myeloid cells or myeloid suppressor cells can further inhibit antitumor T cells via production of NO by the enzyme arginase. [16,17].
- ●
- Regulatory T cells are an important inhibitor of antitumor immunity. T cell activation in the absence of appropriate co-stimulatory signals leads to T cell anergy and generation of induced regulatory T cells (Treg). Treg, characterized by the FoxP3 transcription factor, upregulate a number of cell membrane molecules, including Lag3, CTLA4, GITR, and neuropilin. Treg can inhibit effector T cell activation and function via T-T inhibition or inhibition of antigen presenting cells [17].
- ●
- Finally, cancers avoid recognition by the immune system by means of defective antigen presentation. This can be achieved by reduced expression of HLA type I (MHC I), which is a common event in colorectal cancer [18], as well as, reduced expression of antigen-processing machinery or tumour-associated antigen itself.
2.4. The Ways of Enhancing Immune Response to Peptide Vaccination
2.5. Why Peptide Vaccine?
2.6. Monitoring of Response to Peptide Vaccine Therapy
- ●
- ELISPOT assays are used to assess secretion of proteins by CTLs which are indicative of an activated state, such as, INF-γ, granzyme B, IL-2 [24]. The assays are performed using peripheral blood mononuclear cells (PBMC) isolated from patient blood sample and the result is expressed as the number of reactive CTLs per 100,000 PBMC [23].
- ●
- Following T-cell receptor recognition of antigenic peptide–MHC class I complexes on the surface of target cells, CTLs induce target cell apoptosis through directed exocytosis of perforin and granzymes. The cytotoxic signalling leads to the activation of the caspase cascade that can be measured using flow cytometer. The assay involved labelling of P815, EL4 and T2 lymphoma cells with a cell tracker dye DDAO-SE and staining permeabilized cells with antibody against cleaved (activated) caspase-3. This assay proved to be robust and reliable in evaluating antigen-specific CTL [25].
- ●
- Another approach is to use flow cytometry for detection of apoptosis in CTL target cells (for example cytomegalovirus bearing appropriate HLA molecule). During apoptosis, phosphatidylserine (PS) is externalized on the surface of the cells and is available for binding of annexin V. Both granule marker and annexin V assays allow evaluation of the cytotoxic potential of tumour-specific CTLs [8].
- ●
- By contrast, HLA/epitope tetramer assay is used for mere enumeration of the antigen-specific CTLs. However, when combined with intracellular cytokine staining it gives a complete picture of quantity and function of the TCLs.
- ●
- Lastly, RT-PCR has been used to evaluate cytokine gene expression, including INF-gamma and granzyme B, by CTLs [25,26]. Cells are harvesting from patients who have been administered peptide vaccination followed by RNA extracted from it. cDNA is synthesised and RT-PCR analysis are performed using forward and reverse primers for IFNγ or CD8, The synthesised cDNA is further validated by the measurement of Gene expression using ABI Prism 7700 Sequence Detection System.
