Aronia melanocarpa Fruit Bioactive Fraction Attenuates LPS-Induced Inflammatory Response in Human Bronchial Epithelial Cells




Abstract
:
1. Introduction
2. Materials and Methods
2.1. A. Melanocarpa Fruit Extracts and Chemicals
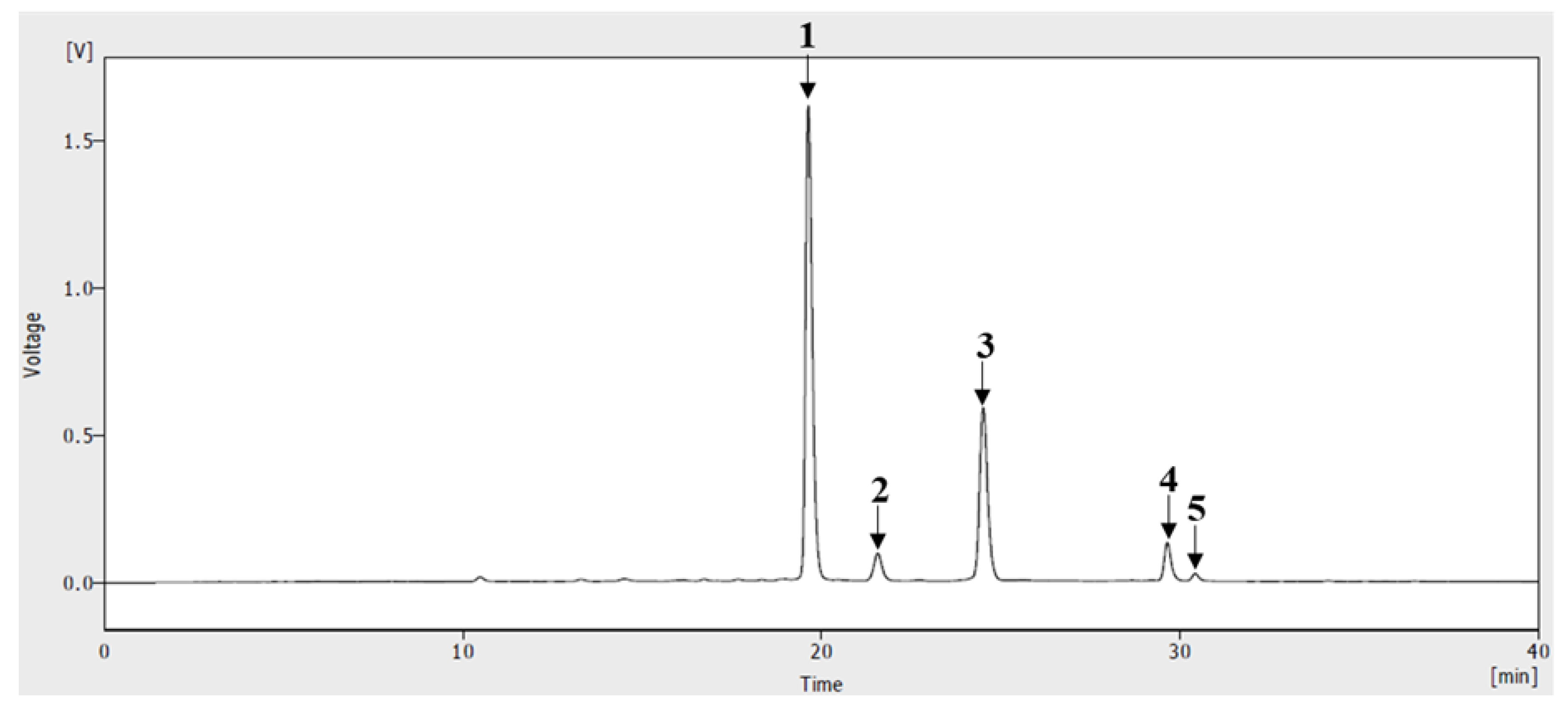
2.2. HPLC of ABF®
2.3. Cell Culture
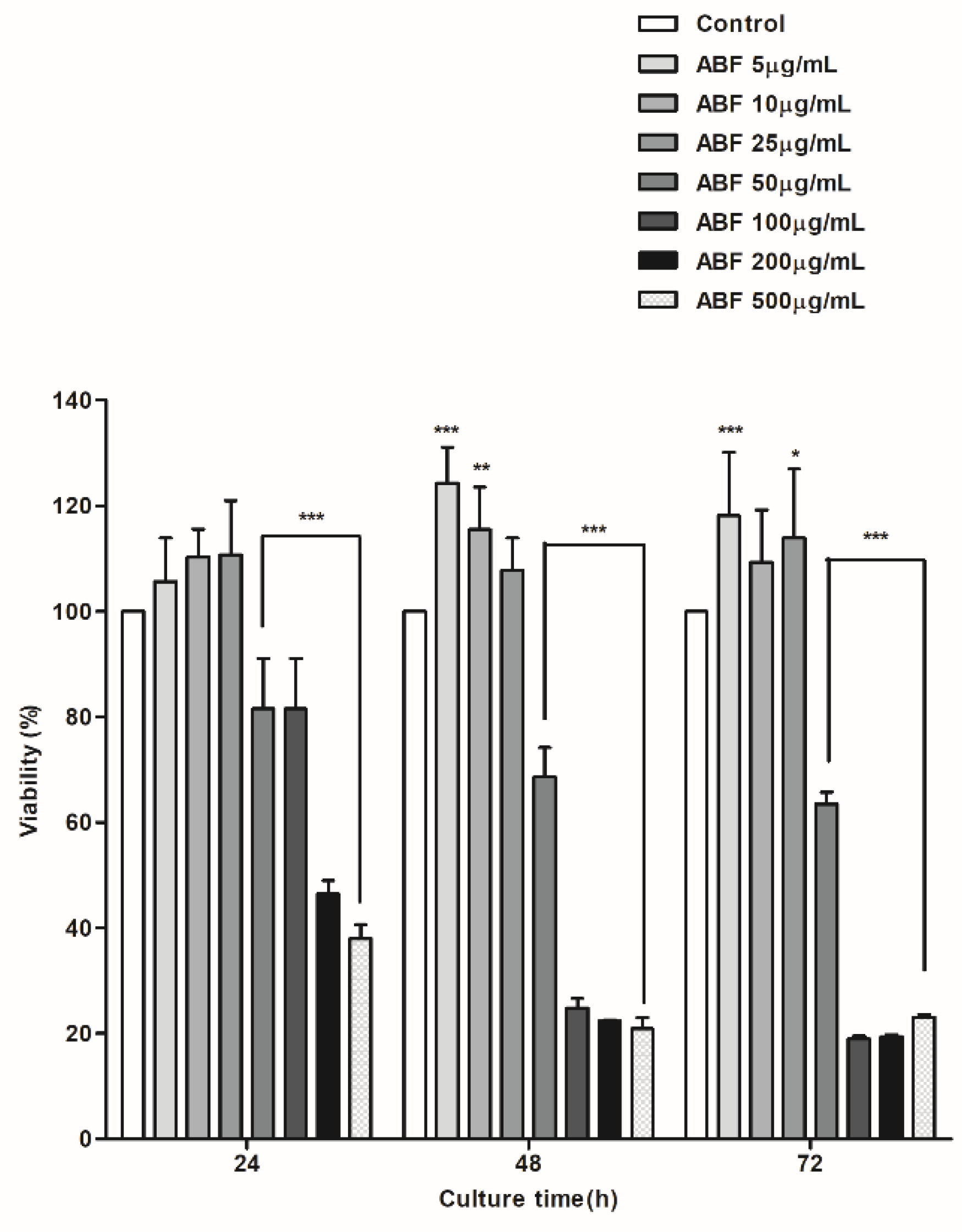
2.4. Cell Viability Assay
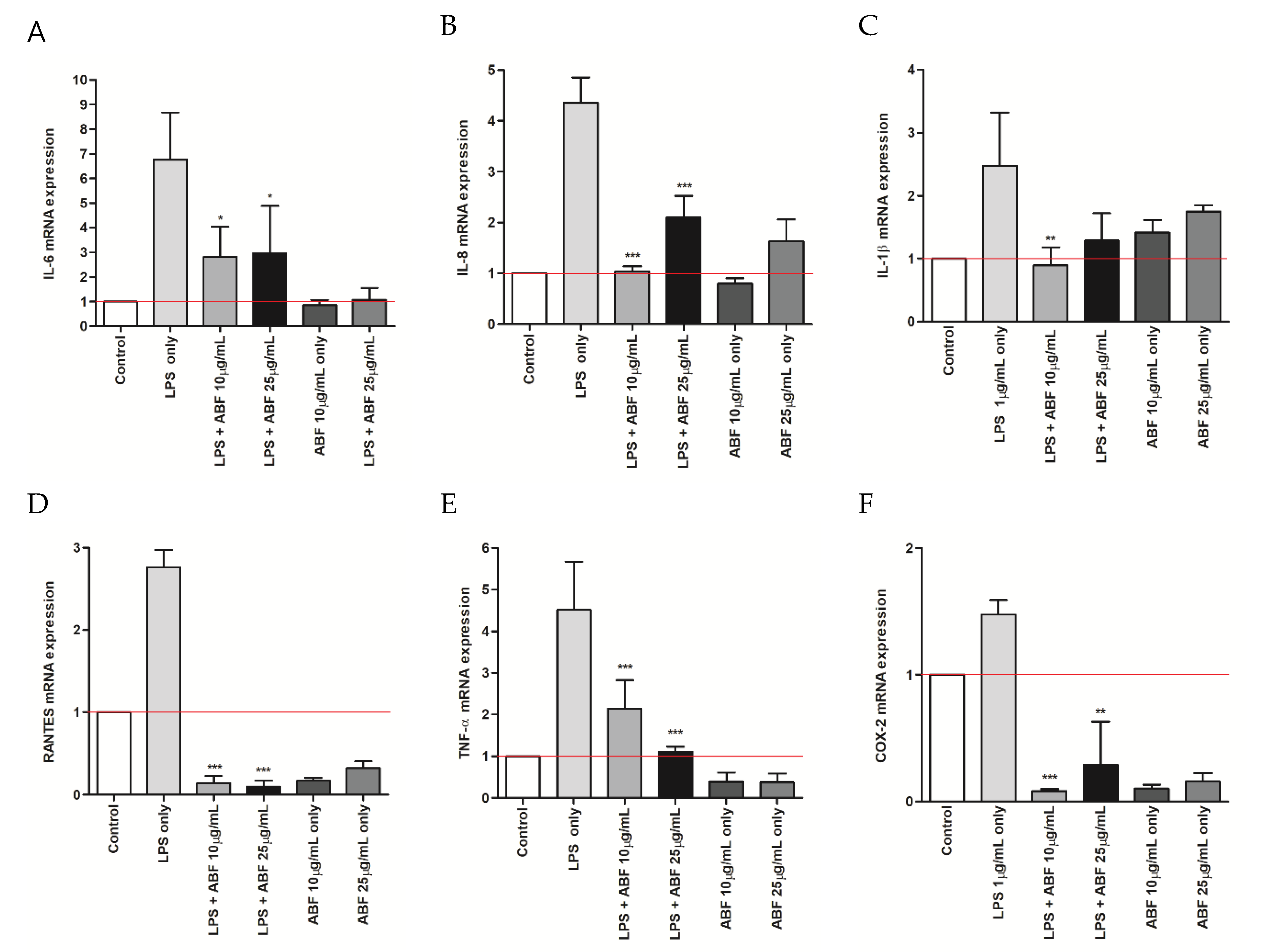
2.5. Quantitative Real-Time PCR (qRT-PCR)
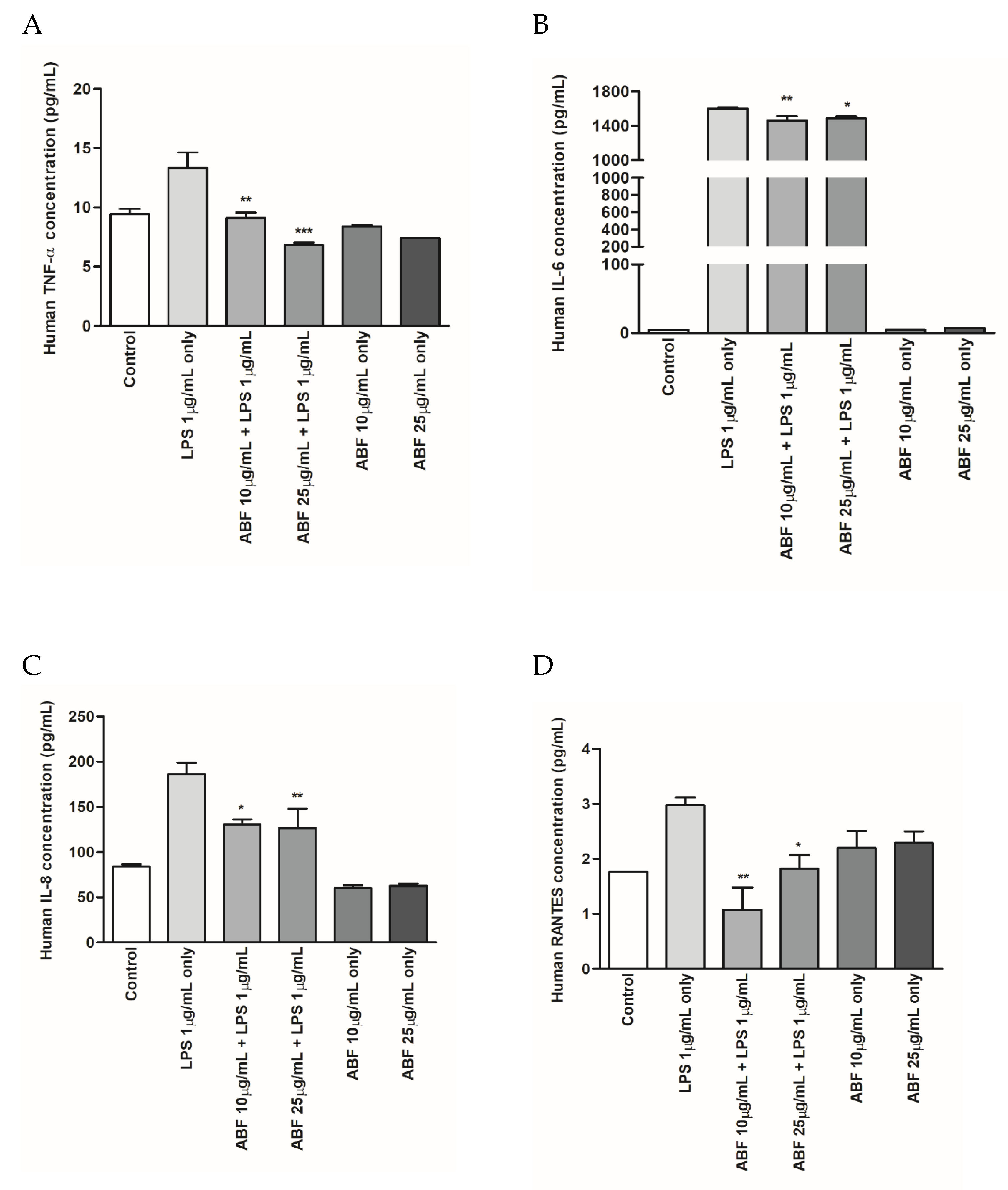
2.6. Inflammatory Cytokine Determination in Cell Supernatant
2.7. Western Blot Analysis
2.8. Intracellular Reactive Oxygen Species (ROS) Determination
2.9. Cell Cycle Analysis
2.10. Statistical Analyses
3. Results
3.1. HPLC of ABF®
3.2. Cytotoxic Effects of ABF® in BEAS-2B Cells
3.3. Effects of ABF® on the mRNA Expression of Inflammatory Cytokines
3.4. Effects of ABF® on the Protein Expression of Inflammatory Cytokines
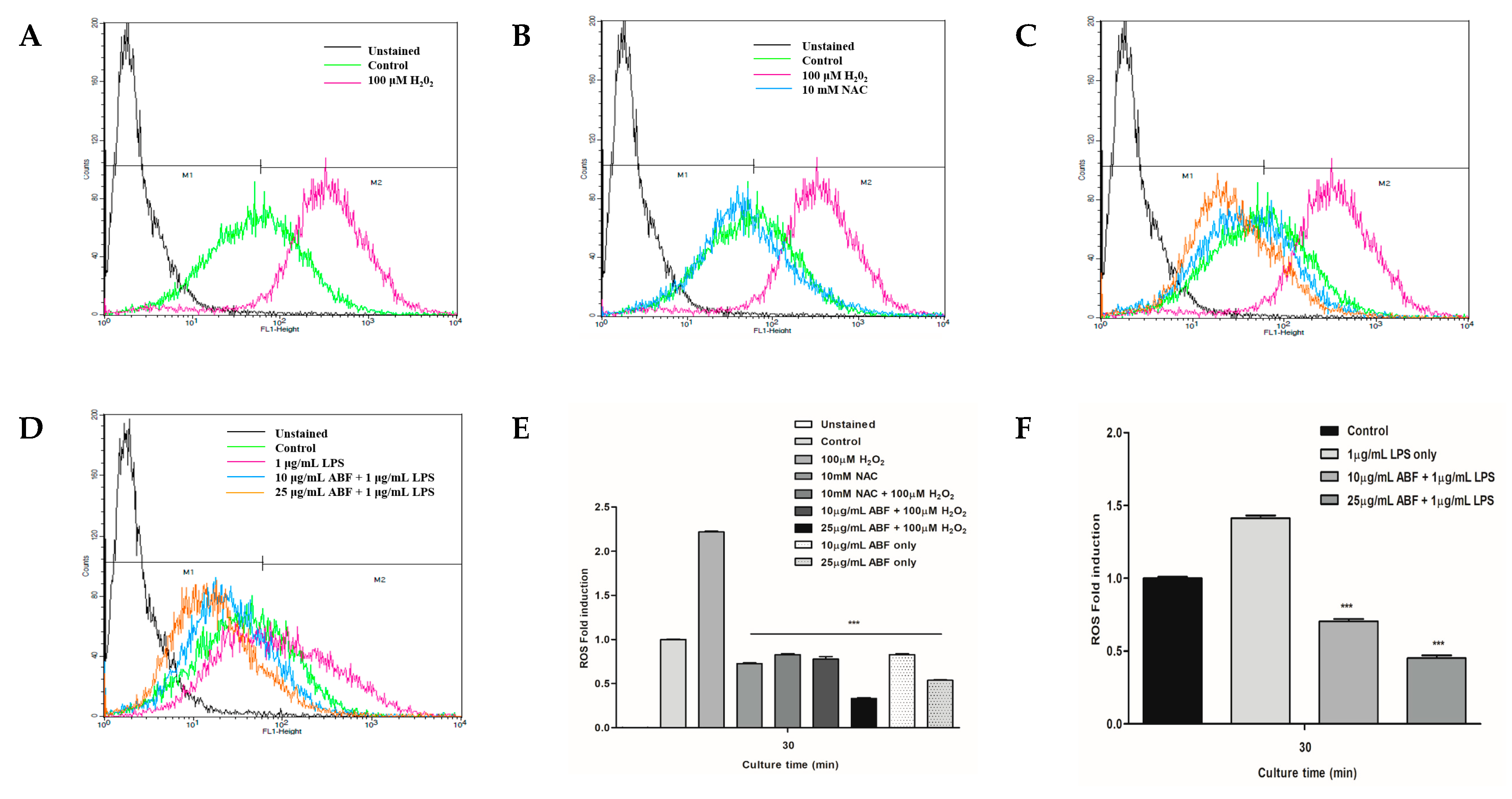
3.5. Effects of ABF® on LPS-Induced ROS Generation
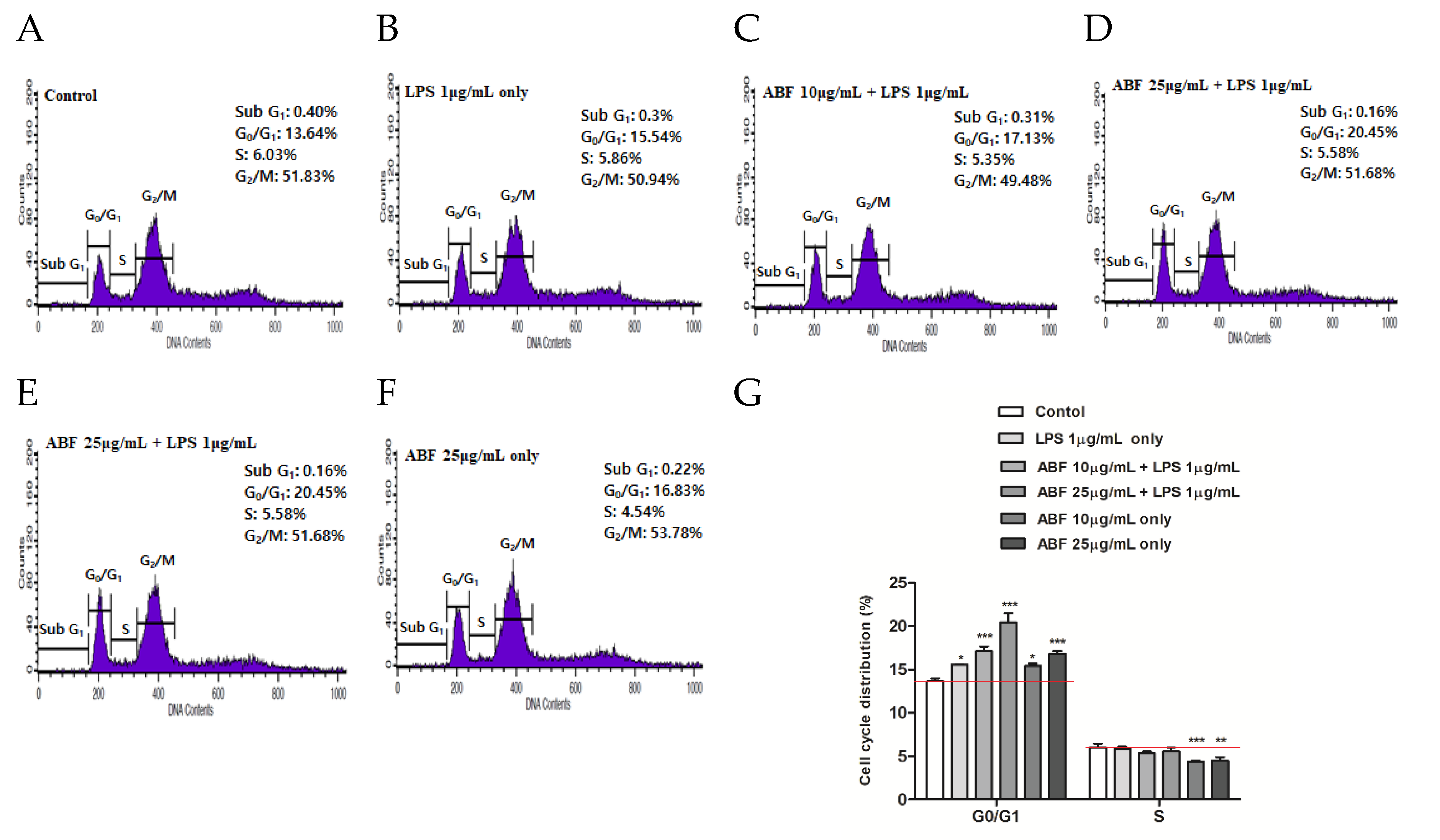
3.6. Effect of ABF® on Induction of Cell Cycle Arrest
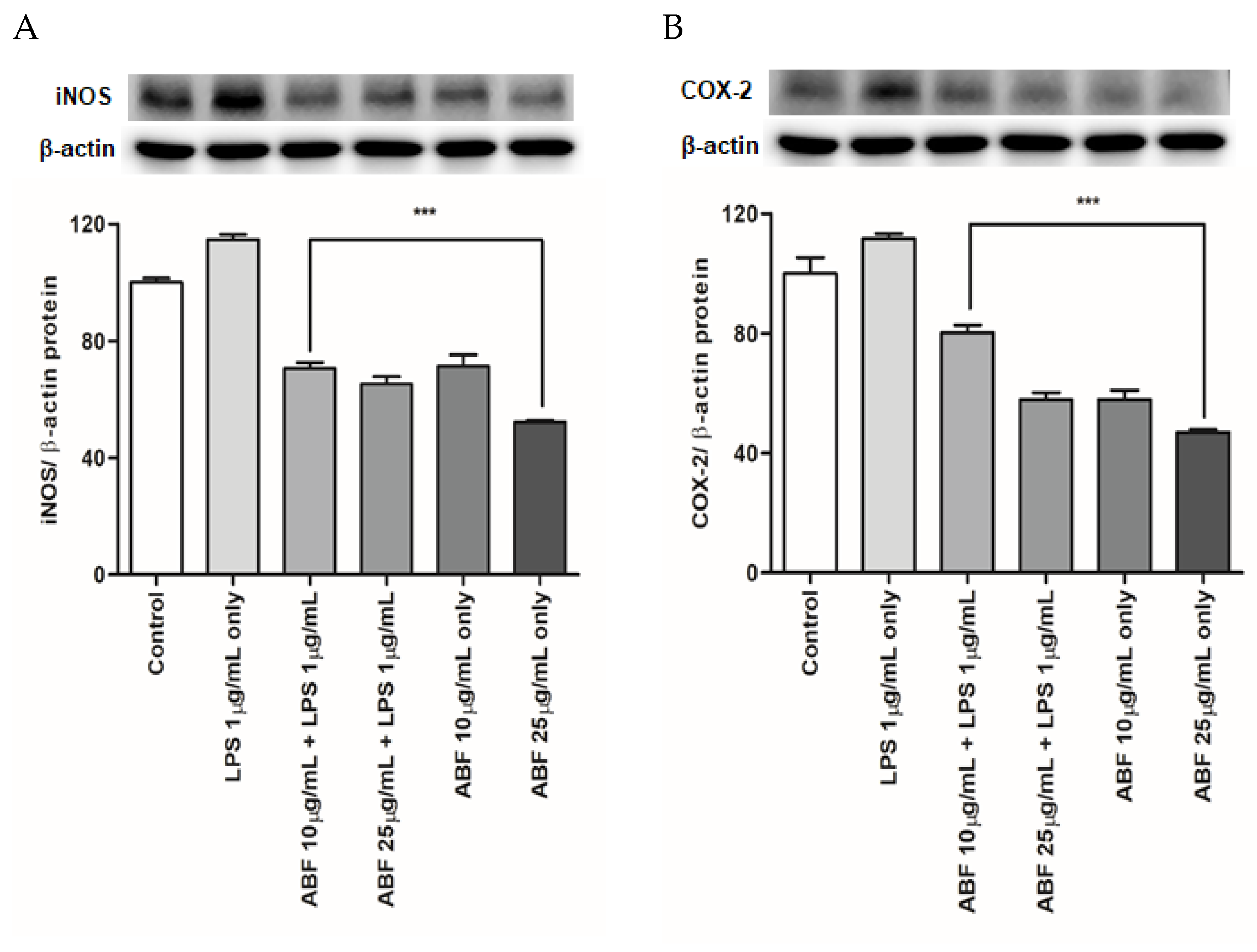
3.7. Effects of ABF® on the Protein Expression of COX-2 and iNOS
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aghasafari, P.; George, U.; Pidaparti, R. A review of inflammatory mechanism in airway diseases. Inflamm. Res. 2019, 68, 59–74. [Google Scholar] [CrossRef]
