Antioxidant Effects of PS5, a Peptidomimetic of Suppressor of Cytokine Signaling 1, in Experimental Atherosclerosis

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis
2.2. Cell Cultures
2.3. Cell Viability, Proliferation and Migration
2.4. Superoxide Measurement by DHE
2.5. Mouse Model of Atherosclerosis and Treatments
2.6. Histological Analysis and Quantification
2.7. mRNA Expression Analysis
2.8. Statistical Analyses
3. Results
3.1. In Vitro Effects of PS5 Compound
3.1.1. Cellular Viability, Proliferation and Migration
3.1.2. Cellular ROS Production
3.2. Biodistribution and In Vivo Effects of PS5 Compound
3.2.1. Atheroma Plaque Development
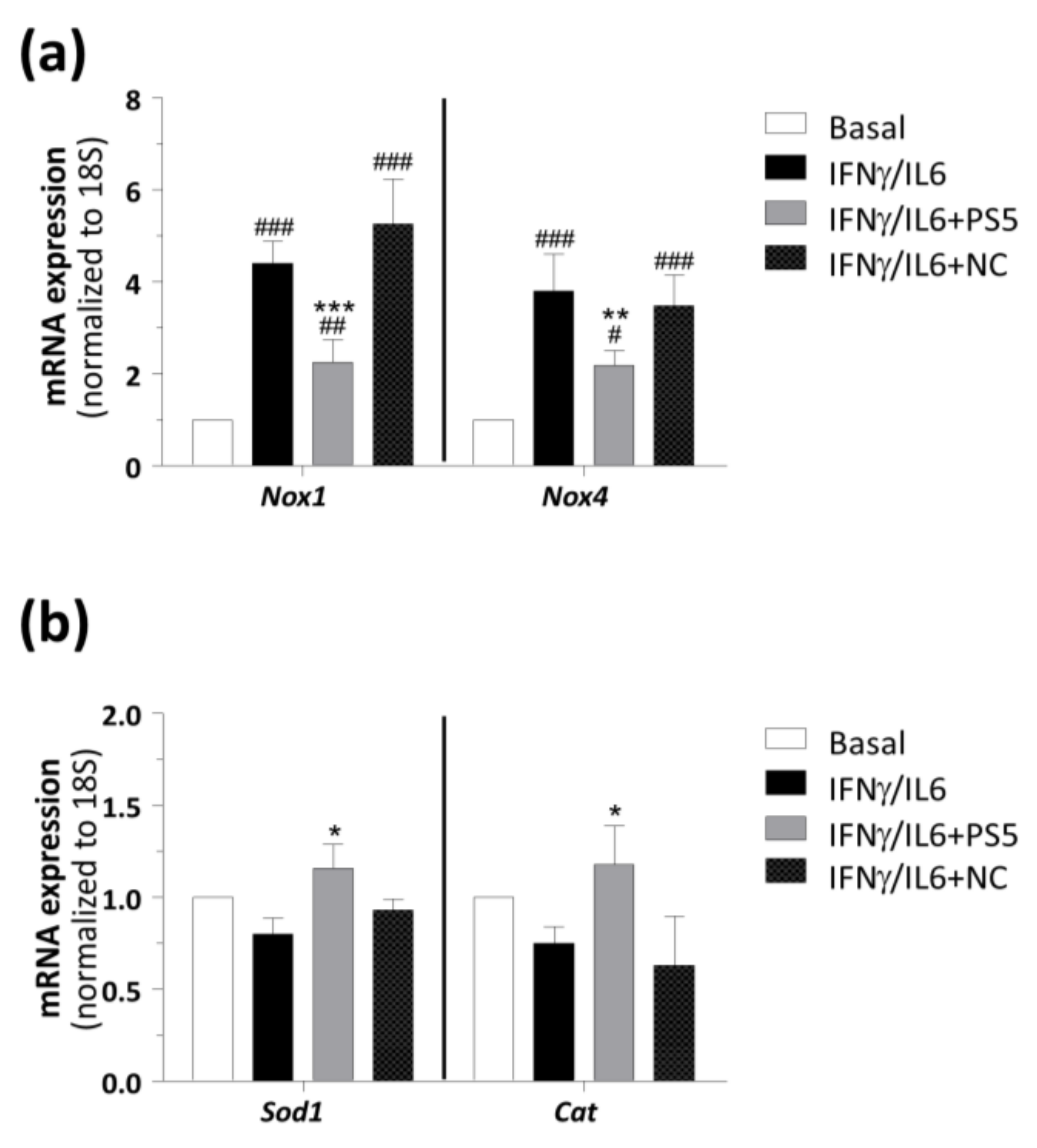
3.2.2. JAK/STAT Regulated and Oxidative Genes in Aortic Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pahwa, R.; Jialal, I. Chronic Inflammation; StatPearls: Treasure Island, FL, USA, 2019. [Google Scholar]
- Kleemann, R.; Zadelaar, S.; Kooistra, T. Cytokines and atherosclerosis: A comprehensive review of studies in mice. Cardiovasc. Res. 2008, 79, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ventura, J.L.; Rodrigues-Diez, R.; Martinez-Lopez, D.; Salaices, M.; Blanco-Colio, L.M.; Briones, A.M. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2315. [Google Scholar] [CrossRef]
- Brandes, R.P.; Schroder, K. Differential vascular functions of Nox family NADPH oxidases. Curr. Opin. Lipidol. 2008, 19, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function—role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants (Basel) 2017, 6, 90. [Google Scholar] [CrossRef]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from In Vivo models and clinical studies. Basic Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef]
- Boengler, K.; Hilfiker-Kleiner, D.; Drexler, H.; Heusch, G.; Schulz, R. The myocardial JAK/STAT pathway: From protection to failure. Pharmacol. Ther. 2008, 120, 172–185. [Google Scholar] [CrossRef]
- Gharavi, N.M.; Alva, J.A.; Mouillesseaux, K.P.; Lai, C.; Yeh, M.; Yeung, W.; Johnson, J.; Szeto, W.L.; Hong, L.; Fishbein, M.; et al. Role of the Jak/STAT pathway in the regulation of interleukin-8 transcription by oxidized phospholipids In Vitro and in atherosclerosis In Vivo. J. Biol. Chem. 2007, 282, 31460–31468. [Google Scholar] [CrossRef]
- Lim, W.S.; Timmins, J.M.; Seimon, T.A.; Sadler, A.; Kolodgie, F.D.; Virmani, R.; Tabas, I. Signal transducer and activator of transcription-1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress In Vitro and in advanced atherosclerotic lesions In Vivo. Circulation 2008, 117, 940–951. [Google Scholar] [CrossRef]