2.7. Targets for Peptide Vaccine Therapy in Colon Cancer

{kind=link}
| Peptides | Targets | Mechanism | Type of Study | Results | Side Effects | Comments | Reference |
|---|---|---|---|---|---|---|---|
| EphA2-derived peptide | EphaA2 | EphA2-specific CTL | In vivo: colon cancer liver metastasis mouse model | Prevents progression of tumour in the liver | No liver or kidney toxicity | Safe to apply clinically to treat colon cancer liver metastases | [21] |
| RNF43-721 | Phase 1 clinical trial in colorectal cancer | Vaccinations were well tolerated | [31] | ||||
| ABT-737 | Bcl-2 small molecule inhibitor | Inhibition of anti-apoptotic Bcl-2 family | In vivo: mouse colon cancer model | Sensitized cancer cells to the antitumor effect of antigen-specific immunotherapy | Improve survival rate | [32] | |
| Multi- peptide cocktail:Epitomes of HER2, MVF, GMP and n-MDP | Multiple targets:HER2, MVF, GMP and n-MDP | Inhibition of EGF-2 | Phase 1 clinical trial in solid cancers including 4 colorectal | 25% SD | No serious adverse events, autoimmune disease, or cardiotoxicity | [33] | |
| Endoglin | Endoglin | Inhibition of angiogenesis | CT26 colon carcinoma mouse model | Inhibition of tumour growth and angiogenesis | [34] | ||
| CEA691 | Carcinoembryonic antigen | Induction of tumor-specific CTLs | Colon carcinoma mouse model |
| Potential for future clinical applications | [35] | |
| MUC1, MHC class II helper peptides | A cell surface associated protein:Mucin 1 | Stimulation of IFN-gamma-producing CD4 (+) helper cells,Induction of CTLs specific to MUC1 and other undefined MC38 tumour antigens | A MUC1-tolerant colon cancer mouse model |
| Potential for future clinical applications | [36] | |
| CEA526–533, NP52–59 | Carcinoembryonic antigen | Activation of tumor-specific CTLs | Murine colon adenocarcinoma mouse model | ||||
| OX40L | TNF family protein | CT26 colon cancer mouse model |
| Potential use for colon metastasis treatment | [37] | ||
| Heat-Shock Protein Gp96 | Heat-Shock Protein | Induction of tumour-specific CTLs | Clinical trial in colorectal cancer liver metastases after tumour resection |
| No significant toxicity | Possible clinical benefit for CRC liver metastatic patients | [38] |
| SART3109–118 SART3315–323 | SART | Induction of tumour-specific CTLs | Clinical trial in patients with advanced colorectal cancer |
| No serious adverse events | Encourage further development of SART3 peptide vaccine for colorectal cancer patients | [39] |
| Lck-derived peptides | Induction of tumor-specific CTLs | [40] | |||||
| CEA605–613 and Flt3L | CEA | Induction of tumor-specific CTLs | Clinical trial metastatic or recurrent colorectal cancer | [41] |
2.8. EphA2
2.9. Survivin
2.10. SART3
2.11. CEA
2.12. MUC-1
3. Conclusions
Conflict of Interest
References
- Ferlay, J.; Autier, P.; Boniol, M.; Heanue, M.; Colombet, M.; Boyle, P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann. Oncol. 2007, 18, 581–592. [Google Scholar]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.Q.; Thun, M.J. Cancer statistics 2007. CA Cancer J. Clin. 2007, 57, 43–66. [Google Scholar] [CrossRef]
- American Cancer Society (ACS), Cancer Facts and Figures 2008; ACS: Atlanta, GA, USA, 2008.
- Parkin, D.M.; Bray, F.I.; Devesa, S.S. Cancer burden in the year 2000. The global picture. Eur. J. Cancer 2001, 37, S4–S66. [Google Scholar]