- Hendaus, M.A.; Jomha, F.A.; Alhammadi, A.H. Virus-induced secondary bacterial infection: A concise review. Ther. Clin. Risk Manag. 2015, 11, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Opal, S.M.; DePalo, V.A. Anti-Inflammatory Cytokines. Chest 2000, 117, 1162–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulling, S.E.; Rawel, H.M. Chokeberry (Aronia melanocarpa)—A review on the characteristic components and potential health effects. Planta Med. 2008, 74, 1625–1634. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Gu, L.; Prior, R.L.; McKay, S. Characterization of Anthocyanins and Proanthocyanidins in Some Cultivars of Ribes, Aronia, and Sambucus and Their Antioxidant Capacity. J. Agric. Food Chem. 2004, 52, 7846–7856. [Google Scholar] [CrossRef]
- Wu, X.; Beecher, G.R.; Holden, J.M.; Haytowitz, D.B.; Gebhardt, S.E.; Prior, R.L. Concentrations of Anthocyanins in Common Foods in the United States and Estimation of Normal Consumption. J. Agric. Food Chem. 2006, 54, 4069–4075. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-H.; Zhu, Y.-N.; Liu, J.; Ren, Y.-X.; Xu, J.-Y.; Yang, Y.-F.; Li, X.-Y.; Zou, J.-P. Differential regulation of resveratrol on lipopolysacchride-stimulated human macrophages with or without IFN-γ pre-priming. Int. Immunopharmacol. 2004, 4, 713–720. [Google Scholar] [CrossRef]
