Current Landscape of NRF2 Biomarkers in Clinical Trials


Abstract
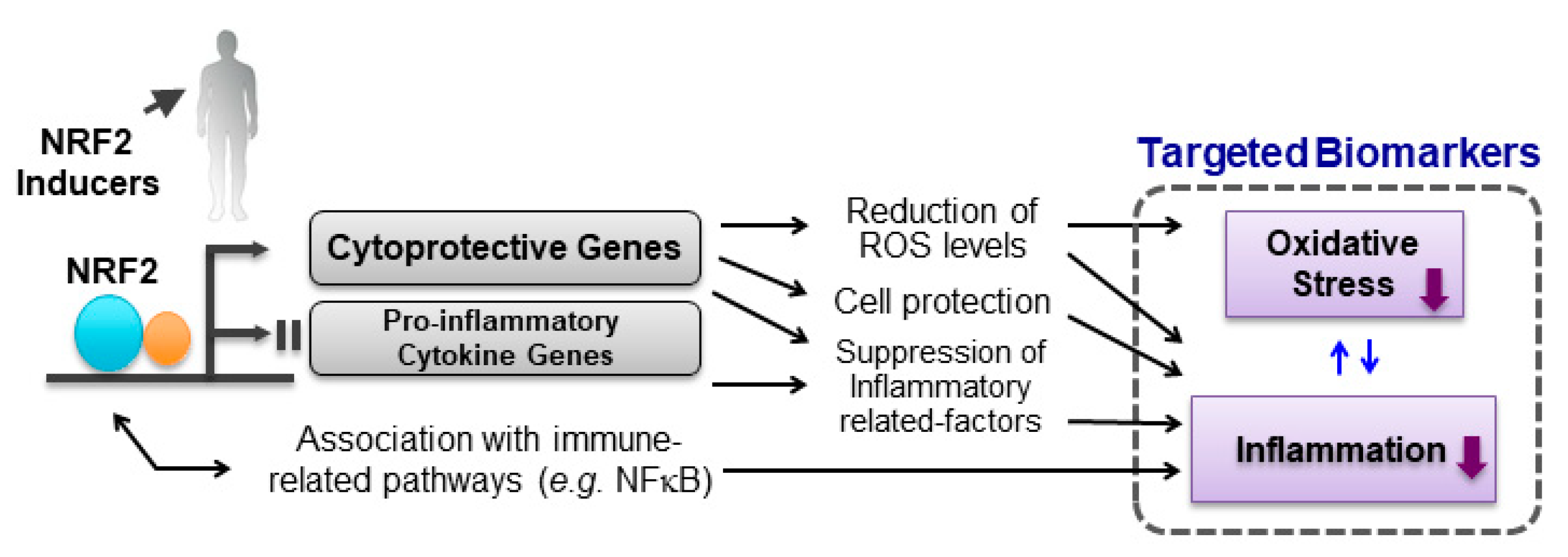
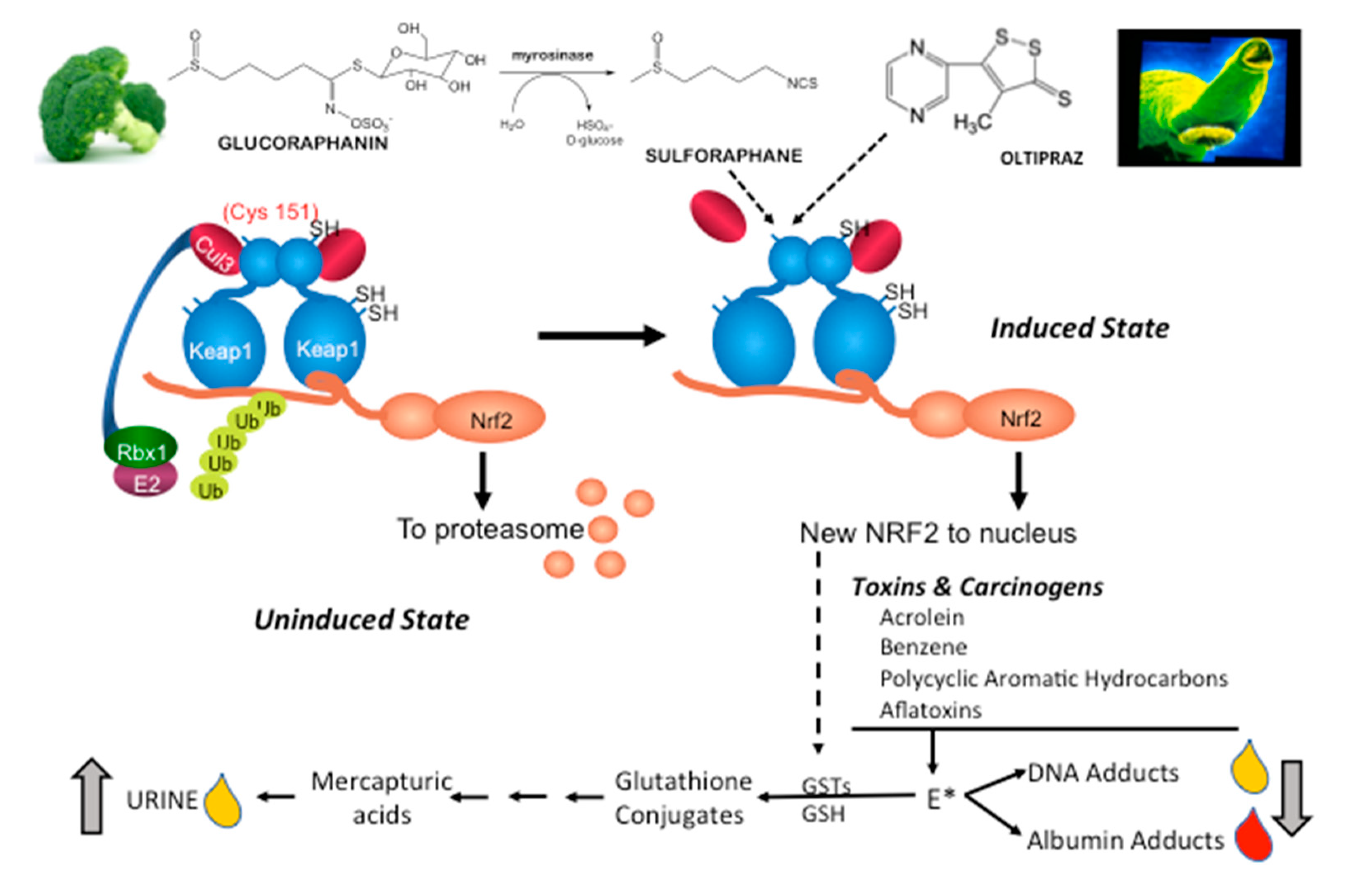
1. The KEAP1-NRF2 System
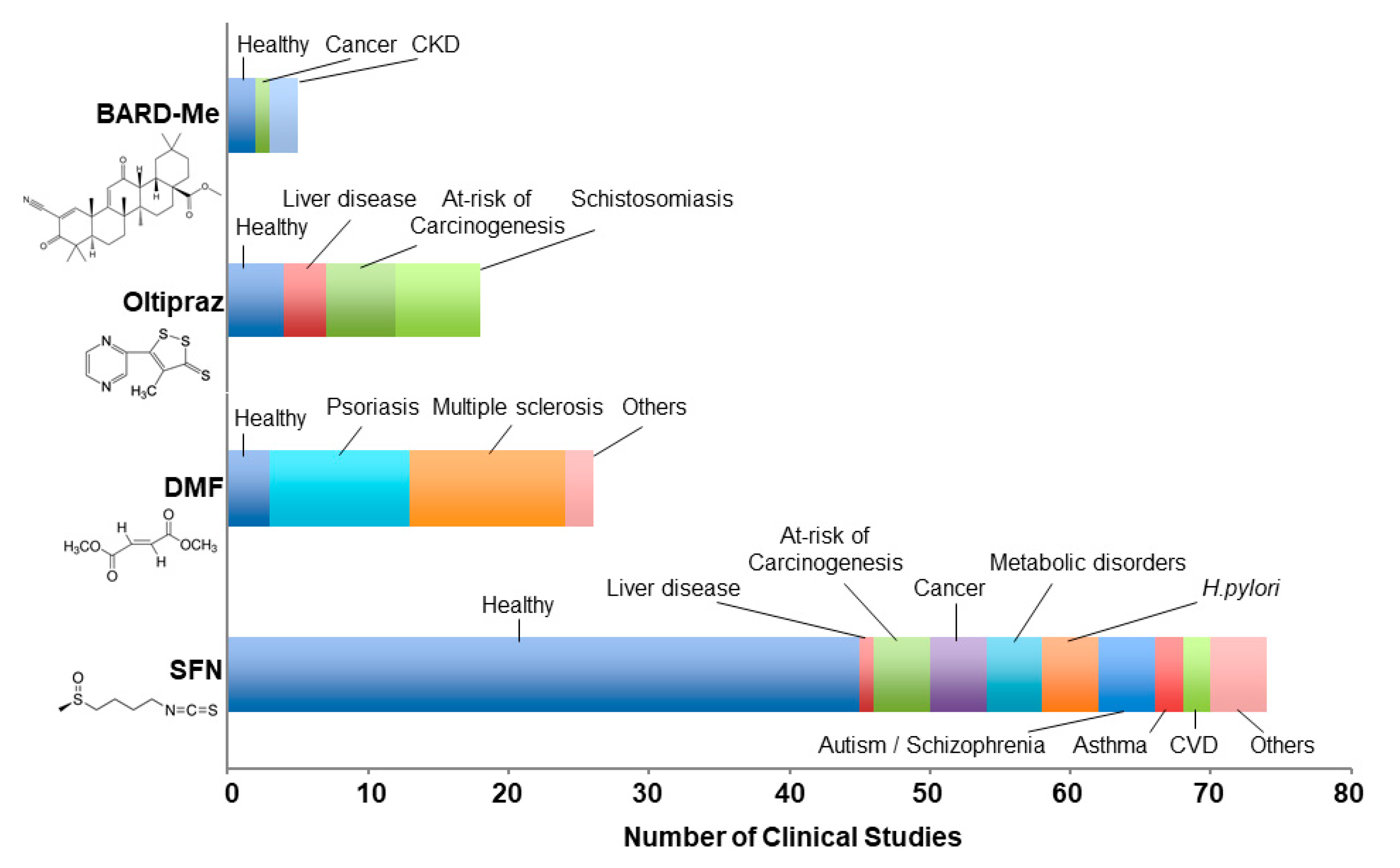
2. Pharmacological Inducers of KEAP1-NRF2 Signaling
2.1. Dimethyl Fumarate (DMF, BG-12, Tecfidera)
2.2. Bardoxolone-methyl (BARD-Me; CDDO-Me)
2.3. Oltipraz
2.4. Sulforaphane (SFN)
2.5. Other Natural Product Inducers
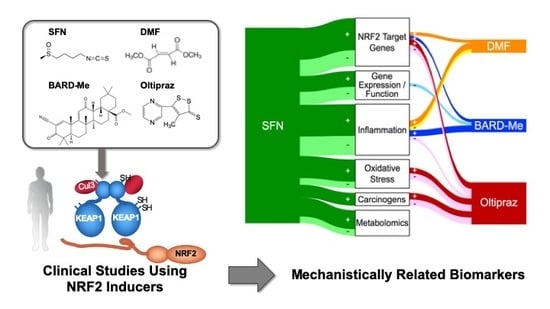
3. Biomarker-Based Clinical Studies and NRF2 Inducers
3.1. Nrf2 Target Genes
3.2. Gene Expression and Function
3.3. Oxidative-Stress-Mediated Biomarkers
3.4. Inflammation-Mediated Biomarkers
3.5. Carcinogen Metabolites/DNA Adducts
3.6. Metabolomics
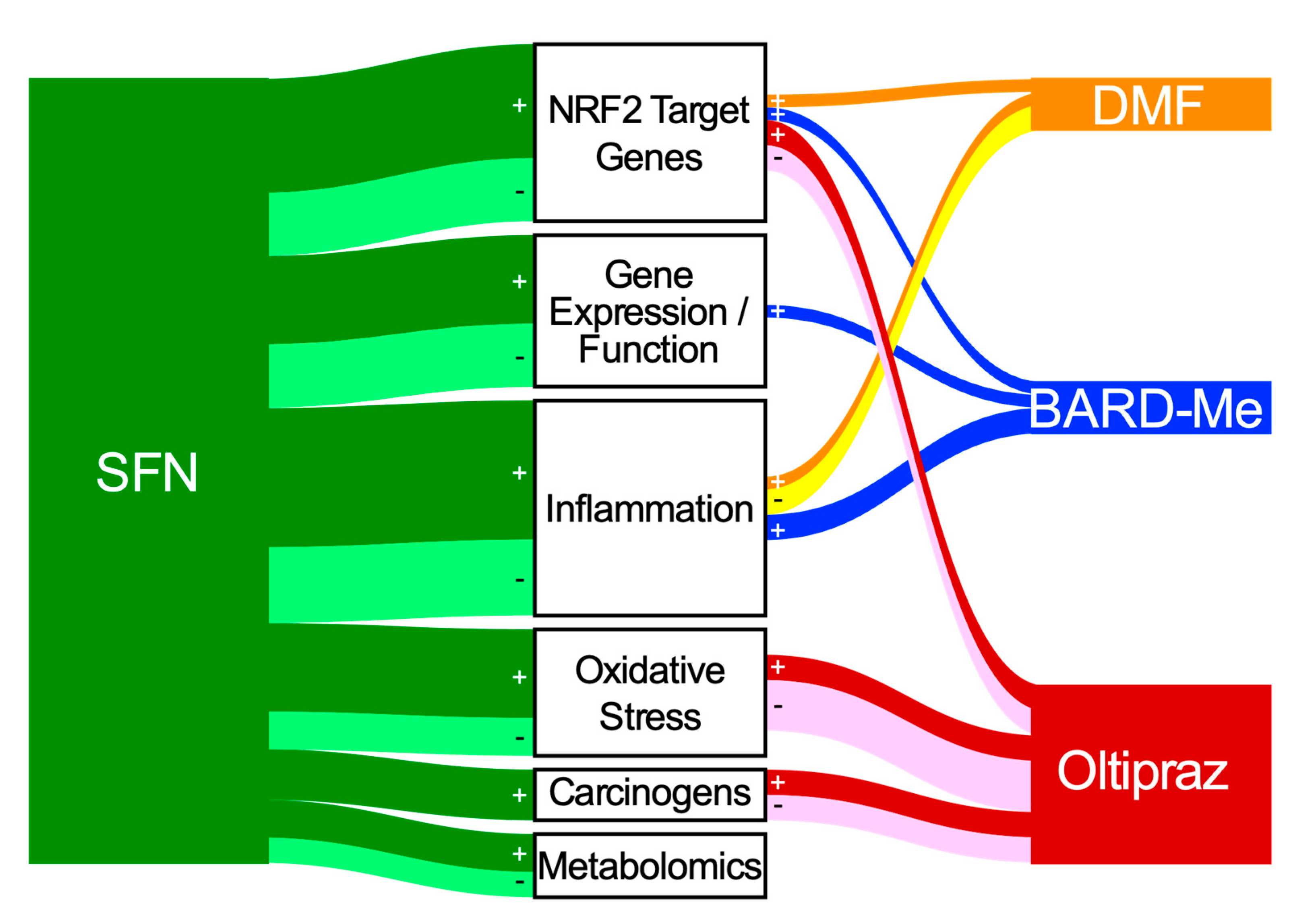
4. Integrated Assessment of Biomarker Outcomes
5. Conclusions
5.1. Critical Path for Biomarkers in NRF2 Drug Development?
5.2. Metrics of Success and Confounders
5.3. Lessons from Dose-Response
5.4. Take-Home Messages
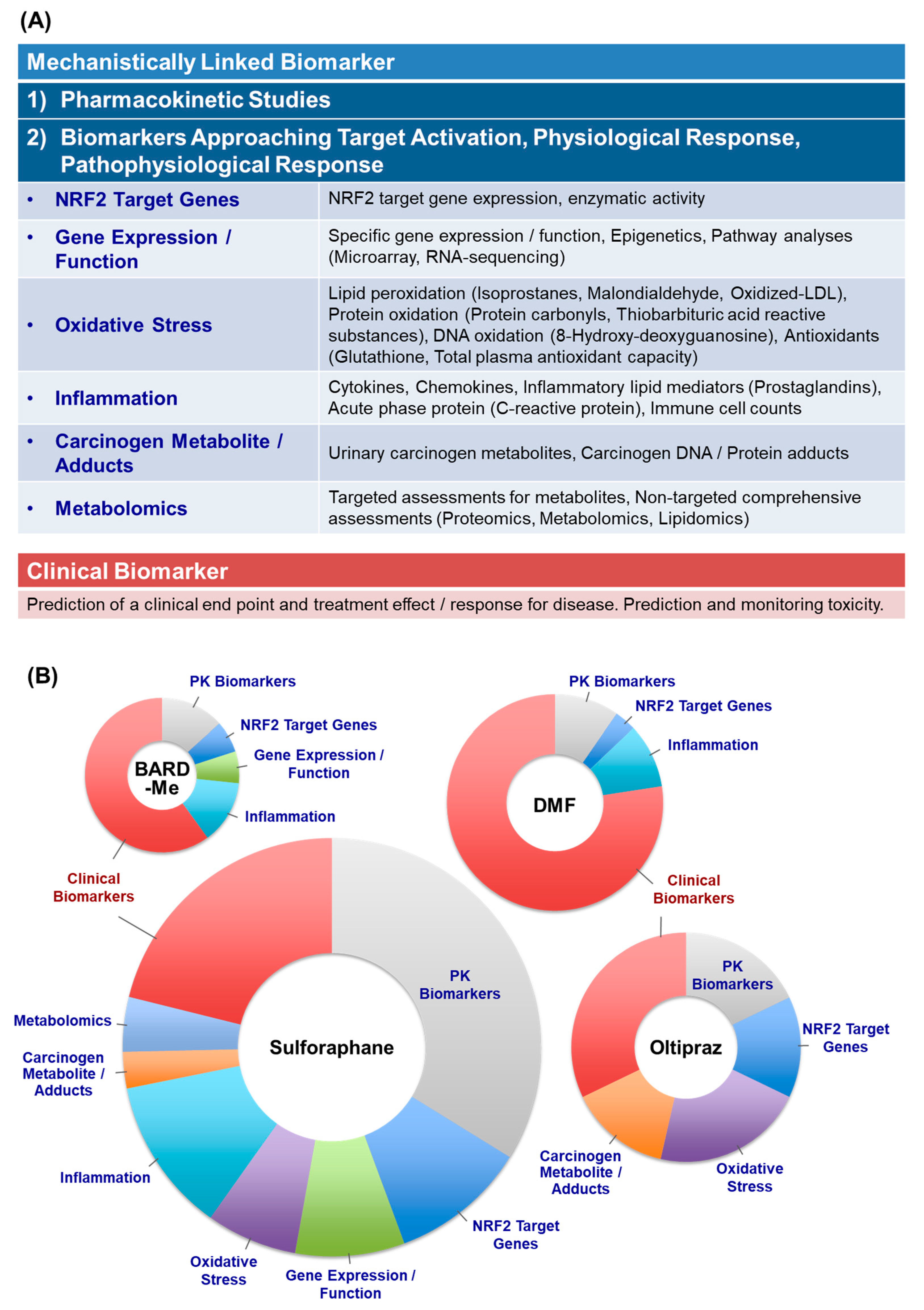
- NRF2 Target Genes: Given the preclinical evidence that all 4 of the agents can activate Nrf2 signaling, it is comforting that all four increased activities or transcript levels of classic NRF2 target genes in clinical trials. NQO1 was most studied and showed reasonable consistency across trials. Worryingly, in most studies the induction of NQO1 transcripts exhibited a limited dynamic range (~ < 2-fold). In an oltipraz study, concordance between expression in surrogate (e.g., PBMCs) and target tissues (e.g., colonic mucosa) was reported. Limited studies suggest possible merit for PCA or clustering analyses to characterize induction “signatures” that may be more revealing than single candidate genes;
- Gene Expression/Function: Most studies here have focused on pathways affecting cancer development and progression. The most promising outcomes (with SFN) have centered on modulation of epigenetic regulators such as histone deacetylase (HDAC) and histone acetyltransferase activities. One cancer-related gene expression panel was largely unaltered in a BARD-Me study and the other two agents were not evaluated in this context;
- Oxidative Stress: Many of the workhorse biomarkers of oxidative stress have been applied to clinical studies with SFN and oltipraz, but not the other two drugs. Oxidized DNA products along with DNA strand breaks have shown protective responses in some of the interventions. Studies using the oxidation products of lipids and proteins have been more variable in their responses, although MDA looks promising. The more integrated measures of TAC, TOS, and OSI have not been revealing in limited studies. Cellular GSH levels have been measured frequently and show repeated, albeit still inconsistent, modulation by intervention;
- Inflammation: In the aggregate over 35 individual inflammation biomarkers have been measured, while barely half evoked a significant response in any study; very few have been evaluated in multiple studies. The context for selection of candidate biomarkers is rarely presented in these studies. The NRF2 target gene Il-6 [227] shows some responsiveness, while other cytokines such as IL-1, IL-8 IL-13, TNFα, and IFNγ have been null in multiple studies. Lipid mediators including PGD2, tetranor-PGEM, 11β-PGF2α, and 11-dehydro-TXB2 offer some promise. Subgroup analyses of responders only within a DMF trial exhibited significant reductions in intracellular NF-κB signaling molecules [50];
- Carcinogen Metabolism/Adducts: Monitoring detoxication metabolites following interventions in study populations provides strong links to canonical NRF2 mechanisms of action. Multiple studies in settings of unavoidable exposures to air pollution and dietary carcinogens highlight successful interventions with oltipraz and SFN. However, such studies require sophisticated mass spectrometry methodologies for metabolite, DNA adduct, and protein adduct quantification. Moreover, interception of all classes of carcinogens and toxins is not achievable. Perhaps phase 0 “microdosing” trials with small, safe amounts of heavy-isotope-labeled substrates can provide an effective means to prioritize tractable exposures [242]. As with all reviewed biomarkers, extrapolation from biomarker change to extent of risk reduction has not been realized;
- Metabolomics: Targeted and nontargeted metabolomics are beginning to be applied successfully as biomarkers in clinical trials of NRF2 inducers, albeit exclusively to date with SFN. Recent studies in mice have shown the power of these tools to define the impact of modulation of Nrf2 signaling on cancer cell metabolism [243] and the maintenance of health in space flight [244]. Combinations of omics approaches are likely to provide more integrated pictures of the actions of targeted NRF2 activation on early, intermediate, and later events on the pathways of disease prevention and mitigation.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. A Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Stewart, D.; Hu, B.; Li, N.; Cook, J.; Nel, A.; Alam, J. Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Delta(12,14)-prostaglandin J(2) is mediated by the stress response elements and transcription factor Nrf2. Antioxid. Redox Signal. 2002, 4, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Yates, M.S.; Kensler, T.W. Chemopreventive promise of targeting the Nrf2 pathway. Drug News Perspect. 2007, 20, 109–117. [Google Scholar] [CrossRef]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of NRF2: A review of their potential for clinical development. Oxid Med. Cell Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or sulforaphane: Is it the source or dose that matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Chapman, E. The role of natural products in revealing NRF2 function. Nat. Prod. Rep. 2020, 37, 797–826. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Dimethyl fumarate: A review in moderate to severe plaque psoriasis. Drugs 2018, 78, 123–130. [Google Scholar] [CrossRef]
- Meissner, M.; Valesky, E.M.; Kippenberger, S.; Kaufmann, R. Dimethyl fumarate-only an anti-psoriatic medication? J. Dtsch. Dermatol. Ges. 2012, 10, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef]
- Mrowietz, U.; Morrison, P.J.; Suhrkamp, I.; Kumanova, M.; Clement, B. The pharmacokinetics of fumaric acid esters reveal their in vivo effects. Trends Pharmacol. Sci. 2018, 39, 1–12. [Google Scholar] [CrossRef]
- Nieboer, C.; de Hoop, D.; Langendijk, P.N.; van Loenen, A.C.; Gubbels, J. Fumaric acid therapy in psoriasis: A double-blind comparison between fumaric acid compound therapy and monotherapy with dimethylfumaric acid ester. Dermatologica 1990, 181, 33–37. [Google Scholar] [CrossRef]
- Altmeyer, P.J.; Matthes, U.; Pawlak, F.; Hoffmann, K.; Frosch, P.J.; Ruppert, P.; Wassilew, S.W.; Horn, T.; Kreysel, H.W.; Lutz, G.; et al. Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J. Am. Acad. Dermatol. 1994, 30, 977–981. [Google Scholar] [CrossRef]
- Mrowietz, U.; Christophers, E.; Altmeyer, P. Treatment of psoriasis with fumaric acid esters: Results of a prospective multicentre study. German Multicentre Study. Br. J. Dermatol. 1998, 138, 456–460. [Google Scholar] [CrossRef]
- Friedrich, M.; Sterry, W.; Klein, A.; Ruckert, R.; Docke, W.D.; Asadullah, K. Addition of pentoxifylline could reduce the side effects of fumaric acid esters in the treatment of psoriasis. Acta Derm. Venereol. 2001, 81, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, H.; Altmeyer, P.; Kaufmann, R.; Ring, J.; Christophers, E.; Pavel, S.; Ziegler, J. Topical calcipotriol plus oral fumaric acid is more effective and faster acting than oral fumaric acid monotherapy in the treatment of severe chronic plaque psoriasis vulgaris. Dermatology 2002, 205, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Stander, H.; Stadelmann, A.; Luger, T.; Traupe, H. Efficacy of fumaric acid ester monotherapy in psoriasis pustulosa palmoplantaris. Br. J. Dermatol. 2003, 149, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Carboni, I.; De Felice, C.; De Simoni, I.; Soda, R.; Chimenti, S. Fumaric acid esters in the treatment of psoriasis: An Italian experience. J. Dermatolog. Treat. 2004, 15, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Venten, I.; Hess, N.; Hirschmuller, A.; Altmeyer, P.; Brockmeyer, N. Treatment of therapy-resistant Alopecia areata with fumaric acid esters. Eur. J. Med. Res. 2006, 11, 300–305. [Google Scholar] [PubMed]