- Holt, P.R.; Kozuch, P.; Mewar, S. Colon cancer and the elderly: From screening to treatment in management of GI disease in the elderly. Best Pract. Res. Clin. Gastroenterol. 2009, 23, 889–907. [Google Scholar] [CrossRef]
- Ragnhammar, P.; Hafstrom, L.; Nygren, P.; Glimelius, B. A systematic overview of chemotherapy effects in colorectal cancer. Acta Oncol. 2001, 40, 282–308. [Google Scholar]
- Van der, B.P.; Traversari, C.; Chomez, P.; Lurquin, C.; de, P.E.; van den Eynde, B.J.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. J. Immunol. 2007, 178, 2617–2621. [Google Scholar]
- Burkett, M.W.; Shafer-Weaver, K.A.; Strobl, S.; Baseler, M.; Malyguine, A. A novel flow cytometric assay for evaluating cell-mediated cytotoxicity. J. Immunother. 2005, 28, 396–402. [Google Scholar] [CrossRef]
- Tsuruma, T.; Hata, F.; Furuhata, T.; Ohmura, T.; Katsuramaki, T.; Yamaguchi, K.; Kimura, Y.; Torigoe, T.; Sato, N.; Hirata, K. Peptide-Based vaccination for colorectal cancer. Expert Opin. Biol. Ther. 2005, 5, 799–807. [Google Scholar] [CrossRef]
- Clemente, C.G.; Mihm, M.C.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Mihm, M.C., Jr.; Clemente, C.G.; Cascinelli, N. Tumor infiltrating lymphocytes in lymph node melanoma metastases: A histopathologic prognostic indicator and an expression of local immune response. Lab. Invest. 1996, 74, 43–47. [Google Scholar]
- Rivoltini, L.; Carrabba, M.; Huber, V.; Castelli, C.; Novellino, L.; Dalerba, P.; Mortarini, R.; Arancia, G.; Anichini, A.; Fais, S.; et al. Immunity to cancer: Attack and escape in T lymphocyte-tumor cell interaction. Immunol. Rev. 2002, 188, 97–113. [Google Scholar] [CrossRef]
- Keane, M.M.; Ettenberg, S.A.; Lowrey, G.A.; Russell, E.K.; Lipkowitz, S. Fas expression and function in normal and malignant breast cell lines. Cancer Res. 1996, 56, 4791–4798. [Google Scholar]
- Evavold, B.D.; Sloan-Lancaster, J.; Allen, P.M. Tickling the TCR: Selective T cell functions stimulated by altered peptide ligands. Immunol. Today 1993, 14, 602–609. [Google Scholar] [CrossRef]
- Sloan-Lancaster, J.; Allen, P.M. Altered peptide ligand-induced partial T cell activation: Molecularmechanisms and role in T cell biology. Annu. Rev. Immunol. 1996, 14, 1–27. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Charles, G.; Elizabeth Jaffee, D.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar] [CrossRef]
- Cabrera, T.; Collado, A.; Fernandez, M.A.; Ferron, A.; Sancho, J.; Ruiz-Cabello, F.; Garrido, F. High frequency of altered HLA class I phenotypes in invasive colorectal carcinomas. Tissue Antigens 1998, 52, 114–123. [Google Scholar] [CrossRef]
- Zaremba, S.; Barzaga, E.; Zhu, M.; Soares, N.; Tsang, K.Y.; Schlom, J. Identification of an enhancer agonist cytotoxic T lymphocyte peptide from human carcinoembryonic antigen. Cancer Res. 1997, 57, 4570–4577. [Google Scholar]
- Schwendener, R.A.; Ludewig, B.; Cerny, A.; Engler, O. Liposome-Based vaccines. Methods Mol. Biol. 2010, 605, 163–175. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Tatsumi, T.; Takehara, T.; Sasakawa, A.; Yamamoto, M.; Kohga, K.; Miyagi, T.; Kanto, T.; Hiramastu, N.; Akagi, T.; et al. EphA2-Derived peptide vaccine with amphiphilic poly(gamma-glutamic acid) nanoparticles elicits an anti-tumor effect against mouse liver tumor. Cancer Immunol. Immunother. 2010, 59, 759–767. [Google Scholar] [CrossRef]