- Chung, E.Y.; Kim, B.H.; Hong, J.-T.; Lee, C.-K.; Ahn, B.; Nam, S.-Y.; Han, S.-B.; Kim, Y. Resveratrol down-regulates interferon-γ-inducible inflammatory genes in macrophages: Molecular mechanism via decreased STAT-1 activation. J. Nutr. Biochem. 2011, 22, 902–909. [Google Scholar] [CrossRef]
- Ohgami, K.; Ilieva, I.; Shiratori, K.; Koyama, Y.; Jin, X.-H.; Yoshida, K.; Kase, S.; Kitaichi, N.; Suzuki, Y.; Tanaka, T.; et al. Anti-inflammatory Effects of Aronia Extract on Rat Endotoxin-Induced Uveitis. Investig. Ophthalmol. Vis. Sci. 2005, 46, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Oszmianski, J.; Sapis, J.C. Anthocyanins in Fruits of Aronia melanocarpa (Chokeberry). J. Food Sci. 1988, 53, 1241–1242. [Google Scholar] [CrossRef]
- Du, X.; Shi, Z.; Peng, Z.; Zhao, C.; Zhang, Y.; Wang, Z.; Li, X.; Liu, G.; Li, X. Acetoacetate induces hepatocytes apoptosis by the ROS-mediated MAPKs pathway in ketotic cows. J. Cell. Physiol. 2017, 232, 3296–3308. [Google Scholar] [CrossRef] [PubMed]
- Afri, M.; Ehrenberg, B.; Talmon, Y.; Schmidt, J.; Cohen, Y.; Frimer, A.A. Active oxygen chemistry within the liposomal bilayer: Part III: Locating Vitamin E, ubiquinol and ubiquinone and their derivatives in the lipid bilayer. Chem. Phys. Lipids 2004, 131, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef]