- Ortiz-Munoz, G.; Martin-Ventura, J.L.; Hernandez-Vargas, P.; Mallavia, B.; Lopez-Parra, V.; Lopez-Franco, O.; Munoz-Garcia, B.; Fernandez-Vizarra, P.; Ortega, L.; Egido, J.; et al. Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arter. Thromb. Vasc. Biol. 2009, 29, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Huang, Q.; Zhang, H.; Liu, R.; Tellides, G.; Min, W.; Yu, L. SOCS1 prevents graft arteriosclerosis by preserving endothelial cell function. J. Am. Coll. Cardiol. 2014, 63, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S.; Romain, M.; Ramkhelawon, B.; Uyttenhove, C.; Pasterkamp, G.; Herbin, O.; Esposito, B.; Perez, N.; Yasukawa, H.; Van Snick, J.; et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J. Exp. Med. 2009, 206, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Manea, S.A.; Manea, A.; Heltianu, C. Inhibition of JAK/STAT signaling pathway prevents high-glucose-induced increase in endothelin-1 synthesis in human endothelial cells. Cell Tissue Res. 2010, 340, 71–79. [Google Scholar] [CrossRef]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arter. Thromb. Vasc. Biol. 2010, 30, 105–112. [Google Scholar] [CrossRef]
- Fenyo, I.M.; Florea, I.C.; Raicu, M.; Manea, A. Tyrphostin AG490 reduces NAPDH oxidase activity and expression in the aorta of hypercholesterolemic apolipoprotein E-deficient mice. Vasc. Pharmacol. 2011, 54, 100–106. [Google Scholar] [CrossRef]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef]
- Gan, A.M.; Pirvulescu, M.M.; Stan, D.; Simion, V.; Calin, M.; Manduteanu, I.; Butoi, E. Monocytes and smooth muscle cells cross-talk activates STAT3 and induces resistin and reactive oxygen species production [corrected]. J. Cell. Biochem. 2013, 114, 2273–2283. [Google Scholar] [CrossRef]
- Barrett, T.J.; Schlegel, M.; Zhou, F.; Gorenchtein, M.; Bolstorff, J.; Moore, K.J.; Fisher, E.A.; Berger, J.S. Platelet regulation of myeloid suppressor of cytokine signaling 3 accelerates atherosclerosis. Sci. Transl. Med. 2019, 11, eaax0481. [Google Scholar] [CrossRef]
- Jung, S.H.; Kim, S.M.; Lee, C.E. Mechanism of suppressors of cytokine signaling 1 inhibition of epithelial-mesenchymal transition signaling through ROS regulation in colon cancer cells: Suppression of Src leading to thioredoxin up-regulation. Oncotarget 2016, 7, 62559–62571. [Google Scholar] [CrossRef]
- Schuett, J.; Kreutz, J.; Grote, K.; Vlacil, A.K.; Schuett, H.; Oberoi, R.; Schmid, A.; Witten, A.; Stoll, M.; Schieffer, B.; et al. Suppressor of Cytokine Signaling 1 is Involved in Gene Regulation Which Controls the Survival of Ly6C(low) Monocytes in Mice. Cell. Physiol. Biochem. 2019, 52, 336–353. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug. Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef]