- Wang, R.F. Human tumor antigens: Implications for cancer vaccine development. J. Mol. Med. 1999, 77, 640–655. [Google Scholar] [CrossRef]
- Shafer-Weaver, K.A.; Sayers, T.; Kuhns, D.B.; Strobl, S.L.; Burkett, M.W.; Baseler, M.; Malyguine, A. Evaluating the cytotoxicity of innate immune effector cells using the GrB ELISPOT assay. J. Transl. Med. 2004, 2, 31. [Google Scholar] [CrossRef]
- Umansky, V.; Malyguine, A.; Shurin, M. New perspectives in cancer immunotherapy and immunomonitoring. Future Oncol. 2009, 5, 941–944. [Google Scholar] [CrossRef]
- He, L.; Hakimi, J.; Salha, D.; Miron, I.; Dunn, P.; Radvanyi, L. A sensitive flow cytometry-based cytotoxic T-lymphocyte assay through detection of cleaved caspase 3 in target cells. J. Immunol. Methods 2005, 304, 43–59. [Google Scholar] [CrossRef]
- Xu, Y.; Theobald, V.; Sung, C.; DePalma, K.; Atwater, L.; Seiger, K.; Perricone, M.A.; Richards, S.M. Validation of a HLA-A2 tetramer flow cytometric method, IFNgamma real time RT-PCR, and IFNgamma ELISPOT for detection of immunologic response to gp100 and MelanA/MART-1 in melanoma patients. J. Transl. Med. 2008, 6, 61. [Google Scholar] [CrossRef]
- Rappu, P.; Nylund, C.; Ristiniemi, N.; Kulpakko, J.; Vihinen, P.; Hernberg, M.; Mirtti, T.; Alanen, K.; Kallajoki, M.; Vuoristo, M.S.; et al. Detection of melanoma-derived cancer-testis antigen CT16 in patient sera by a novel immunoassay. Int. J. Cancer 2011, 128, 2382–2392. [Google Scholar]
- Goydos, J.S.; Elder, E.; Whiteside, T.L.; Finn, O.J.; Lotze, M.T. A phase I trial of a synthetic mucin peptide vaccine. Induction of specific immune reactivity in patients with adenocarcinoma. J. Surg. Res. 1996, 63, 298–304. [Google Scholar] [CrossRef]
- Tsuruma, T.; Hata, F.; Torigoe, T.; Furuhatal, T.; Idenoue, S.; Kurotaki, T.; Yamamoto, M.; Yagihashi, A.; Ohmura, T.; Yamaguchi, K.; et al. Phase I clinical study of anti-apoptosis protein, survivin-derived peptide vaccine therapy for patients with advanced or recurrent colorectal cancer. J. Transl. Med. 2004, 2, 19. [Google Scholar] [CrossRef]
- Karanikas, V.; Colau, D.; Baurain, J.F.; Chiari, R.; Thonnard, J.; Gutierrez-Roelens, I.; Goffinet, C.; van Schaftingen, E.V.; Weynants, P.; Boon, T.; et al. High frequency of cytolytic T lymphocytes directed against a tumor-specific mutated antigen detectable with HLA tetramers in the blood of a lung carcinoma patient with long survival. Cancer Res. 2001, 61, 3718–3724. [Google Scholar]
- Yoshimatsu, K.; Yokomizo, H.; Osawa, G.; Fujimoto, T.; Otani, T.; Tsunoda, T.; Nakamura, Y.; Ogawa, K. Phase I study of combination therapy with peptide vaccine and anti-cancer drug for colorectal cancer. Gan To Kagaku Ryoho 2008, 35, 2268–2270. [Google Scholar]
- Begley, J.; Vo, D.D.; Morris, L.F.; Bruhn, K.W.; Prins, R.M.; Mok, S.; Koya, R.C.; Garban, H.J.; Comin-Anduix, B.; Craft, N.; et al. Immunosensitization with a Bcl-2 small molecule inhibitor. Cancer Immunol. Immunother. 2009, 58, 699–708. [Google Scholar] [CrossRef]
- Kaumaya, P.T.; Foy, K.C.; Garrett, J.; Rawale, S.V.; Vicari, D.; Thurmond, J.M.; Lamb, T.; Mani, A.; Kane, Y.; Balint, C.R.; et al. Phase I active immunotherapy with combination of two chimeric, human epidermal growth factor receptor 2, B-cell epitopes fused to a promiscuous T-cell epitope in patients with metastatic and/or recurrent solid tumors. J. Clin. Oncol. 2009, 27, 5270–5277. [Google Scholar] [CrossRef]