- Diamond, G.; Legarda, D.; Ryan, L.K. The innate immune response of the respiratory epithelium. Immunol. Rev. 2000, 173, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Teran, L.M. CCL Chemokines and asthma. Immunol. Today 2000, 21, 235–242. [Google Scholar] [CrossRef]
- Mitchell, P.D.; O’Byrne, P.M. Epithelial-Derived Cytokines in Asthma. Chest 2017, 151, 1338–1344. [Google Scholar] [CrossRef]
- Beck, L.A.; Dalke, S.; Leiferman, K.M.; Bickel, C.A.; Hamilton, R.; Rosen, H.; Bochner, B.S.; Schleimer, R.P. Cutaneous injection of RANTES causes eosinophil recruitment: Comparison of nonallergic and allergic human subjects. J. Immunol. 1997, 159, 2962–2972. [Google Scholar] [CrossRef]
- Nickel, R.; Beck, L.A.; Stellato, C.; Schleimer, R.P. Chemokines and allergic disease. J. Allergy Clin. Immunol. 1999, 104, 723–742. [Google Scholar] [CrossRef]
- Stellato, C.; Beck, L.A.; Gorgone, G.A.; Proud, D.; Schall, T.J.; Ono, S.J.; Lichtenstein, L.M.; Schleimer, R.P. Expression of the chemokine RANTES by a human bronchial epithelial cell line. Modulation by cytokines and glucocorticoids. J. Immunol. 1995, 155, 410–418. [Google Scholar]
- Rahmani, J.; Clark, C.; Kord Varkaneh, H.; Lakiang, T.; Vasanthan, L.T.; Onyeche, V.; Mousavi, S.M.; Zhang, Y. The effect of Aronia consumption on lipid profile, blood pressure, and biomarkers of inflammation: A systematic review and meta-analysis of randomized controlled trials. Phytother. Res. 2019, 33, 1981–1990. [Google Scholar] [CrossRef]