- Kappos, L.; Gold, R.; Miller, D.H.; Macmanus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Yang, M.; et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008, 372, 1463–1472. [Google Scholar] [CrossRef]
- MacManus, D.G.; Miller, D.H.; Kappos, L.; Gold, R.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Eraksoy, M.; et al. BG-12 reduces evolution of new enhancing lesions to T1-hypointense lesions in patients with multiple sclerosis. J. Neurol. 2011, 258, 449–456. [Google Scholar] [CrossRef]
- Kappos, L.; Gold, R.; Miller, D.H.; MacManus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Eraksoy, M.; et al. Effect of BG-12 on contrast-enhanced lesions in patients with relapsing--remitting multiple sclerosis: Subgroup analyses from the phase 2b study. Mult. Scler. 2012, 18, 314–321. [Google Scholar] [CrossRef]
- Sheikh, S.I.; Nestorov, I.; Russell, H.; O’Gorman, J.; Huang, R.; Milne, G.L.; Scannevin, R.H.; Novas, M.; Dawson, K.T. Tolerability and pharmacokinetics of delayed-release dimethyl fumarate administered with and without aspirin in healthy volunteers. Clin. Ther. 2013, 35, 1582–1594. [Google Scholar] [CrossRef]
- Bar-Or, A.; Gold, R.; Kappos, L.; Arnold, D.L.; Giovannoni, G.; Selmaj, K.; O’Gorman, J.; Stephan, M.; Dawson, K.T. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: Subgroup analyses of the DEFINE study. J. Neurol. 2013, 260, 2297–2305. [Google Scholar] [CrossRef]
- Hutchinson, M.; Fox, R.J.; Miller, D.H.; Phillips, J.T.; Kita, M.; Havrdova, E.; O’Gorman, J.; Zhang, R.; Novas, M.; Viglietta, V.; et al. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: Subgroup analyses of the CONFIRM study. J. Neurol. 2013, 260, 2286–2296. [Google Scholar] [CrossRef]
- Arnold, D.L.; Gold, R.; Kappos, L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Yang, M.; Zhang, R.; Stephan, M.; Sheikh, S.I.; et al. Magnetization transfer ratio in the delayed-release dimethyl fumarate DEFINE study. J. Neurol. 2014, 261, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.L.; Gold, R.; Kappos, L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Yang, M.; Zhang, R.; Stephan, M.; Sheikh, S.I.; et al. Effects of delayed-release dimethyl fumarate on MRI measures in the Phase 3 DEFINE study. J. Neurol. 2014, 261, 1794–1802. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Gold, R.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Sarda, S.P.; Agarwal, S.; Zhang, A.; Sheikh, S.I.; et al. Quality of life outcomes with BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: The Define study. Mult. Scler. 2014, 20, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Kita, M.; Fox, R.J.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Sarda, S.P.; Agarwal, S.; Kong, J.; Zhang, A.; Viglietta, V.; et al. Effects of BG-12 (dimethyl fumarate) on health-related quality of life in patients with relapsing-remitting multiple sclerosis: Findings from the CONFIRM study. Mult. Scler. 2014, 20, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Fox, R.J.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Wheeler-Kingshott, C.A.; Tozer, D.J.; MacManus, D.G.; Yousry, T.A.; et al. Effects of delayed-release dimethyl fumarate on MRI measures in the phase 3 CONFIRM study. Neurology 2015, 84, 1145–1152. [Google Scholar] [CrossRef]
- Kappos, L.; Giovannoni, G.; Gold, R.; Phillips, J.T.; Arnold, D.L.; Hotermans, C.; Zhang, A.; Viglietta, V.; Fox, R.J.; DEFINE and CONFIRM study investigators. Time course of clinical and neuroradiological effects of delayed-release dimethyl fumarate in multiple sclerosis. Eur. J. Neurol. 2015, 22, 664–671. [Google Scholar] [CrossRef]
- Freedman, M.S.; Montalban, X.; Miller, A.E.; Dive-Pouletty, C.; Hass, S.; Thangavelu, K.; Leist, T.P. Comparing outcomes from clinical studies of oral disease-modifying therapies (dimethyl fumarate, fingolimod, and teriflunomide) in relapsing MS: Assessing absolute differences using a number needed to treat analysis. Mult. Scler. Relat. Disord. 2016, 10, 204–212. [Google Scholar] [CrossRef][Green Version]
- Lijnen, R.; Otters, E.; Balak, D.; Thio, B. Long-term safety and effectiveness of high-dose dimethylfumarate in the treatment of moderate to severe psoriasis: A prospective single-blinded follow-up study. J. Dermatolog. Treat. 2016, 27, 31–36. [Google Scholar] [CrossRef]
- Zhu, B.; Nestorov, I.; Zhao, G.; Meka, V.; Leahy, M.; Kam, J.; Sheikh, S.I. Evaluation of potential drug-drug interaction between delayed-release dimethyl fumarate and a commonly used oral contraceptive (norgestimate/ethinyl estradiol) in healthy women. Clin. Pharmacol. Drug Dev. 2017, 6, 604–613. [Google Scholar] [CrossRef]
- Fernandez, O.; Giovannoni, G.; Fox, R.J.; Gold, R.; Phillips, J.T.; Potts, J.; Okwuokenye, M.; Marantz, J.L. Efficacy and safety of delayed-release dimethyl fumarate for relapsing-remitting multiple sclerosis in prior interferon users: An integrated nalysis of DEFINE and CONFIRM. Clin. Ther. 2017, 39, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Cutter, G.; Fox, R.J.; Xiao, J.; Lewin, J.B.; Edwards, M.R. Comparative effectiveness of delayed-release dimethyl fumarate versus glatiramer acetate in multiple sclerosis patients: Results of a matching-adjusted indirect comparison. J. Comp. Eff. Res. 2017, 6, 313–323. [Google Scholar] [CrossRef]
- Havrdova, E.; Giovannoni, G.; Gold, R.; Fox, R.J.; Kappos, L.; Phillips, J.T.; Okwuokenye, M.; Marantz, J.L. Effect of delayed-release dimethyl fumarate on no evidence of disease activity in relapsing-remitting multiple sclerosis: Integrated analysis of the phase III DEFINE and CONFIRM studies. Eur. J. Neurol. 2017, 24, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the Phase 3 DEFINE and CONFIRM studies. Mult. Scler. 2017, 23, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Weisenseel, P.; Reich, K.; Griemberg, W.; Merten, K.; Groschel, C.; Gomez, N.N.; Taipale, K.; Brau, B.; Zschocke, I. Efficacy and safety of fumaric acid esters in combination with phototherapy in patients with moderate-to-severe plaque psoriasis (FAST). J. Dtsch. Dermatol. Ges. 2017, 15, 180–186. [Google Scholar] [CrossRef]
- Mrowietz, U.; Szepietowski, J.C.; Loewe, R.; van de Kerkhof, P.; Lamarca, R.; Ocker, W.G.; Tebbs, V.M.; Pau-Charles, I. Efficacy and safety of LAS41008 (dimethyl fumarate) in adults with moderate-to-severe chronic plaque psoriasis: A randomized, double-blind, Fumaderm((R))-and placebo-controlled trial (BRIDGE). Br. J. Dermatol. 2017, 176, 615–623. [Google Scholar] [CrossRef]
- Gold, R.; Arnold, D.L.; Bar-Or, A.; Hutchinson, M.; Kappos, L.; Havrdova, E.; MacManus, D.G.; Yousry, T.A.; Pozzilli, C.; Selmaj, K.; et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult. Scler. 2017, 23, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Koulinska, I.; Riester, K.; Chalkias, S.; Edwards, M.R. Effect of bismuth subsalicylate on gastrointestinal tolerability in healthy volunteers receiving oral delayed-release dimethyl fumarate: PREVENT, a randomized, multicenter, double-blind, placebo-controlled study. Clin. Ther. 2018, 40, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Ochi, H.; Niino, M.; Onizuka, Y.; Hiramatsu, K.; Hase, M.; Yun, J.; Matta, A.; Torii, S. 72-week safety and tolerability of dimethyl fumarate in Japanese patients with relapsing-remitting multiple sclerosis: Analysis of the randomised, double blind, placebo-controlled, phase III APEX study and its open-label extension. Adv. Ther. 2018, 35, 1598–1611. [Google Scholar] [CrossRef]
- Braley, T.J.; Huber, A.K.; Segal, B.M.; Kaplish, N.; Saban, R.; Washnock-Schmid, J.M.; Chervin, R.D. A randomized, subject and rater-blinded, placebo-controlled trial of dimethyl fumarate for obstructive sleep apnea. Sleep 2018, 41. [Google Scholar] [CrossRef]