- Tan, G.H.; Li, Y.N.; Huang, F.Y.; Wang, H.; Bai, R.Z.; Jang, J. Combination of recombinant xenogeneic endoglin DNA and protein vaccination enhances anti-tumor effects. Immunol. Invest 2007, 36, 423–440. [Google Scholar] [CrossRef]
- Saha, A.; Chatterjee, S.K.; Foon, K.A.; Celis, E.; Bhattacharya-Chatterjee, M. Therapy of established tumors in a novel murine model transgenic for human carcinoembryonic antigen and HLA-A2 with a combination of anti-idiotype vaccine and CTL peptides of carcinoembryonic antigen. Cancer Res. 2007, 67, 2881–2892. [Google Scholar] [CrossRef]
- Mukherjee, P.; Pathangey, L.B.; Bradley, J.B.; Tinder, T.L.; Basu, G.D.; Akporiaye, E.T.; Gendler, S.J. MUC1-Specific immune therapy generates a strong anti-tumor response in a MUC1-tolerant colon cancer model. Vaccine 2007, 25, 1607–1618. [Google Scholar]
- Ali, S.A.; Ahmad, M.; Lynam, J.; McLean, C.S.; Entwisle, C.; Loudon, P.; Choolun, E.; McArdle, S.E.B.; Li, G.; Mian, S.; et al. Anti-Tumour therapeutic efficacy of OX40L in murine tumour model. Vaccine 2004, 22, 3585–3594. [Google Scholar] [CrossRef]
- Mazzaferro, V.; Coppa, J.; Carrabba, M.G.; Rivoltini, L.; Schiavo, M.; Regalia, E.; Mariani, L.; Camerini, T.; Marchianò, A.; Andreola, S.; et al. Vaccination with autologous tumor-derived heat-shock protein gp96 after liver resection for metastatic colorectal cancer. Clin. Cancer Res. 2003, 9, 3235–3245. [Google Scholar]
- Miyagi, Y.; Imai, N.; Sasatomi, T.; Yamada, A.; Mine, T.; Katagiri, K.; Nakagawa, M.; Muto, A.; Okouchi, S.; Isomoto, H.; et al. Induction of cellular immune responses to tumor cells and peptides in colorectal cancer patients by vaccination with SART3 peptides. Clin. Cancer Res. 2001, 7, 3950–3962. [Google Scholar]
- Imai, N.; Harashima, N.; Ito, M.; Miyagi, Y.; Harada, M.; Yamada, A.; Itoh, K. Identification of Lck-derived peptides capable of inducing HLA-A2-restricted and tumor-specific CTLs in cancer patients with distant metastases. Int. J. Cancer 2001, 94, 237–242. [Google Scholar] [CrossRef]
- Fong, L.; Hou, Y.; Rivas, A.; Benike, C.; Yuen, A.; Fisher, G.A.; Davis, M.M.; Engleman, E.G. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc. Natl. Acad. Sci. USA 2001, 98, 8809–8814. [Google Scholar]
- Oba, S.M.; Wang, Y.J.; Song, J.P.; Li, Z.Y.; Kobayashi, K.; Tsugane, S.; Hamada, G.S.; Tanaka, M.; Sugimura, H. Genomic structure and loss of heterozygosity of EPHB2 in colorectal cancer. Cancer Lett. 2001, 164, 97–104. [Google Scholar] [CrossRef]
- Cheng, N.; Brantley, D.M.; Chen, J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002, 13, 75–85. [Google Scholar] [CrossRef]
- Saito, T.; Masuda, N.; Miyazaki, T.; Kanoh, K.; Suzuki, H.; Shimura, T.; Asao, T.; Kuwano, H. Expression of EphA2 and E-cadherin in colorectal cancer: Correlation with cancer metastasis. Oncol. Rep. 2004, 11, 605–611. [Google Scholar]
- Yamaguchi, S.; Tatsumi, T.; Takehara, T.; Sakamori, R.; Uemura, A.; Mizushima, T.; Ohkawa, K.; Storkus, W.J.; Hayashi, N. Immunotherapy of murine colon cancer using receptor tyrosine kinase EphA2-derived peptide-pulsed dendritic cell vaccines. Cancer 2007, 110, 1469–1477. [Google Scholar] [CrossRef]