- Jeon, Y.D.; Kang, S.H.; Moon, K.H.; Lee, J.H.; Kim, D.G.; Kim, W.; Kim, J.S.; Ahn, B.Y.; Jin, J.S. The Effect of Aronia Berry on Type 1 Diabetes In Vivo and In Vitro. J. Med. Food 2018, 21, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Valcheva-Kuzmanova, S.; Stavreva, G.; Dancheva, V.; Terziev, L.; Atanasova, M.; Stoyanova, A.; Dimitrova, A.; Shopova, V. Effect of Aronia melanocarpa fruit juice on amiodarone-induced pneumotoxicity in rats. Pharmacogn. Mag. 2014, 10, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Appel, K.; Meiser, P.; Millán, E.; Collado, J.A.; Rose, T.; Gras, C.C.; Carle, R.; Muñoz, E. Chokeberry (Aronia melanocarpa (Michx.) Elliot) concentrate inhibits NF-κB and synergizes with selenium to inhibit the release of pro-inflammatory mediators in macrophages. Fitoterapia 2015, 105, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Thi, N.; Hwang, E.-S. Anti-cancer and anti-inflammatory activities of aronia (Aronia melanocarpa) leaves. Asian Pac. J. Trop. Biomed. 2018, 8, 586–592. [Google Scholar] [CrossRef]
- Borissova, P.; Valcheva, S.; Belcheva, A. Antiinflammatory effect of flavonoids in the natural juice from Aronia melanocarpa, rutin and rutin-magnesium complex on an experimental model of inflammation induced by histamine and serotonin. Acta Physiol. Pharmacol. Bulg. 1994, 20, 25–30. [Google Scholar]