- Prosperini, L.; Lucchini, M.; Haggiag, S.; Bellantonio, P.; Bianco, A.; Buscarinu, M.C.; Buttari, F.; Centonze, D.; Cortese, A.; De Giglio, L.; et al. Fingolimod vs. dimethyl fumarate in multiple sclerosis: A real-world propensity score-matched study. Neurology 2018, 91, e153–e161. [Google Scholar] [CrossRef] [PubMed]
- Alroughani, R.; Das, R.; Penner, N.; Pultz, J.; Taylor, C.; Eraly, S. Safety and efficacy of delayed-release dimethyl fumarate in pediatric patients with relapsing multiple sclerosis (FOCUS). Pediatr. Neurol. 2018, 83, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Cohan, S.L.; Moses, H.; Calkwood, J.; Tornatore, C.; LaGanke, C.; Smoot, K.E.; Meka, V.; Okwuokenye, M.; Hotermans, C.; Mendoza, J.P.; et al. Clinical outcomes in patients with relapsing-remitting multiple sclerosis who switch from natalizumab to delayed-release dimethyl fumarate: A multicenter retrospective observational study (STRATEGY). Mult. Scler. Relat. Disord. 2018, 22, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Sator, P.; Loewe, R.; Zamani, O.; Holzer, G.; Wolf, P.; Mlynek, A.; Berger, T.; Richter, L.; Schuller, E. Dimethyl fumarate is efficacious in severe plaque psoriasis: Post hoc analysis from the BRIDGE trial in Austria. Wien. Klin. Wochenschr. 2019, 131, 485–492. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef]
- Saida, T.; Yamamura, T.; Kondo, T.; Yun, J.; Yang, M.; Li, J.; Mahadavan, L.; Zhu, B.; Sheikh, S.I. A randomized placebo-controlled trial of delayed-release dimethyl fumarate in patients with relapsing-remitting multiple sclerosis from East Asia and other countries. BMC Neurol. 2019, 19, 5. [Google Scholar] [CrossRef]
- Spencer, C.A.; Groudine, M. Molecular analysis of the c-myc transcription elongation block. Implications for the generation of Burkitt’s lymphoma. Ann. N. Y. Acad. Sci. 1990, 599, 12–28. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef]
- Ahuja, M.; Ammal Kaidery, N.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 signaling mechanisms of fumaric acid esters and their role in neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced experimental Parkinson’s-like disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T.; Sporn, M.B. Synthetic oleanane triterpenoids: Multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef] [PubMed]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and anticancer activity of CDDO and CDDO-Me, two derivatives of natural triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Krauth, M.; Huff, J.W.; Ferguson, D.A.; Ruiz, S.; Meyer, C.J.; Warnock, D.G. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am. J. Nephrol. 2011, 33, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef]
- Lambers Heerspink, H.J.; Chertow, G.M.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Goldsberry, A.; Krauth, M.; Linde, P.; McMurray, J.J.; Meyer, C.J.; et al. Baseline characteristics in the Bardoxolone methyl EvAluation in patients with Chronic kidney disease and type 2 diabetes mellitus: The Occurrence of renal eveNts (BEACON) trial. Nephrol. Dial. Transplant. 2013, 28, 2841–2850. [Google Scholar] [CrossRef]
- Teuscher, N.S.; Kelley, R.J.; Dumas, E.O.; Klein, C.E.; Awni, W.M.; Meyer, C.J. A food effect study and dose proportionality study to assess the pharmacokinetics and safety of bardoxolone methyl in healthy volunteers. Clin. Pharmacol. Drug Dev. 2014, 3, 314–320. [Google Scholar] [CrossRef]
- Chin, M.P.; Reisman, S.A.; Bakris, G.L.; O’Grady, M.; Linde, P.G.; McCullough, P.A.; Packham, D.; Vaziri, N.D.; Ward, K.W.; Warnock, D.G.; et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 2014, 39, 499–508. [Google Scholar] [CrossRef]
- Chin, M.P.; Wrolstad, D.; Bakris, G.L.; Chertow, G.M.; de Zeeuw, D.; Goldsberry, A.; Linde, P.G.; McCullough, P.A.; McMurray, J.J.; Wittes, J.; et al. Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J. Card. Fail. 2014, 20, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Appel, G.B.; Block, G.A.; Chin, M.P.; Coyne, D.W.; Goldsberry, A.; Kalantar-Zadeh, K.; Meyer, C.J.; Molitch, M.E.; Pergola, P.E.; et al. Effects of bardoxolone methyl on body weight, waist circumference and glycemic control in obese patients with type 2 diabetes mellitus and stage 4 chronic kidney disease. J. Diabetes Complicat. 2018, 32, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Bakris, G.L.; Block, G.A.; Chertow, G.M.; Goldsberry, A.; Inker, L.A.; Heerspink, H.J.L.; O’Grady, M.; Pergola, P.E.; Wanner, C.; et al. Bardoxolone methyl improves kidney function in patients with chronic kidney disease stage 4 and type 2 diabetes: Post-hoc analyses from bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes study. Am. J. Nephrol. 2018, 47, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Rizk, D.V.; Silva, A.L.; Pergola, P.E.; Toto, R.; Warnock, D.G.; Chin, M.P.; Goldsberry, A.; O’Grady, M.; Meyer, C.J.; McCullough, P.A. Effects of bardoxolone methyl on magnesium in patients with type 2 diabetes mellitus and chronic kidney disease. Cardiorenal Med. 2019, 9, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Rich, S.; Goldsberry, A.; Grady, M.O.A.; Meyer, C.J. Effects of bardoxolone methyl on QT interval in healthy volunteers. Cardiorenal Med. 2019, 9, 326–333. [Google Scholar] [CrossRef]
- Yamawaki, K.; Kanda, H.; Shimazaki, R. Nrf2 activator for the treatment of kidney diseases. Toxicol. Appl. Pharmacol. 2018, 360, 30–37. [Google Scholar] [CrossRef]
- Liu, M.; Reddy, N.M.; Higbee, E.M.; Potteti, H.R.; Noel, S.; Racusen, L.; Kensler, T.W.; Sporn, M.B.; Reddy, S.P.; Rabb, H. The Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys from ischemia-reperfusion injury in mice. Kidney Int. 2014, 85, 134–141. [Google Scholar] [CrossRef]
- Wu, J.; Liu, X.; Fan, J.; Chen, W.; Wang, J.; Zeng, Y.; Feng, X.; Yu, X.; Yang, X. Bardoxolone methyl (BARD) ameliorates aristolochic acid (AA)-induced acute kidney injury through Nrf2 pathway. Toxicology 2014, 318, 22–31. [Google Scholar] [CrossRef]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef]
- Johnson, N.M.; Egner, P.A.; Baxter, V.K.; Sporn, M.B.; Wible, R.S.; Sutter, T.R.; Groopman, J.D.; Kensler, T.W.; Roebuck, B.D. Complete protection against aflatoxin B(1)-induced liver cancer with a triterpenoid: DNA adduct dosimetry, molecular signature, and genotoxicity threshold. Cancer Prev. Res. 2014, 7, 658–665. [Google Scholar] [CrossRef]
- Taguchi, K.; Takaku, M.; Egner, P.A.; Morita, M.; Kaneko, T.; Mashimo, T.; Kensler, T.W.; Yamamoto, M. Generation of a new model rat: Nrf2 knockout rats are sensitive to aflatoxin B1 toxicity. Toxicol. Sci. 2016, 152, 40–52. [Google Scholar] [CrossRef] [PubMed]
- To, C.; Ringelberg, C.S.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Sporn, M.B.; Liby, K.T. Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis 2015, 36, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.S.; Bhandari, R.; Torres, G.M.; Martyanov, V.; ElTanbouly, M.A.; Archambault, K.; Whitfield, M.L.; Liby, K.T.; Pioli, P.A. CDDO-Me alters the tumor microenvironment in estrogen receptor negative breast cancer. Sci. Rep. 2020, 10, 6560. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.S.; Shipman, E.P.; Kim, H.; Liby, K.T.; Pioli, P.A. CDDO-Me redirects activation of breast tumor associated macrophages. PLoS ONE 2016, 11, e0149600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar] [CrossRef]
- Bella, H.; Rahim, A.G.; Mustafa, M.D.; Ahmed, M.A.; Wasfi, S.; Bennett, J.L. Oltipraz—Antischistosomal efficacy in Sudanese infected with Schistosoma mansoni. Am. J. Trop. Med. Hyg. 1982, 31, 775–778. [Google Scholar] [CrossRef]
- Burchard, G.D.; Kern, P.; Baltes, R.; Dietrich, M. Comparative trial of oltipraz versus praziquantel in the treatment of urinary schistosomiasis in the Gabon. Tropenmed. Parasitol. 1984, 35, 91–94. [Google Scholar]
- Kern, P.; Burchard, G.D.; Dietrich, M. Comparative study of oltipraz versus praziquantel for treatment of schistosomiasis with intestinal manifestation in the Gabon (Schistosoma intercalatum and S. haematobium). Tropenmed. Parasitol. 1984, 35, 95–99. [Google Scholar]
- el Igail, A.B.; el Tayeb, M.; Kardaman, M.W.; Daffalla, A.A.; Dixon, H.G.; Fenwick, A. Dose-finding trial using Oltipraz to treat schoolchildren infected with Schistosoma mansoni in Gezira, Sudan. J. Trop. Med. Hyg. 1985, 88, 101–104. [Google Scholar]