- Torella, D.; Curcio, A.; Gasparri, C.; Galuppo, V.; De Serio, D.; Surace, F.C.; Cavaliere, A.L.; Leone, A.; Coppola, C.; Ellison, G.M.; et al. Fludarabine prevents smooth muscle proliferation In Vitro and neointimal hyperplasia In Vivo through specific inhibition of STAT-1 activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2935–H2943. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Oguiza, A.; Mallavia, B.; Lazaro, I.; Ortiz-Munoz, G.; Lopez-Franco, O.; Egido, J.; Gomez-Guerrero, C. Gene delivery of suppressors of cytokine signaling (SOCS) inhibits inflammation and atherosclerosis development in mice. Basic Res. Cardiol. 2015, 110, 8. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanz, L.; Bernal, S.; Recio, C.; Lazaro, I.; Oguiza, A.; Melgar, A.; Jimenez-Castilla, L.; Egido, J.; Gomez-Guerrero, C. SOCS1-targeted therapy ameliorates renal and vascular oxidative stress in diabetes via STAT1 and PI3K inhibition. Lab. Invest. 2018, 98, 1276–1290. [Google Scholar] [CrossRef]
- Hernandez-Vargas, P.; Lopez-Franco, O.; Sanjuan, G.; Ruperez, M.; Ortiz-Munoz, G.; Suzuki, Y.; Aguado-Roncero, P.; Perez-Tejerizo, G.; Blanco, J.; Egido, J.; et al. Suppressors of cytokine signaling regulate angiotensin II-activated Janus kinase-signal transducers and activators of transcription pathway in renal cells. J. Am. Soc. Nephrol. 2005, 16, 1673–1683. [Google Scholar] [CrossRef]
- Galic, S.; Sachithanandan, N.; Kay, T.W.; Steinberg, G.R. Suppressor of cytokine signalling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem. J. 2014, 461, 177–188. [Google Scholar] [CrossRef]
- Yoshimura, A.; Yasukawa, H. JAK’s SOCS: A mechanism of inhibition. Immunity 2012, 36, 157–159. [Google Scholar] [CrossRef]
- Flowers, L.O.; Subramaniam, P.S.; Johnson, H.M. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene 2005, 24, 2114–2120. [Google Scholar] [CrossRef]
- Ahmed, C.M.; Dabelic, R.; Waiboci, L.W.; Jager, L.D.; Heron, L.L.; Johnson, H.M. SOCS-1 mimetics protect mice against lethal poxvirus infection: Identification of a novel endogenous antiviral system. J. Virol. 2009, 83, 1402–1415. [Google Scholar] [CrossRef]
- Flowers, L.O.; Johnson, H.M.; Mujtaba, M.G.; Ellis, M.R.; Haider, S.M.; Subramaniam, P.S. Characterization of a peptide inhibitor of Janus kinase 2 that mimics suppressor of cytokine signaling 1 function. J. Immunol. 2004, 172, 7510–7518. [Google Scholar] [CrossRef]