- Rose-John, S.; Winthrop, K.; Calabrese, L. The role of IL-6 in host defence against infections: Immunobiology and clinical implications. Nat. Rev. Rheumatol. 2017, 13, 399–409. [Google Scholar] [CrossRef]
- Jones, B.E.; Maerz, M.D.; Buckner, J.H. IL-6: A cytokine at the crossroads of autoimmunity. Curr. Opin. Immunol. 2018, 55, 9–14. [Google Scholar] [CrossRef]
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Matsukura, S.; Kokubu, F.; Noda, H.; Tokunaga, H.; Adachi, M. Expression of IL-6, IL-8, and RANTES on human bronchial epithelial cells, NCI-H292, induced by influenza virus A. J. Allergy Clin. Immunol. 1996, 98, 1080–1087. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, B.; Pendyala, S.; Hecker, L.; Lee, P.J.; Natarajan, V.; Thannickal, V.J. NOX Enzymes and Pulmonary Disease. Antioxid. Redox Signal. 2009, 11, 2505–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendyala, S.; Natarajan, V. Redox regulation of Nox proteins. Respir. Physiol. Neurobiol. 2010, 174, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Parker, D.; Prince, A. Innate Immunity in the Respiratory Epithelium. Am. J. Respir. Cell Mol. Biol. 2011, 45, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yin, S.; Chen, Y.; Wu, Y.; Zheng, W.; Dong, H.; Bai, Y.; Qin, Y.; Li, J.; Feng, S.; et