- Kardaman, M.W.; Fenwick, A.; el Igail, A.B.; el Tayeb, M.; Bennett, J.L.; Daffalla, A.A. Field trials with Oltipraz against Schistosoma mansoni in the Gezira Irrigated Area, Sudan. J. Trop. Med. Hyg. 1985, 88, 95–100. [Google Scholar]
- el Tayeb, M.; Daffalla, A.A.; Kardaman, M.W.; See, R.; Fenwick, A. Praziquantel and oltipraz: The treatment of schoolchildren infected with Schistosoma mansoni and/or Schistosoma haematobium in Gezira, Sudan. Ann. Trop. Med. Parasitol. 1988, 82, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Bueding, E.; Dolan, P.; Leroy, J.P. The antischistosomal activity of oltipraz. Res. Commun. Chem. Pathol. Pharmacol. 1982, 37, 293–303. [Google Scholar] [PubMed]
- Ansher, S.S.; Dolan, P.; Bueding, E. Biochemical effects of dithiolthiones. Food Chem. Toxicol. 1986, 24, 405–415. [Google Scholar] [CrossRef]
- Ansher, S.S.; Dolan, P.; Bueding, E. Chemoprotective effects of two dithiolthiones and of butylhydroxyanisole against carbon tetrachloride and acetaminophen toxicity. Hepatology 1983, 3, 932–935. [Google Scholar] [CrossRef]
- Kelloff, G.J.; Boone, C.W.; Steele, V.E.; Fay, J.R.; Lubet, R.A.; Crowell, J.A.; Sigman, C.C. Mechanistic considerations in chemopreventive drug development. J. Cell Biochem. Suppl. 1994, 20, 1–24. [Google Scholar] [CrossRef]
- Kensler, T.W.; Groopman, J.D.; Sutter, T.R.; Curphey, T.J.; Roebuck, B.D. Development of cancer chemopreventive agents: Oltipraz as a paradigm. Chem. Res. Toxicol. 1999, 12, 113–126. [Google Scholar] [CrossRef]
- Ali, H.M.; Homeida, M.M.; Sulaiman, S.M.; Bennett, J.L. Diet-controlled blood levels of oltipraz in healthy male subjects. J. Antimicrob. Chemother. 1984, 13, 465–470. [Google Scholar] [CrossRef]
- O’Dwyer, P.J.; Szarka, C.E.; Yao, K.S.; Halbherr, T.C.; Pfeiffer, G.R.; Green, F.; Gallo, J.M.; Brennan, J.; Frucht, H.; Goosenberg, E.B.; et al. Modulation of gene expression in subjects at risk for colorectal cancer by the chemopreventive dithiolethione oltipraz. J. Clin. Investig. 1996, 98, 1210–1217. [Google Scholar] [CrossRef]
- Kensler, T.W.; He, X.; Otieno, M.; Egner, P.A.; Jacobson, L.P.; Chen, B.; Wang, J.S.; Zhu, Y.R.; Zhang, B.C.; Wang, J.B.; et al. Oltipraz chemoprevention trial in Qidong, People’s Republic of China: Modulation of serum aflatoxin albumin adduct biomarkers. Cancer Epidemiol. Biomark. Prev. 1998, 7, 127–134. [Google Scholar]
- Wang, J.S.; Shen, X.; He, X.; Zhu, Y.R.; Zhang, B.C.; Wang, J.B.; Qian, G.S.; Kuang, S.Y.; Zarba, A.; Egner, P.A.; et al. Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China. J. Natl. Cancer Inst. 1999, 91, 347–354. [Google Scholar] [CrossRef]
- Benson, A.B., III; Olopade, O.I.; Ratain, M.J.; Rademaker, A.; Mobarhan, S.; Stucky-Marshall, L.; French, S.; Dolan, M.E. Chronic daily low dose of 4-methyl-5-(2-pyrazinyl)-1,2-dithiole-3-thione (Oltipraz) in patients with previously resected colon polyps and first degree female relatives of breast cancer patients. Clin. Cancer Res. 2000, 6, 3870–3877. [Google Scholar] [PubMed]
- O’Dwyer, P.J.; Szarka, C.; Brennan, J.M.; Laub, P.B.; Gallo, J.M. Pharmacokinetics of the chemopreventive agent oltipraz and of its metabolite M3 in human subjects after a single oral dose. Clin. Cancer Res. 2000, 6, 4692–4696. [Google Scholar]
- Camoirano, A.; Bagnasco, M.; Bennicelli, C.; Cartiglia, C.; Wang, J.B.; Zhang, B.C.; Zhu, Y.R.; Qian, G.S.; Egner, P.A.; Jacobson, L.P.; et al. Oltipraz chemoprevention trial in Qidong, People’s Republic of China: Results of urine genotoxicity assays as related to smoking habits. Cancer Epidemiol. Biomark. Prev. 2001, 10, 775–783. [Google Scholar]
- Pendyala, L.; Schwartz, G.; Bolanowska-Higdon, W.; Hitt, S.; Zdanowicz, J.; Murphy, M.; Lawrence, D.; Creaven, P.J. Phase I/pharmacodynamic study of N-acetylcysteine/oltipraz in smokers: Early termination due to excessive toxicity. Cancer Epidemiol. Biomark. Prev. 2001, 10, 269–272. [Google Scholar]
- Dimitrov, N.V.; Leece, C.M.; Tompkins, E.R.; Seymour, E.; Bennink, M.; Gardiner, J.; Crowell, J.; Hawk, E.; Nashawaty, M.; Bennett, J.L. Oltipraz concentrations in plasma, buccal mucosa cells, and lipids: Pharmacological studies. Cancer Epidemiol. Biomark. Prev. 2001, 10, 201–207. [Google Scholar]
- Szarka, C.E.; Yao, K.S.; Pfeiffer, G.R.; Balshem, A.M.; Litwin, S.; Frucht, H.; Goosenberg, E.B.; Engstrom, P.F.; Clapper, M.L.; O’Dwyer, P.J. Chronic dosing of oltipraz in people at increased risk for colorectal cancer. Cancer Detect. Prev. 2001, 25, 352–361. [Google Scholar]
- Kelley, M.J.; Glaser, E.M.; Herndon, J.E., II; Becker, F.; Bhagat, R.; Zhang, Y.J.; Santella, R.M.; Carmella, S.G.; Hecht, S.S.; Gallot, L.; et al. Safety and efficacy of weekly oral oltipraz in chronic smokers. Cancer Epidemiol. Biomark. Prev. 2005, 14, 892–899. [Google Scholar] [CrossRef]
- Glintborg, B.; Weimann, A.; Kensler, T.W.; Poulsen, H.E. Oltipraz chemoprevention trial in Qidong, People’s Republic of China: Unaltered oxidative biomarkers. Free Radic. Biol. Med. 2006, 41, 1010–1014. [Google Scholar] [CrossRef]
- Kim, S.G.; Kim, Y.M.; Choi, Y.H.; Lee, M.G.; Choi, J.Y.; Han, J.Y.; Cho, S.H.; Jang, J.W.; Um, S.H.; Chon, C.Y.; et al. Pharmacokinetics of oltipraz and its major metabolite (RM) in patients with liver fibrosis or cirrhosis: Relationship with suppression of circulating TGF-beta1. Clin. Pharmacol Ther. 2010, 88, 360–368. [Google Scholar] [CrossRef]
- Kim, S.G.; Kim, Y.M.; Choi, J.Y.; Han, J.Y.; Jang, J.W.; Cho, S.H.; Um, S.H.; Chon, C.Y.; Lee, D.H.; Jang, J.J.; et al. Oltipraz therapy in patients with liver fibrosis or cirrhosis: A randomized, double-blind, placebo-controlled phase II trial. J. Pharm. Pharmacol. 2011, 63, 627–635. [Google Scholar] [CrossRef]
- Kim, W.; Kim, B.G.; Lee, J.S.; Lee, C.K.; Yeon, J.E.; Chang, M.S.; Kim, J.H.; Kim, H.; Yi, S.; Lee, J.; et al. Randomised clinical trial: The efficacy and safety of oltipraz, a liver X receptor alpha-inhibitory dithiolethione in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 45, 1073–1083. [Google Scholar] [CrossRef]
- Egner, P.A.; Kensler, T.W.; Prestera, T.; Talalay, P.; Libby, A.H.; Joyner, H.H.; Curphey, T.J. Regulation of phase 2 enzyme induction by oltipraz and other dithiolethiones. Carcinogenesis 1994, 15, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Interactive effects of Nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis 2003, 24, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. [Google Scholar] [CrossRef]
- Fahey, J.W.; Zhang, Y.; Talalay, P. Broccoli sprouts: An exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc. Natl. Acad. Sci. USA 1997, 94, 10367–10372. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol. Biomark. Prev. 1998, 7, 1091–1100. [Google Scholar]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and done? Targeting the NRF2 pathway with sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef]
- Conaway, C.C.; Getahun, S.M.; Liebes, L.L.; Pusateri, D.J.; Topham, D.K.; Botero-Omary, M.; Chung, F.L. Disposition of glucosinolates and sulforaphane in humans after ingestion of steamed and fresh broccoli. Nutr. Cancer 2000, 38, 168–178. [Google Scholar] [CrossRef]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta 2002, 316, 43–53. [Google Scholar] [CrossRef]
- Murashima, M.; Watanabe, S.; Zhuo, X.G.; Uehara, M.; Kurashige, A. Phase 1 study of multiple biomarkers for metabolism and oxidative stress after one-week intake of broccoli sprouts. Biofactors 2004, 22, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Galan, M.V.; Kishan, A.A.; Silverman, A.L. Oral broccoli sprouts for the treatment of Helicobacter pylori infection: A preliminary report. Dig. Dis. Sci. 2004, 49, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Gasper, A.V.; Al-Janobi, A.; Smith, J.A.; Bacon, J.R.; Fortun, P.; Atherton, C.; Taylor, M.A.; Hawkey, C.J.; Barrett, D.A.; Mithen, R.F. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am. J. Clin. Nutr. 2005, 82, 1283–1291. [Google Scholar] [CrossRef]