- Waiboci, L.W.; Ahmed, C.M.; Mujtaba, M.G.; Flowers, L.O.; Martin, J.P.; Haider, M.I.; Johnson, H.M. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: Implications for the development of a SOCS-1 antagonist. J. Immunol. 2007, 178, 5058–5068. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Lazaro, I.; Oguiza, A.; Lopez-Sanz, L.; Bernal, S.; Blanco, J.; Egido, J.; Gomez-Guerrero, C. Suppressor of Cytokine Signaling-1 Peptidomimetic Limits Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 575–585. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Di Natale, C.; Florio, D.; Marasco, D. Peptides as Therapeutic Agents for Inflammatory-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2714. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Lee, E.; Ouzounova, M.; Di Natale, C.; Novellino, E.; Merlino, A.; Korkaya, H.; Marasco, D. Mimetics of suppressor of cytokine signaling 3: Novel potential therapeutics in triple breast cancer. Int. J. Cancer 2018, 143, 2177–2186. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Lopez-Sanz, L.; Mercurio, F.A.; Fortuna, S.; Leone, M.; Gomez-Guerrero, C.; Marasco, D. Chimeric Peptidomimetics of SOCS 3 Able to Interact with JAK2 as Anti-inflammatory Compounds. ACS Med. Chem. Lett. 2020, 11, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Viparelli, F.; Cassese, A.; Doti, N.; Paturzo, F.; Marasco, D.; Dathan, N.A.; Monti, S.M.; Basile, G.; Ungaro, P.; Sabatella, M.; et al. Targeting of PED/PEA-15 molecular interaction with phospholipase D1 enhances insulin sensitivity in skeletal muscle cells. J. Biol. Chem. 2008, 283, 21769–21778. [Google Scholar] [CrossRef]
- La Manna, S.; Scognamiglio, P.L.; Di Natale, C.; Leone, M.; Mercurio, F.A.; Malfitano, A.M.; Cianfarani, F.; Madonna, S.; Caravella, S.; Albanesi, C.; et al. Characterization of linear mimetic peptides of Interleukin-22 from dissection of protein interfaces. Biochimie 2017, 138, 106–115. [Google Scholar] [CrossRef]
- Madonna, S.; Scarponi, C.; Morelli, M.; Sestito, R.; Scognamiglio, P.L.; Marasco, D.; Albanesi, C. SOCS3 inhibits the pathological effects of IL-22 in non-melanoma skin tumor-derived keratinocytes. Oncotarget 2017, 8, 24652–24667. [Google Scholar] [CrossRef]
- Russo, A.; Aiello, C.; Grieco, P.; Marasco, D. Targeting “Undruggable” Proteins: Design of Synthetic Cyclopeptides. Curr. Med. Chem. 2016, 23, 748–762. [Google Scholar] [CrossRef]
- Canesi, F.; Mateo, V.; Couchie, D.; Karabina, S.; Negre-Salvayre, A.; Rouis, M.; El Hadri, K. A thioredoxin-mimetic peptide exerts potent anti-inflammatory, antioxidant, and atheroprotective effects in ApoE2.Ki mice fed high fat diet. Cardiovasc. Res. 2019, 115, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Tabet, F.; Remaley, A.T.; Segaliny, A.I.; Millet, J.; Yan, L.; Nakhla, S.; Barter, P.J.; Rye, K.A.; Lambert, G. The 5A apolipoprotein A-I mimetic peptide displays antiinflammatory and antioxidant properties In Vivo and In Vitro. Arter. Thromb. Vasc. Biol. 2010, 30, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Marasco, D.; Perretta, G.; Sabatella, M.; Ruvo, M. Past and future perspectives of synthetic peptide libraries. Curr. Protein Pept. Sci. 2008, 9, 447–467. [Google Scholar] [CrossRef] [PubMed]