al. LPS-induced proinflammatory cytokine expression in human airway epithelial cells and macrophages via NF-κB, STAT3 or AP-1 activation. Mol. Med. Rep. 2018, 17, 5484–5491. [Google Scholar] [CrossRef] [Green Version]
- Rietschel, E.T.; Brade, H. Bacterial Endotoxins. Sci. Am. 1992, 267, 54–61. [Google Scholar] [CrossRef]
- Britt, R.D., Jr.; Locy, M.L.; Tipple, T.E.; Nelin, L.D.; Rogers, L.K. Lipopolysaccharide-induced Cyclooxygenase-2 Expression in Mouse Transformed Clara Cells. Cell. Physiol. Biochem. 2012, 29, 213–222. [Google Scholar] [CrossRef]
- Schmidt, L.M.; Belvisi, M.G.; Bode, K.A.; Bauer, J.; Schmidt, C.; Suchy, M.-T.; Tsikas, D.; Scheuerer, J.; Lasitschka, F.; Gröne, H.-J.; et al. Bronchial Epithelial Cell-Derived Prostaglandin E2 Dampens the Reactivity of Dendritic Cells. J. Immunol. 2011, 186, 2095–2105. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Accession Number | Sequence | Product (bp) |
|---|---|---|---|
| IL-6 | NM000600.5 | 5′-GTGTTGCCTGCTGCCTTC-3′ 5′-AGTGCCTCTTTGCTGCTTTC-3′ | 194 |
| IL-8 | NM000584 | 5′-GACATACTCCAAACCTTTCCAC-3′ 5′- CTTCTCCACAACCCTCTGC-3′ | 160 |
| RANTES | M21121 | 5′-TTTGCCTACATTGCCCGC-3′ 5′-TTTCGGGTGACAAAGACGACT-3′ | 370 |
| TNF-α | NM000594 | 5′-ATCTTCTCGAACCCCGAGTG-3′ 5′-GGGTTTGCTACAACATGGGC-3′ | 51 |