- Kensler, T.W.; Chen, J.G.; Egner, P.A.; Fahey, J.W.; Jacobson, L.P.; Stephenson, K.K.; Ye, L.; Coady, J.L.; Wang, J.B.; Wu, Y.; et al. Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People’s Republic of China. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2605–2613. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Fahey, J.W.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Stephenson, K.K.; Wade, K.L.; Ye, L.; Talalay, P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: A clinical phase I study. Nutr. Cancer 2006, 55, 53–62. [Google Scholar] [CrossRef]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.S.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef]
- Gasper, A.V.; Traka, M.; Bacon, J.R.; Smith, J.A.; Taylor, M.A.; Hawkey, C.J.; Barrett, D.A.; Mithen, R.F. Consuming broccoli does not induce genes associated with xenobiotic metabolism and cell cycle control in human gastric mucosa. J. Nutr. 2007, 137, 1718–1724. [Google Scholar] [CrossRef]
- Myzak, M.C.; Tong, P.; Dashwood, W.M.; Dashwood, R.H.; Ho, E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp. Biol. Med. 2007, 232, 227–234. [Google Scholar]
- Rungapamestry, V.; Duncan, A.J.; Fuller, Z.; Ratcliffe, B. Effect of meal composition and cooking duration on the fate of sulforaphane following consumption of broccoli by healthy human subjects. Br. J. Nutr. 2007, 97, 644–652. [Google Scholar] [CrossRef]
- Traka, M.; Gasper, A.V.; Melchini, A.; Bacon, J.R.; Needs, P.W.; Frost, V.; Chantry, A.; Jones, A.M.; Ortori, C.A.; Barrett, D.A.; et al. Broccoli consumption interacts with GSTM1 to perturb oncogenic signalling pathways in the prostate. PLoS ONE 2008, 3, e2568. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M.; Klopping-Ketelaars, I.W.; van den Berg, R.; Vaes, W.H. Bioavailability and kinetics of sulforaphane in humans after consumption of cooked versus raw broccoli. J. Agric. Food Chem. 2008, 56, 10505–10509. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, N.; Coldham, N.; Gielbert, A.; Sauer, M.J.; Ioannides, C. Repeated intake of broccoli does not lead to higher plasma levels of sulforaphane in human volunteers. Cancer Lett. 2009, 284, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Riedl, M.A.; Saxon, A.; Diaz-Sanchez, D. Oral sulforaphane increases Phase II antioxidant enzymes in the human upper airway. Clin. Immunol. 2009, 130, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Riso, P.; Martini, D.; Visioli, F.; Martinetti, A.; Porrini, M. Effect of broccoli intake on markers related to oxidative stress and cancer risk in healthy smokers and nonsmokers. Nutr. Cancer 2009, 61, 232–237. [Google Scholar] [CrossRef]
- Yanaka, A.; Fahey, J.W.; Fukumoto, A.; Nakayama, M.; Inoue, S.; Zhang, S.; Tauchi, M.; Suzuki, H.; Hyodo, I.; Yamamoto, M. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori-infected mice and humans. Cancer Prev. Res. 2009, 2, 353–360. [Google Scholar] [CrossRef]
- Christiansen, B.; Bellostas Muguerza, N.; Petersen, A.M.; Kveiborg, B.; Madsen, C.R.; Thomas, H.; Ihlemann, N.; Sorensen, J.C.; Kober, L.; Sorensen, H.; et al. Ingestion of broccoli sprouts does not improve endothelial function in humans with hypertension. PLoS ONE 2010, 5, e12461. [Google Scholar] [CrossRef]
- Bahadoran, Z.; Mirmiran, P.; Hosseinpanah, F.; Hedayati, M.; Hosseinpour-Niazi, S.; Azizi, F. Broccoli sprouts reduce oxidative stress in type 2 diabetes: A randomized double-blind clinical trial. Eur. J. Clin. Nutr. 2011, 65, 972–977. [Google Scholar] [CrossRef]
- Hauder, J.; Winkler, S.; Bub, A.; Rufer, C.E.; Pignitter, M.; Somoza, V. LC-MS/MS quantification of sulforaphane and indole-3-carbinol metabolites in human plasma and urine after dietary intake of selenium-fortified broccoli. J. Agric. Food Chem. 2011, 59, 8047–8057. [Google Scholar] [CrossRef]
- Clarke, J.D.; Riedl, K.; Bella, D.; Schwartz, S.J.; Stevens, J.F.; Ho, E. Comparison of isothiocyanate metabolite levels and histone deacetylase activity in human subjects consuming broccoli sprouts or broccoli supplement. J. Agric. Food Chem. 2011, 59, 10955–10963. [Google Scholar] [CrossRef]
- Egner, P.A.; Chen, J.G.; Wang, J.B.; Wu, Y.; Sun, Y.; Lu, J.H.; Zhu, J.; Zhang, Y.H.; Chen, Y.S.; Friesen, M.D.; et al. Bioavailability of sulforaphane from two broccoli sprout beverages: Results of a short-term, cross-over clinical trial in Qidong, China. Cancer Prev. Res. 2011, 4, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Healy, Z.R.; Liu, H.; Holtzclaw, W.D.; Talalay, P. Inactivation of tautomerase activity of macrophage migration inhibitory factor by sulforaphane: A potential biomarker for anti-inflammatory intervention. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Bahadoran, Z.; Tohidi, M.; Nazeri, P.; Mehran, M.; Azizi, F.; Mirmiran, P. Effect of broccoli sprouts on insulin resistance in type 2 diabetic patients: A randomized double-blind clinical trial. Int. J. Food Sci. Nutr. 2012, 63, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.M.; Teran-Garcia, M.; Jeffery, E.H. Enhancing sulforaphane absorption and excretion in healthy men through the combined consumption of fresh broccoli sprouts and a glucoraphanin-rich powder. Br. J. Nutr. 2012, 107, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Wehage, S.L.; Holtzclaw, W.D.; Kensler, T.W.; Egner, P.A.; Shapiro, T.A.; Talalay, P. Protection of humans by plant glucosinolates: Efficiency of conversion of glucosinolates to isothiocyanates by the gastrointestinal microflora. Cancer Prev. Res. 2012, 5, 603–611. [Google Scholar] [CrossRef]
- Kensler, T.W.; Ng, D.; Carmella, S.G.; Chen, M.; Jacobson, L.P.; Munoz, A.; Egner, P.A.; Chen, J.G.; Qian, G.S.; Chen, T.Y.; et al. Modulation of the metabolism of airborne pollutants by glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages in Qidong, China. Carcinogenesis 2012, 33, 101–107. [Google Scholar] [CrossRef]
- Mirmiran, P.; Bahadoran, Z.; Hosseinpanah, F.; Keyzad, A.; Azizi, F. Effects of broccoli sprout with high sulforaphane concentration on inflammatory markers in type 2 diabetic patients: A randomized double-blind placebo-controlled clinical trial. J. Funct. Foods 2012, 4, 837–841. [Google Scholar] [CrossRef]
- Saha, S.; Hollands, W.; Teucher, B.; Needs, P.W.; Narbad, A.; Ortori, C.A.; Barrett, D.A.; Rossiter, J.T.; Mithen, R.F.; Kroon, P.A. Isothiocyanate concentrations and interconversion of sulforaphane to erucin in human subjects after consumption of commercial frozen broccoli compared to fresh broccoli. Mol. Nutr. Food Res. 2012, 56, 1906–1916. [Google Scholar] [CrossRef]
- Armah, C.N.; Traka, M.H.; Dainty, J.R.; Defernez, M.; Janssens, A.; Leung, W.; Doleman, J.F.; Potter, J.F.; Mithen, R.F. A diet rich in high-glucoraphanin broccoli interacts with genotype to reduce discordance in plasma metabolite profiles by modulating mitochondrial function. Am. J. Clin. Nutr. 2013, 98, 712–722. [Google Scholar] [CrossRef]
- Meyer, M.; Kesic, M.J.; Clarke, J.; Ho, E.; Simmen, R.C.; Diaz-Sanchez, D.; Noah, T.L.; Jaspers, I. Sulforaphane induces SLPI secretion in the nasal mucosa. Respir. Med. 2013, 107, 472–475. [Google Scholar] [CrossRef]
- Poulton, E.J.; Levy, L.; Lampe, J.W.; Shen, D.D.; Tracy, J.; Shuhart, M.C.; Thummel, K.E.; Eaton, D.L. Sulforaphane is not an effective antagonist of the human pregnane X-receptor in vivo. Toxicol. Appl. Pharmacol. 2013, 266, 122–131. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bahadoran, Z.; Mirmiran, P.; Yeganeh, M.Z.; Hosseinpanah, F.; Zojaji, H.; Azizi, F. Complementary and alternative medicinal effects of broccoli sprouts powder on Helicobacter pylori eradication rate in type 2 diabetic patients: A randomized clinical trial. J. Funct. Foods 2014, 7, 390–397. [Google Scholar] [CrossRef]
- Baier, S.R.; Zbasnik, R.; Schlegel, V.; Zempleni, J. Off-target effects of sulforaphane include the derepression of long terminal repeats through histone acetylation events. J. Nutr. Biochem. 2014, 25, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Chen, J.G.; Zarth, A.T.; Ng, D.K.; Wang, J.B.; Kensler, K.H.; Jacobson, L.P.; Munoz, A.; Johnson, J.L.; Groopman, J.D.; et al. Rapid and sustainable detoxication of airborne pollutants by broccoli sprout beverage: Results of a randomized clinical trial in China. Cancer Prev. Res. 2014, 7, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Heber, D.; Li, Z.; Garcia-Lloret, M.; Wong, A.M.; Lee, T.Y.; Thames, G.; Krak, M.; Zhang, Y.; Nel, A. Sulforaphane-rich broccoli sprout extract attenuates nasal allergic response to diesel exhaust particles. Food Funct. 2014, 5, 35–41. [Google Scholar] [CrossRef]