- Doti, N.; Scognamiglio, P.L.; Madonna, S.; Scarponi, C.; Ruvo, M.; Perretta, G.; Albanesi, C.; Marasco, D. New mimetic peptides of the kinase-inhibitory region (KIR) of SOCS1 through focused peptide libraries. Biochem. J. 2012, 443, 231–240. [Google Scholar] [CrossRef]
- La Manna, S.; Lopez-Sanz, L.; Leone, M.; Brandi, P.; Scognamiglio, P.L.; Morelli, G.; Novellino, E.; Gomez-Guerrero, C.; Marasco, D. Structure-activity studies of peptidomimetics based on kinase-inhibitory region of suppressors of cytokine signaling 1. Biopolymers 2017, 110, e23082. [Google Scholar] [CrossRef]
- Madonna, S.; Scarponi, C.; Doti, N.; Carbone, T.; Cavani, A.; Scognamiglio, P.L.; Marasco, D.; Albanesi, C. Therapeutical potential of a peptide mimicking the SOCS1 kinase inhibitory region in skin immune responses. Eur. J. Immunol. 2013, 43, 1883–1895. [Google Scholar] [CrossRef]
- Fik-Jaskolka, M.A.; Mkrtchyan, A.F.; Saghyan, A.S.; Palumbo, R.; Belter, A.; Hayriyan, L.A.; Simonyan, H.; Roviello, V.; Roviello, G.N. Biological macromolecule binding and anticancer activity of synthetic alkyne-containing L-phenylalanine derivatives. Amino Acids 2020, 52, 755–769. [Google Scholar] [CrossRef]
- Fik-Jaskolka, M.A.; Mkrtchyan, A.F.; Saghyan, A.S.; Palumbo, R.; Belter, A.; Hayriyan, L.A.; Simonyan, H.; Roviello, V.; Roviello, G.N. Spectroscopic and SEM evidences for G4-DNA binding by a synthetic alkyne-containing amino acid with anticancer activity. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 229, 117884. [Google Scholar] [CrossRef]
- Hu, D.; Yin, C.; Luo, S.; Habenicht, A.J.R.; Mohanta, S.K. Vascular Smooth Muscle Cells Contribute to Atherosclerosis Immunity. Front. Immunol. 2019, 10, 1101. [Google Scholar] [CrossRef]
- Xu, H.; Jiang, J.; Chen, W.; Li, W.; Chen, Z. Vascular Macrophages in Atherosclerosis. J. Immunol. Res. 2019, 2019, 4354786. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: Another inconvenient truth. Free. Radic. Biol. Med. 2010, 48, 983–1001. [Google Scholar] [CrossRef] [PubMed]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Kang, S.W. Targeting cellular antioxidant enzymes for treating atherosclerotic vascular disease. Biomol. Ther. (Seoul) 2013, 21, 89–96. [Google Scholar] [CrossRef]
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef]
- Lim, S.; Park, S. Role of vascular smooth muscle cell in the inflammation of atherosclerosis. BMB Rep. 2014, 47, 1–7. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seica, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef]