| IL-1β | NM000576 | 5′-TGATGGCTTATTACAGTGGCAATG-3′ 5′-GTAGTGGTGGTCGGAGATTCG-3 | 140 |
| TGF-β1 | NM000660.5 | 5-TGAACCGGCCTTTCCTGCTTCTCATG -3′ 5′-GCGGAAGTCAATGTACAGCTGCCGC-3′ | 152 |
| COX-2 | U04636 | 5′-CAAATCCTTGCTGTTCCCACCCAT-3′ 5′-GTGCACTGTGTTTGGAGTGGGTTT-3′ | 173 |
| β-actin | NM001101 | 5′-GCGAGAAGATGACCCAGATC-3′ 5′-GGATAGCACAGCCTGGATAG-3′ | 77 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, B.-K.; Lee, J.-W.; Choi, H.; Yim, S.-V. Aronia melanocarpa Fruit Bioactive Fraction Attenuates LPS-Induced Inflammatory Response in Human Bronchial Epithelial Cells. Antioxidants 2020, 9, 816. https://doi.org/10.3390/antiox9090816
Jang B-K, Lee J-W, Choi H, Yim S-V. Aronia melanocarpa Fruit Bioactive Fraction Attenuates LPS-Induced Inflammatory Response in Human Bronchial Epithelial Cells. Antioxidants. 2020; 9(9):816. https://doi.org/10.3390/antiox9090816
Chicago/Turabian StyleJang, Bong-Keun, Jin-Woo Lee, Hyun Choi, and Sung-Vin Yim. 2020. "Aronia melanocarpa Fruit Bioactive Fraction Attenuates LPS-Induced Inflammatory Response in Human Bronchial Epithelial Cells" Antioxidants 9, no. 9: 816. https://doi.org/10.3390/antiox9090816
APA StyleJang, B.-K., Lee, J.-W., Choi, H., & Yim, S.-V. (2020). Aronia melanocarpa Fruit Bioactive Fraction Attenuates LPS-Induced Inflammatory Response in Human Bronchial Epithelial Cells. Antioxidants, 9(9), 816. https://doi.org/10.3390/antiox9090816