- Noah, T.L.; Zhang, H.; Zhou, H.; Glista-Baker, E.; Muller, L.; Bauer, R.N.; Meyer, M.; Murphy, P.C.; Jones, S.; Letang, B.; et al. Effect of broccoli sprouts on nasal response to live attenuated influenza virus in smokers: A randomized, double-blind study. PLoS ONE 2014, 9, e98671. [Google Scholar] [CrossRef]
- Oliviero, T.; Verkerk, R.; Vermeulen, M.; Dekker, M. In vivo formation and bioavailability of isothiocyanates from glucosinolates in broccoli as affected by processing conditions. Mol. Nutr. Food Res. 2014, 58, 1447–1456. [Google Scholar] [CrossRef]
- Singh, K.; Connors, S.L.; Macklin, E.A.; Smith, K.D.; Fahey, J.W.; Talalay, P.; Zimmerman, A.W. Sulforaphane treatment of autism spectrum disorder (ASD). Proc. Natl. Acad. Sci. USA 2014, 111, 15550–15555. [Google Scholar] [CrossRef]
- Atwell, L.L.; Hsu, A.; Wong, C.P.; Stevens, J.F.; Bella, D.; Yu, T.W.; Pereira, C.B.; Lohr, C.V.; Christensen, J.M.; Dashwood, R.H.; et al. Absorption and chemopreventive targets of sulforaphane in humans following consumption of broccoli sprouts or a myrosinase-treated broccoli sprout extract. Mol. Nutr. Food Res. 2015, 59, 424–433. [Google Scholar] [CrossRef]
- Alumkal, J.J.; Slottke, R.; Schwartzman, J.; Cherala, G.; Munar, M.; Graff, J.N.; Beer, T.M.; Ryan, C.W.; Koop, D.R.; Gibbs, A.; et al. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Investig. New Drugs 2015, 33, 480–489. [Google Scholar] [CrossRef]
- Armah, C.N.; Derdemezis, C.; Traka, M.H.; Dainty, J.R.; Doleman, J.F.; Saha, S.; Leung, W.; Potter, J.F.; Lovegrove, J.A.; Mithen, R.F. Diet rich in high glucoraphanin broccoli reduces plasma LDL cholesterol: Evidence from randomised controlled trials. Mol. Nutr. Food Res. 2015, 59, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Atwell, L.L.; Zhang, Z.; Mori, M.; Farris, P.; Vetto, J.T.; Naik, A.M.; Oh, K.Y.; Thuillier, P.; Ho, E.; Shannon, J. Sulforaphane bioavailability and chemopreventive activity in women scheduled for breast biopsy. Cancer Prev. Res. 2015, 8, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Reynolds, C.; Brooker, A.; Talalay, P.; Fahey, J.W. Sulforaphane improves the bronchoprotective response in asthmatics through Nrf2-mediated gene pathways. Respir. Res. 2015, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Jang, J.Y.; Kim, Y.H.; Kim, J.W.; Shim, J.J. The effects of broccoli sprout extract containing sulforaphane on lipid peroxidation and Helicobacter pylori infection in the gastric mucosa. Gut Liver 2015, 9, 486–493. [Google Scholar] [CrossRef]
- Cipolla, B.G.; Mandron, E.; Lefort, J.M.; Coadou, Y.; Della Negra, E.; Corbel, L.; Le Scodan, R.; Azzouzi, A.R.; Mottet, N. Effect of sulforaphane in men with biochemical recurrence after radical prostatectomy. Cancer Prev. Res. 2015, 8, 712–719. [Google Scholar] [CrossRef]
- Fahey, J.W.; Holtzclaw, W.D.; Wehage, S.L.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Sulforaphane bioavailability from glucoraphanin-rich broccoli: Control by active endogenous myrosinase. PLoS ONE 2015, 10, e0140963. [Google Scholar] [CrossRef]
- Kikuchi, M.; Ushida, Y.; Shiozawa, H.; Umeda, R.; Tsuruya, K.; Aoki, Y.; Suganuma, H.; Nishizaki, Y. Sulforaphane-rich broccoli sprout extract improves hepatic abnormalities in male subjects. World J. Gastroenterol. 2015, 21, 12457–12467. [Google Scholar] [CrossRef]
- Medina, S.; Dominguez-Perles, R.; Moreno, D.A.; Garcia-Viguera, C.; Ferreres, F.; Gil, J.I.; Gil-Izquierdo, A. The intake of broccoli sprouts modulates the inflammatory and vascular prostanoids but not the oxidative stress-related isoprostanes in healthy humans. Food Chem. 2015, 173, 1187–1194. [Google Scholar] [CrossRef]
- Shiina, A.; Kanahara, N.; Sasaki, T.; Oda, Y.; Hashimoto, T.; Hasegawa, T.; Yoshida, T.; Iyo, M.; Hashimoto, K. An open study of sulforaphane-rich broccoli sprout extract in patients with schizophrenia. Clin. Psychopharmacol. Neurosci. 2015, 13, 62–67. [Google Scholar] [CrossRef]
- Rajendran, P.; Dashwood, W.M.; Li, L.; Kang, Y.; Kim, E.; Johnson, G.; Fischer, K.A.; Lohr, C.V.; Williams, D.E.; Ho, E.; et al. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clin. Epigenetics 2015, 7, 102. [Google Scholar] [CrossRef]
- Bauman, J.E.; Zang, Y.; Sen, M.; Li, C.; Wang, L.; Egner, P.A.; Fahey, J.W.; Normolle, D.P.; Grandis, J.R.; Kensler, T.W.; et al. Prevention of carcinogen-induced oral cancer by sulforaphane. Cancer Prev. Res. 2016, 9, 547–557. [Google Scholar] [CrossRef]
- Doss, J.F.; Jonassaint, J.C.; Garrett, M.E.; Ashley-Koch, A.E.; Telen, M.J.; Chi, J.T. Phase 1 study of a sulforaphane-containing broccoli sprout homogenate for sickle cell disease. PLoS ONE 2016, 11, e0152895. [Google Scholar] [CrossRef] [PubMed]
- Duran, C.G.; Burbank, A.J.; Mills, K.H.; Duckworth, H.R.; Aleman, M.M.; Kesic, M.J.; Peden, D.B.; Pan, Y.; Zhou, H.; Hernandez, M.L. A proof-of-concept clinical study examining the NRF2 activator sulforaphane against neutrophilic airway inflammation. Respir. Res. 2016, 17, 89. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Meyer, M.; Bauer, R.N.; Zhou, H.; Zhang, H.; Jones, S.; Robinette, C.; Noah, T.L.; Jaspers, I. Effect of broccoli sprouts and live attenuated influenza virus on peripheral blood natural killer cells: A randomized, double-blind study. PLoS ONE 2016, 11, e0147742. [Google Scholar] [CrossRef] [PubMed]
- Sudini, K.; Diette, G.B.; Breysse, P.N.; McCormack, M.C.; Bull, D.; Biswal, S.; Zhai, S.; Brereton, N.; Peng, R.D.; Matsui, E.C. A randomized controlled trial of the effect of broccoli sprouts on antioxidant gene expression and airway inflammation in asthmatics. J. Allergy Clin. Immunol. Pract. 2016, 4, 932–940. [Google Scholar] [CrossRef]
- Wise, R.A.; Holbrook, J.T.; Criner, G.; Sethi, S.; Rayapudi, S.; Sudini, K.R.; Sugar, E.A.; Burke, A.; Thimmulappa, R.; Singh, A.; et al. Lack of effect of oral sulforaphane administration on Nrf2 expression in COPD: A randomized, double-blind, placebo controlled trial. PLoS ONE 2016, 11, e0163716. [Google Scholar] [CrossRef]
- Axelsson, A.S.; Tubbs, E.; Mecham, B.; Chacko, S.; Nenonen, H.A.; Tang, Y.; Fahey, J.W.; Derry, J.M.J.; Wollheim, C.B.; Wierup, N.; et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Davidson, R.; Gardner, S.; Jupp, O.; Bullough, A.; Butters, S.; Watts, L.; Donell, S.; Traka, M.; Saha, S.; Mithen, R.; et al. Isothiocyanates are detected in human synovial fluid following broccoli consumption and can affect the tissues of the knee joint. Sci. Rep. 2017, 7, 3398. [Google Scholar] [CrossRef]
- Fahey, J.W.; Wade, K.L.; Wehage, S.L.; Holtzclaw, W.D.; Liu, H.; Talalay, P.; Fuchs, E.; Stephenson, K.K. Stabilized sulforaphane for clinical use: Phytochemical delivery efficiency. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Nucifora, L.G.; Koga, M.; Shaffer, L.S.; Higgs, C.; Tanaka, T.; Wang, A.M.; Coughlin, J.M.; Barker, P.B.; Fahey, J.W.; et al. Sulforaphane augments glutathione and influences brain metabolites in human subjects: A clinical pilot study. Mol. Neuropsychiatry 2018, 3, 214–222. [Google Scholar] [CrossRef]
- Tahata, S.; Singh, S.V.; Lin, Y.; Hahm, E.R.; Beumer, J.H.; Christner, S.M.; Rao, U.N.; Sander, C.; Tarhini, A.A.; Tawbi, H.; et al. Evaluation of biodistribution of sulforaphane after administration of oral broccoli sprout extract in melanoma patients with multiple atypical nevi. Cancer Prev. Res. 2018, 11, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Bent, S.; Lawton, B.; Warren, T.; Widjaja, F.; Dang, K.; Fahey, J.W.; Cornblatt, B.; Kinchen, J.M.; Delucchi, K.; Hendren, R.L. Identification of urinary metabolites that correlate with clinical improvements in children with autism treated with sulforaphane from broccoli. Mol. Autism 2018, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Housley, L.; Magana, A.A.; Hsu, A.; Beaver, L.M.; Wong, C.P.; Stevens, J.F.; Choi, J.; Jiang, Y.; Bella, D.; Williams, D.E.; et al. Untargeted metabolomic screen reveals changes in human plasma metabolite profiles following consumption of fresh broccoli sprouts. Mol. Nutr. Food Res. 2018, 62, e1700665. [Google Scholar] [CrossRef] [PubMed]