- Altieri, D.C. The molecular basis and potential role of survivin in cancer diagnosis and therapy. Trends Mol. Med. 2001, 7, 542–547. [Google Scholar] [CrossRef]
- Kawasaki, H.; Altieri, D.C.; Lu, C.D.; Toyoda, M.; Tenjo, T.; Tanigawa, N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998, 58, 5071–5074. [Google Scholar]
- Hirohashi, Y.; Torigoe, T.; Maeda, A.; Nabeta, Y.; Kamiguchi, K.; Sato, T.; Yoda, J.; Ikeda, H.; Hirata, K.; Yamanaka, N.; et al. An HLA-A24-restricted cytotoxic T lymphocyte epitope of a tumor-associated protein, survivin. Clin. Cancer Res. 2002, 8, 1731–1739. [Google Scholar]
- Idenoue, S.; Hirohashi, Y.; Torigoe, T.; Sato, Y.; Tamura, Y.; Hariu, H.; Yamamoto, M.; Kurotaki, T.; Tsuruma, T.; Asanuma, H.; et al. A potent immunogenic general cancer vaccine that targets survivin, an inhibitor of apoptosis proteins. Clin. Cancer Res. 2005, 11, 1474–1482. [Google Scholar]
- Tsuruma, T.; Iwayama, Y.; Ohmura, T.; Katsuramaki, T.; Hata, F.; Furuhata, T.; Yamaguchi, K.; Kimura, Y.; Torigoe, T.; Toyota, N.; et al. Clinical and immunological evaluation of anti-apoptosis protein, survivin-derived peptide vaccine in phase I clinical study for patients with advanced or recurrent breast cancer. J. Transl. Med. 2008, 6, 24. [Google Scholar] [CrossRef]
- Sasatomi, T.; Suefuji, Y.; Matsunaga, K.; Yamana, H.; Miyagi, Y.; Araki, Y.; Ogata, Y.; Itoh, K.; Shirouzu, K. Expression of tumor rejection antigens in colorectal carcinomas. Cancer 2002, 94, 1636–1641. [Google Scholar] [CrossRef]
- Miyagi, Y.; Imai, N.; Sasatomi, T.; Yamada, A.; Mine, T.; Katagiri, K.; Nakagawa, M.; Muto, A.; Okouchi, S.; Isomoto, H.; et al. Induction of cellular immune responses to tumor cells and peptides in colorectal cancer patients by vaccination with SART3 peptides. Clin. Cancer Res. 2001, 7, 3950–3962. [Google Scholar]
- Miyamoto, S.; Nakamura, M.; Shitara, K.; Nakamura, K.; Ohki, Y.; Ishii, G.; Goya, M.; Kodama, K.; Sangai, T.; Maeda, H.; et al. Blockade of paracrine supply of insulin-like growth factors using neutralizing antibodies suppresses the liver metastasis of human colorectal cancers. Clin. Cancer Res. 2005, 11, 3494–3502. [Google Scholar]
- Saha, A.; Chatterjee, S.K.; Foon, K.A.; Celis, E.; Bhattacharya-Chatterjee, M. Therapy of established tumors in a novel murine model transgenic for human carcinoembryonic antigen and HLA-A2 with a combination of anti-idiotype vaccine and CTL peptides of carcinoembryonic antigen. Cancer Res. 2007, 67, 2881–2892. [Google Scholar] [CrossRef]
- Niv, Y. MUC1 and colorectal cancer pathophysiology considerations. World J. Gastroenterol. 2008, 14, 2139–2141. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bartnik, A.; Nirmal, A.J.; Yang, S.-Y. Peptide Vaccine Therapy in Colorectal Cancer. Vaccines 2013, 1, 1-16. https://doi.org/10.3390/vaccines1010001
Bartnik A, Nirmal AJ, Yang S-Y. Peptide Vaccine Therapy in Colorectal Cancer. Vaccines. 2013; 1(1):1-16. https://doi.org/10.3390/vaccines1010001
Chicago/Turabian StyleBartnik, Aleksandra, Ajit Johnson Nirmal, and Shi-Yu Yang. 2013. "Peptide Vaccine Therapy in Colorectal Cancer" Vaccines 1, no. 1: 1-16. https://doi.org/10.3390/vaccines1010001
APA StyleBartnik, A., Nirmal, A. J., & Yang, S.-Y. (2013). Peptide Vaccine Therapy in Colorectal Cancer. Vaccines, 1(1), 1-16. https://doi.org/10.3390/vaccines1010001