- Bhatt, S.R.; Lokhandwala, M.F.; Banday, A.A. Vascular oxidative stress upregulates angiotensin II type I receptors via mechanisms involving nuclear factor kappa B. Clin. Exp. Hypertens. 2014, 36, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Roberts, L.J.; Shi, M.J.; Zhou, L.C.; Ballard, B.R.; Richardson, A.; Guo, Z.M. Retardation of atherosclerosis by overexpression of catalase or both Cu/Zn-superoxide dismutase and catalase in mice lacking apolipoprotein E. Circ. Res. 2004, 95, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Grothusen, C.; Schuett, H.; Hillmer, A.; Lumpe, S.; Grote, K.; Ballmaier, M.; Bleich, A.; Glage, S.; Tietge, U.J.; Luchtefeld, M.; et al. Role of suppressor of cytokine signaling-1 in murine atherosclerosis. PLoS ONE 2012, 7, e51608. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gen Taqman | Primer Code | Gen Taqman | Primer Code | |
|---|---|---|---|---|
| Ccl2 | Mm00441242_m1 | Sod1 | Mm01344233_g1 | |
| Ccl5 | Mm01302428_m1 | Catalasa | Mm00437992_m1 | |
| Cxcl10 | Mm00445235_m1 | 18S | 4310893E | |
| Gen SYBR Green | Forward sequence (5’-3’) | Reverse sequence (5’-3’) | ||
| Nox1 | CCAACAGGCCATGGATGGAT | CACTCCAGTAAGCCAGCAA | ||
| Nox4 | CCCTCCTGGCTGCATTAGTC | AACCCTCGAGGCAAAGATCC | ||
| 18S | CCGTCGTAGTTCCGACCATAA | CAGCTTTGCAACCATACTCCC | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Manna, S.; Lopez-Sanz, L.; Bernal, S.; Jimenez-Castilla, L.; Prieto, I.; Morelli, G.; Gomez-Guerrero, C.; Marasco, D. Antioxidant Effects of PS5, a Peptidomimetic of Suppressor of Cytokine Signaling 1, in Experimental Atherosclerosis. Antioxidants 2020, 9, 754. https://doi.org/10.3390/antiox9080754
La Manna S, Lopez-Sanz L, Bernal S, Jimenez-Castilla L, Prieto I, Morelli G, Gomez-Guerrero C, Marasco D. Antioxidant Effects of PS5, a Peptidomimetic of Suppressor of Cytokine Signaling 1, in Experimental Atherosclerosis. Antioxidants. 2020; 9(8):754. https://doi.org/10.3390/antiox9080754
Chicago/Turabian StyleLa Manna, Sara, Laura Lopez-Sanz, Susana Bernal, Luna Jimenez-Castilla, Ignacio Prieto, Giancarlo Morelli, Carmen Gomez-Guerrero, and Daniela Marasco. 2020. "Antioxidant Effects of PS5, a Peptidomimetic of Suppressor of Cytokine Signaling 1, in Experimental Atherosclerosis" Antioxidants 9, no. 8: 754. https://doi.org/10.3390/antiox9080754
APA StyleLa Manna, S., Lopez-Sanz, L., Bernal, S., Jimenez-Castilla, L., Prieto, I., Morelli, G., Gomez-Guerrero, C., & Marasco, D. (2020). Antioxidant Effects of PS5, a Peptidomimetic of Suppressor of Cytokine Signaling 1, in Experimental Atherosclerosis. Antioxidants, 9(8), 754. https://doi.org/10.3390/antiox9080754