- Okunade, O.; Niranjan, K.; Ghawi, S.K.; Kuhnle, G.; Methven, L. Supplementation of the diet by exogenous myrosinase via mustard seeds to increase the bioavailability of sulforaphane in healthy human subjects after the consumption of cooked broccoli. Mol. Nutr. Food Res. 2018, 62, e1700980. [Google Scholar] [CrossRef]
- Sivapalan, T.; Melchini, A.; Saha, S.; Needs, P.W.; Traka, M.H.; Tapp, H.; Dainty, J.R.; Mithen, R.F. Bioavailability of glucoraphanin and sulforaphane from high-glucoraphanin broccoli. Mol. Nutr. Food Res. 2018, 62, e1700911. [Google Scholar] [CrossRef]
- Charron, C.S.; Vinyard, B.T.; Ross, S.A.; Seifried, H.E.; Jeffery, E.H.; Novotny, J.A. Absorption and metabolism of isothiocyanates formed from broccoli glucosinolates: Effects of BMI and daily consumption in a randomised clinical trial. Br. J. Nutr. 2018, 120, 1370–1379. [Google Scholar] [CrossRef]
- Lopez-Chillon, M.T.; Carazo-Diaz, C.; Prieto-Merino, D.; Zafrilla, P.; Moreno, D.A.; Villano, D. Effects of long-term consumption of broccoli sprouts on inflammatory markers in overweight subjects. Clin. Nutr. 2019, 38, 745–752. [Google Scholar] [CrossRef]
- Chen, J.G.; Johnson, J.; Egner, P.; Ng, D.; Zhu, J.; Wang, J.B.; Xue, X.F.; Sun, Y.; Zhang, Y.H.; Lu, L.L.; et al. Dose-dependent detoxication of the airborne pollutant benzene in a randomized trial of broccoli sprout beverage in Qidong, China. Am. J. Clin. Nutr. 2019, 110, 675–684. [Google Scholar] [CrossRef]
- Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Panjwani, A.A.; Liu, H.; Cornblatt, G.; Cornblatt, B.S.; Ownby, S.L.; Fuchs, E.; Holtzclaw, W.D.; et al. Bioavailability of sulforaphane following ingestion of glucoraphanin-rich broccoli sprout and seed extracts with active myrosinase: A pilot study of the effects of proton pump inhibitor administration. Nutrients 2019, 11, 1489. [Google Scholar] [CrossRef]
- Traka, M.H.; Melchini, A.; Coode-Bate, J.; Al Kadhi, O.; Saha, S.; Defernez, M.; Troncoso-Rey, P.; Kibblewhite, H.; O’Neill, C.M.; Bernuzzi, F.; et al. Transcriptional changes in prostate of men on active surveillance after a 12-mo glucoraphanin-rich broccoli intervention-results from the Effect of Sulforaphane on prostate CAncer PrEvention (ESCAPE) randomized controlled trial. Am. J. Clin. Nutr. 2019, 109, 1133–1144. [Google Scholar] [CrossRef]
- Palliyaguru, D.L.; Salvatore, S.R.; Schopfer, F.J.; Cheng, X.; Zhou, J.; Kensler, T.W.; Wendell, S.G. Evaluation of 2-thiothiazolidine-4-carboxylic acid, a common metabolite of isothiocyanates, as a potential biomarker of cruciferous vegetable intake. Mol. Nutr. Food Res. 2019, 63, e1801029. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Ziros, P.G.; Chen, J.G.; Groopman, J.D.; Kensler, T.W.; Sykiotis, G.P. Broccoli sprout beverage is safe for thyroid hormonal and autoimmune status: Results of a 12-week randomized trial. Food Chem. Toxicol. 2019, 126, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Garzotto, M.; Davis, E.W., II; Mori, M.; Stoller, W.A.; Farris, P.E.; Wong, C.P.; Beaver, L.M.; Thomas, G.V.; Williams, D.E.; et al. Sulforaphane bioavailability and chemopreventive activity in men presenting for biopsy of the prostate gland: A randomized controlled trial. Nutr. Cancer 2020, 72, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Langston-Cox, A.; Anderson, D.; Creek, D.J.; Palmer, K.; Wallace, E.M.; Marshall, S.A. Measuring sulforaphane and its metabolites in human plasma: A high throughput method. Molecules 2020, 25, 829. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zimmerman, A.W.; Singh, K.; Connors, S.L.; Diggins, E.; Stephenson, K.K.; Dinkova-Kostova, A.T.; Fahey, J.W. Biomarker exploration in human peripheral blood mononuclear cells for monitoring sulforaphane treatment responses in autism spectrum disorder. Sci. Rep. 2020, 10, 5822. [Google Scholar] [CrossRef]
- Sun, J.; Charron, C.S.; Novotny, J.A.; Peng, B.; Yu, L.; Chen, P. Profiling glucosinolate metabolites in human urine and plasma after broccoli consumption using non-targeted and targeted metabolomic analyses. Food Chem. 2020, 309, 125660. [Google Scholar] [CrossRef]
- Dashwood, R.H.; Ho, E. Dietary histone deacetylase inhibitors: From cells to mice to man. Semin. Cancer Biol. 2007, 17, 363–369. [Google Scholar] [CrossRef]
- Zhang, Y. The molecular basis that unifies the metabolism, cellular uptake and chemopreventive activities of dietary isothiocyanates. Carcinogenesis 2012, 33, 2–9. [Google Scholar] [CrossRef]
- Paunkov, A.; Chartoumpekis, D.V.; Ziros, P.G.; Sykiotis, G.P. A bibliometric review of the Keap1/Nrf2 pathway and its related antioxidant compounds. Antioxidants 2019, 8, 353. [Google Scholar] [CrossRef]
- Shen, T.; Jiang, T.; Long, M.; Chen, J.; Ren, D.M.; Wong, P.K.; Chapman, E.; Zhou, B.; Zhang, D.D. A curcumin derivative that inhibits vinyl carbamate-induced lung carcinogenesis via activation of the Nrf2 protective response. Antioxid. Redox Signal. 2015, 23, 651–664. [Google Scholar] [CrossRef]
- Yang, H.; Xu, W.; Zhou, Z.; Liu, J.; Li, X.; Chen, L.; Weng, J.; Yu, Z. Curcumin attenuates urinary excretion of albumin in type II diabetic patients with enhancing nuclear factor erythroid-derived 2-like 2 (Nrf2) system and repressing inflammatory signaling efficacies. Exp. Clin. Endocrinol. Diabetes 2015, 123, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Osorio, A.S.; Garcia-Nino, W.R.; Gonzalez-Reyes, S.; Alvarez-Mejia, A.E.; Guerra-Leon, S.; Salazar-Segovia, J.; Falcon, I.; Montes de Oca-Solano, H.; Madero, M.; Pedraza-Chaverri, J. The effect of dietary supplementation with curcumin on redox status and Nrf2 activation in patients with nondiabetic or diabetic proteinuric chronic kidney disease: A pilot study. J. Ren. Nutr. 2016, 26, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, J.F.; Leal, V.O.; Rizzetto, F.; Grimmer, G.H.; Ribeiro-Alves, M.; Daleprane, J.B.; Carraro-Eduardo, J.C.; Mafra, D. Effects of resveratrol supplementation in Nrf2 and NF-kappaB expressions in nondialyzed chronic kidney disease patients: A randomized, double-blind, placebo-controlled, crossover clinical trial. J. Ren. Nutr. 2016, 26, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Seyyedebrahimi, S.; Khodabandehloo, H.; Nasli Esfahani, E.; Meshkani, R. The effects of resveratrol on markers of oxidative stress in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled clinical trial. Acta Diabetol. 2018, 55, 341–353. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working, G. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Cohen, A.F.; Burggraaf, J.; van Gerven, J.M.; Moerland, M.; Groeneveld, G.J. The use of biomarkers in human pharmacology (Phase I) studies. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 55–74. [Google Scholar] [CrossRef]
- Devling, T.W.; Lindsay, C.D.; McLellan, L.I.; McMahon, M.; Hayes, J.D. Utility of siRNA against Keap1 as a strategy to stimulate a cancer chemopreventive phenotype. Proc. Natl. Acad. Sci. USA 2005, 102, 7280–7285A. [Google Scholar] [CrossRef]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef]
- Goodman, M.; Bostick, R.M.; Kucuk, O.; Jones, D.P. Clinical trials of antioxidants as cancer prevention agents: Past, present, and future. Free Radic. Biol. Med. 2011, 51, 1068–1084. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and clinical significance of biomarkers of oxidative stress in humans. Oxid Med. Cell Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- Czerska, M.; Mikolajewska, K.; Zielinski, M.; Gromadzinska, J.; Wasowicz, W. Today’s oxidative stress markers. Med. Pr. 2015, 66, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Wakabayashi, N.; Misra, V.; Nilles, T.; Biswal, S.; Trush, M.A.; Kensler, T.W. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch. Biochem. Biophys. 2006, 454, 7–15. [Google Scholar] [CrossRef]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar] [CrossRef]
- Celsus, A.C. De medicina. Self-published 25.
- Ghezzi, P.; Floridi, L.; Boraschi, D.; Cuadrado, A.; Manda, G.; Levic, S.; D’Acquisto, F.; Hamilton, A.; Athersuch, T.J.; Selley, L. Oxidative stress and inflammation induced by environmental and psychological stressors: A biomarker perspective. Antioxid. Redox Signal. 2018, 28, 852–872. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2). Mol. Cell Biol. 2004, 24, 36–45. [Google Scholar] [CrossRef]
- Jung, H.; Kim, T.; Chae, H.Z.; Kim, K.T.; Ha, H. Regulation of macrophage migration inhibitory factor and thiol-specific antioxidant protein PAG by direct interaction. J. Biol. Chem. 2001, 276, 15504–15510. [Google Scholar] [CrossRef]
- Lin, W.; Wu, R.T.; Wu, T.; Khor, T.O.; Wang, H.; Kong, A.N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Brigelius-Flohe, R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid. Redox Signal. 2005, 7, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Tsai, P.Y.; Ka, S.M.; Chang, J.M.; Chen, H.C.; Shui, H.A.; Li, C.Y.; Hua, K.F.; Chang, W.L.; Huang, J.J.; Yang, S.S.; et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Biol. Med. 2011, 51, 744–754. [Google Scholar] [CrossRef]
- Taguchi, K.; Kensler, T.W. Nrf2 in liver toxicology. Arch. Pharm. Res. 2020, 43, 337–349. [Google Scholar] [CrossRef]
- Fahey, J.W.; Haristoy, X.; Dolan, P.M.; Kensler, T.W.; Scholtus, I.; Stephenson, K.K.; Talalay, P.; Lozniewski, A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7610–7615. [Google Scholar] [CrossRef]
- Kensler, T.W.; Groopman, J.D. Carcinogen-DNA and protein adducts: Biomarkers for cohort selection and modifiable endpoints in chemoprevention trials. J. Cell Biochem. Suppl. 1996, 25, 85–91. [Google Scholar] [CrossRef]
- Yuan, J.M.; Stepanov, I.; Murphy, S.E.; Wang, R.; Allen, S.; Jensen, J.; Strayer, L.; Adams-Haduch, J.; Upadhyaya, P.; Le, C.; et al. Clinical trial of 2-phenethyl isothiocyanate as an inhibitor of metabolic activation of a tobacco-specific lung carcinogen in cigarette smokers. Cancer Prev. Res. 2016, 9, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.G.; Young, P.J.; Agus, C.; Knize, M.G.; Boobis, A.R.; Gooderham, N.J.; Lake, B.G. Cruciferous vegetable consumption alters the metabolism of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) in humans. Carcinogenesis 2004, 25, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Tolstikov, V.; Moser, A.J.; Sarangarajan, R.; Narain, N.R.; Kiebish, M.A. Current status of metabolomic biomarker discovery: Impact of study design and demographic characteristics. Metabolites 2020, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Schmidt, M.A.; Kaddurah-Daouk, R. Current concepts in pharmacometabolomics, biomarker discovery, and precision medicine. Metabolites 2020, 10, 129. [Google Scholar] [CrossRef]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef]
- Macari, E.R.; Lowrey, C.H. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood 2011, 117, 5987–5997. [Google Scholar] [CrossRef]
- Campbell, M.R.; Karaca, M.; Adamski, K.N.; Chorley, B.N.; Wang, X.; Bell, D.A. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid Med. Cell Longev. 2013, 2013, 120305. [Google Scholar] [CrossRef]
- Ushida, Y.; Suganuma, H.; Yanaka, A. Low-dose of the sulforaphane precursor glucoraphanin as a dietary supplement induces chemoprotective enzymes in humans. Food Nutr. Sci. 2015, 6, 1603–1612. [Google Scholar] [CrossRef]
- Kensler, T.W.; Groopman, J.D. Is it time to advance the chemoprevention of environmental carcinogenesis with microdosing trials? Cancer Prev. Res. 2009, 2, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Uruno, A.; Yumoto, A.; Taguchi, K.; Suzuki, M.; Harada, N.; Ryoke, R.; Naganuma, E.; Osanai, N.; Goto, A.; et al. Space travel of mice displays contribution of Nrf2 to maintenance of homeostasis. Commun. Biol. 2020, in press. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DMF | BARD-Me | Oltipraz | SFN | TOTAL | Percent Significant Outcomes | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sig. Δ | NS | Sig. Δ | NS | Sig. Δ | NS | Sig. Δ | NS | Sig. Δ | NS | |||
| Nrf2 Target Genes | Activity | |||||||||||
| NQO1 | 1 | 1 | 1 | 1 | 50% | |||||||
| GST | 1 | 2 | 1 | 2 | 2 | 50% | ||||||
| SOD | 1 | 0 | 1 | ALL NULL | ||||||||
| GPX | 1 | 0 | 1 | ALL NULL | ||||||||
| Transcripts | ||||||||||||
| NQO1 | 1 | 1 | 1 | 1 | 4 | 5 | 7 | 6 | 54% | |||
| HMOX1 | 1 | 3 | 6 | 3 | 7 | 30% | ||||||
| GCLC | 1 | 2 | 0 | 3 | ALL NULL | |||||||
| GCLM | 2 | 2 | 2 | 2 | 50% | |||||||
| GSTM | 1 | 1 | 2 | 1 | 3 | 25% | ||||||
| GSTP | 1 | 1 | 1 | 1 | 50% | |||||||
| UGT | 1 | 0 | 1 | ALL NULL | ||||||||
| GPX | 1 | 1 | 0 | 2 | ALL NULL | |||||||
| γGCS | 2 | 2 | 0 | 100% | ||||||||
| TR1 | 1 | 1 | 0 | 100% | ||||||||
| LTB4DH | 1 | 1 | 0 | 100% | ||||||||
| AKR1C1 | 1 | 2 | 1 | 2 | 33% | |||||||
| AKR1C2 | 1 | 1 | 0 | 100% | ||||||||
| AKR1C3 | 1 | 0 | 1 | ALL NULL | ||||||||
| HBG1 | 1 | 0 | 1 | ALL NULL | ||||||||
| CBR1 | 1 | 1 | 1 | 1 | 50% | |||||||
| SLC7A11 | 1 | 0 | 1 | ALL NULL | ||||||||
| PCA cytoprotection/detox/antioxidant | 1 | 1 | 0 | 100% | ||||||||
| Nrf2 related genes (aggregated transcripts) | ||||||||||||
| NQO1, HMOX1, AKR1C1, HSP27, HSP70 | 1 | 1 | 0 | 100% | ||||||||
| Gene Function/Expression | HDAC | 3 | 2 | 3 | 2 | 60% | ||||||
| Histone acetylation | 1 | 1 | 1 | 1 | 50% | |||||||
| CYP3A4 | 1 | 0 | 1 | ALL NULL | ||||||||
| TGFβ pathway | 1 | 1 | 0 | 100% | ||||||||
| Epidermal growth factor receptor | 1 | 1 | 0 | 100% | ||||||||
| Insulin signaling | 1 | 1 | 0 | 100% | ||||||||
| Cancer-related | ||||||||||||
| RNA-seq of prostate cancer genes | 1 | 1 | 0 | 100% | ||||||||
| p21WAF/CIP1 | 1 | 0 | 1 | ALL NULL | ||||||||
| Cyclin D1 | 1 | 1 | 0 | 100% | ||||||||
| STAT3 | 1 | 0 | 1 | ALL NULL | ||||||||
| p-STAT3 | 1 | 0 | 1 | ALL NULL | ||||||||
| p21 | 1 | 0 | 1 | ALL NULL | ||||||||
| Active caspase 3 | 1 | 0 | 1 | ALL NULL | ||||||||
| VEGF | 1 | 0 | 1 | ALL NULL | ||||||||
| HIF1α | 1 | 0 | 1 | ALL NULL | ||||||||
| Decorin | 1 | 1 | 0 | 100% | ||||||||
| Insulin-like growth factor | 1 | 0 | 1 | ALL NULL | ||||||||
| p16 | 1 | 1 | 0 | 100% | ||||||||
| Oxidative Stress | GSH (Glutathione) levels | 2 | 3 | 2 | 1 | 4 | 4 | 50% | ||||
| 8-OHdG and oxidized nucleosides | 1 | 3 | 3 | 1 | 75% | |||||||
| DNA strand breaks | 1 | 1 | 0 | 100% | ||||||||
| PCOOH (phosphatidylcholine hydroperoxide) | 1 | 1 | 0 | 100% | ||||||||
| 8-isoprostane | 1 | 3 | 1 | 3 | 25% | |||||||
| TBARS | 2 | 0 | 2 | ALL NULL | ||||||||
| Protein carbonyls | 1 | 0 | 1 | ALL NULL | ||||||||
| TAC (Total antioxidant capacity) | 1 | 2 | 1 | 2 | 33% | |||||||
| TOS (Total oxidant status) | 1 | 0 | 1 | ALL NULL | ||||||||
| OSI (Oxidative stress index) | 1 | 1 | 0 | 100% | ||||||||
| MDA | 2 | 2 | 0 | 100% | ||||||||
| Oxidized-LDL | 1 | 1 | 0 | 100% | ||||||||
| Inflammation | Cytokine | |||||||||||
| IL-1 | 1 | 2 | 0 | 3 | ALL NULL | |||||||
| IL-4 | 1 | 0 | 1 | ALL NULL | ||||||||
| IL-6 | 1 | 3 | 3 | 3 | 4 | 43% | ||||||
| IL-8 | 1 | 3 | 0 | 4 | ALL NULL | |||||||
| IL-10 | 1 | 0 | 1 | ALL NULL | ||||||||
| IL-12 | 1 | 0 | 1 | ALL NULL | ||||||||
| IL-13 | 1 | 1 | 0 | 2 | ALL NULL | |||||||
| IL-17 | 1 | 0 | 1 | ALL NULL | ||||||||
| TNFα | 1 | 1 | 1 | 1 | 2 | 33% | ||||||
| IFNγ | 1 | 2 | 0 | 3 | ALL NULL | |||||||
| Chemokines | ||||||||||||
| CCL5 | 1 | 0 | 1 | ALL NULL | ||||||||
| MIP-1B (CCL4) | 1 | 0 | 1 | ALL NULL | ||||||||
| MCP-1 (CCL2) | 1 | 2 | 2 | 1 | 67% | |||||||
| CXCL1 | 1 | 0 | 1 | ALL NULL | ||||||||
| IP-10 (CXCL10) | 1 | 1 | 1 | 1 | 50% | |||||||
| MIG | 1 | 1 | 0 | 100% | ||||||||
| Lipid mediators | ||||||||||||
| PGD2 | 1 | 1 | 0 | 100% | ||||||||
| Tetranor-PGEM | 1 | 1 | 0 | 100% | ||||||||
| 11β-PGF2α | 1 | 1 | 0 | 100% | ||||||||
| 11-dehydro-TXB2 | 1 | 1 | 0 | 100% | ||||||||
| NF-kB pathway | 1 | 1 | 0 | 100% | ||||||||
| CRP | 2 | 3 | 2 | 3 | 40% | |||||||
| Immune response | ||||||||||||
| WBC counts | 1 | 1 | 0 | 100% | ||||||||
| Neutrophil counts | 1 | 0 | 1 | ALL NULL | ||||||||
| Monocyte counts | 1 | 0 | 1 | ALL NULL | ||||||||
| Macrophage counts | 1 | 0 | 1 | ALL NULL | ||||||||
| T cell counts | 1 | 0 | 1 | ALL NULL | ||||||||
| NKT cells | 1 | 0 | 1 | ALL NULL | ||||||||
| CD4+ and CD8+ T-lymphocytes | 1 | 1 | 1 | 1 | 50% | |||||||
| Proinflammatory genes (aggregated transcripts) | 1 | 1 | 0 | 100% | ||||||||
| PCA immune-response genes | 1 | 0 | 1 | ALL NULL | ||||||||
| Others | ||||||||||||
| MIF | 1 | 1 | 0 | 100% | ||||||||
| SLPI | 1 | 1 | 0 | 100% | ||||||||
| CD105+ and iNOS+ cells | 1 | 1 | 0 | 100% | ||||||||
| Virus-induced granzyme B production in NK cells | 1 | 1 | 0 | 100% | ||||||||
| Serum pepsinogen I and II | 1 | 1 | 1 | 1 | 50% | |||||||
| Carcinogen Metabolites/Adducts | Aflatoxin-albumin adducts | 1 | 1 | 0 | 100% | |||||||
| Aflatoxin-DNA adducts | 1 | 1 | 0 | 100% | ||||||||
| Aflatoxin mercapturic acid | 1 | 1 | 0 | 100% | ||||||||
| Polycyclic aromatic hydrocarbon-DNA adducts | 1 | 0 | 1 | ALL NULL | ||||||||
| Benzo(a)pyrene-7,8-diol-9,10-epoxide adducts | 1 | 0 | 1 | ALL NULL | ||||||||
| Mutagenicity (urine) | 1 | 0 | 1 | ALL NULL | ||||||||
| Acrolein mercapturic acid | 2 | 2 | 0 | 100% | ||||||||
| Benzene mercapturic acid | 3 | 3 | 0 | 100% | ||||||||
| Crotonaldehyde mercapturic acid | 2 | 2 | 0 | 100% | ||||||||
| Metabolomics | Cystine | 1 | 1 | 0 | 100% | |||||||
| Plasma metabolites | 1 | 1 | 1 | 1 | 50% | |||||||
| Urinary metabolites | 1 | 1 | 0 | 100% | ||||||||
| Metabolites in prostate biopsies | 1 | 0 | 1 | ALL NULL | ||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yagishita, Y.; Gatbonton-Schwager, T.N.; McCallum, M.L.; Kensler, T.W. Current Landscape of NRF2 Biomarkers in Clinical Trials. Antioxidants 2020, 9, 716. https://doi.org/10.3390/antiox9080716
Yagishita Y, Gatbonton-Schwager TN, McCallum ML, Kensler TW. Current Landscape of NRF2 Biomarkers in Clinical Trials. Antioxidants. 2020; 9(8):716. https://doi.org/10.3390/antiox9080716
Chicago/Turabian StyleYagishita, Yoko, Tonibelle N. Gatbonton-Schwager, Melissa L. McCallum, and Thomas W. Kensler. 2020. "Current Landscape of NRF2 Biomarkers in Clinical Trials" Antioxidants 9, no. 8: 716. https://doi.org/10.3390/antiox9080716
APA StyleYagishita, Y., Gatbonton-Schwager, T. N., McCallum, M. L., & Kensler, T. W. (2020). Current Landscape of NRF2 Biomarkers in Clinical Trials. Antioxidants, 9(8), 716. https://doi.org/10.3390/antiox9080716