Abstract
The transcription factor NF-E2 p45-related factor 2 (NRF2; encoded by NFE2L2) plays a critical role in the maintenance of cellular redox and metabolic homeostasis, as well as the regulation of inflammation and cellular detoxication pathways. The contribution of the NRF2 pathway to organismal homeostasis is seen in many studies using cell lines and animal models, raising intense attention towards targeting its clinical promise. Over the last three decades, an expanding number of clinical studies have examined NRF2 inducers targeting an ever-widening range of diseases. Full understanding of the pharmacokinetic and pharmacodynamic properties of drug candidates rely partly on the identification, validation, and use of biomarkers to optimize clinical applications. This review focuses on results from clinical trials with four agents known to target NRF2 signaling in preclinical studies (dimethyl fumarate, bardoxolone methyl, oltipraz, and sulforaphane), and evaluates the successes and limitations of biomarkers focused on expression of NRF2 target genes and others, inflammation and oxidative stress biomarkers, carcinogen metabolism and adduct biomarkers in unavoidably exposed populations, and targeted and untargeted metabolomics. While no biomarkers excel at defining pharmacodynamic actions in this setting, it is clear that these four lead clinical compounds do touch the NRF2 pathway in humans.
1. The KEAP1-NRF2 System
It has been more than 20 years since the initial molecular description of the Keap1-Nrf2 signaling pathway, wherein it was recognized as an adaptive response pathway to exogenous [1], and later as an adaptive response pathway to endogenous stresses [2]. Nuclear factor erythroid 2 related factor 2 (NRF2) is a transcription factor that regulates the expression of over 300 target genes with roles in antioxidant and anti-inflammatory actions, electrophile detoxication, cell metabolism, proliferation and differentiation, and general cytoprotection. Kelch-like ECH-associated protein-1 (KEAP1) is an adaptor protein for the Cullin-3 ubiquitin ligase and a key cytoplasmic repressor of NRF2. KEAP1 interaction with NRF2 leads to NRF2 polyubiquitination and its subsequent proteasomal degradation. In the presence of oxidative stress or inducers, which are often electrophiles, key “sensor” cysteine thiol groups on KEAP1 are modified, disrupting the degradation process and allowing for nascent NRF2 to directly translocate into the nucleus and to target gene transcription. Target genes are defined by the presence of functional antioxidant response elements (AREs) in their regulatory regions. NRF2 heterodimerizes with one of the small musculo-aponeurotic fibrosarcomas (sMAF) to bind to the ARE. Sentinel downstream targets include NAD(P)H: quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HMOX1), glutamate-cysteine ligase catalytic subunit (GCLC), and the glutathione-S-transferases (GSTs). The rapid inducibility of a response based on a de-repression mechanism is an important feature of this cytoprotective KEAP1-NRF2 system. An additional feature is the extensive crosstalk between NRF2 and other transcription factor signaling pathways, allowing for further fine-tuning of physiological responses to stress. There are excellent reviews that document the historic milestones in the characterization of the pathway, the molecular mechanisms governing the functions of KEAP1 and NRF2, as well as their roles in physiology and pathology [3,4,5,6].
2. Pharmacological Inducers of KEAP1-NRF2 Signaling
A plethora of preclinical studies, utilizing cell culture models as well as animals, continues to lead to the discovery and characterization of molecules that could induce (or inhibit) the pathway and mitigate toxicities or diseases driven by exposures to electrophiles, oxidants, and intrinsic inflammatory processes. The profound protective actions of many classes of “Nrf2 inducers” in rodent models of acute and chronic diseases, as well as corresponding actions in genetically manipulated models (ranging from C. elegans, Drosphila, and zebrafish to mice and rats) in which elements of the pathway were disrupted or amplified, led to the notion that NRF2 signaling in humans might be a propitious target for disease prevention and treatment [5,7,8]. As reviewed elsewhere [9,10,11,12], many drugs and natural products, exhibiting remarkable structural diversity, are under development and clinical evaluation for these purposes. One of many features retarding drug development is the lack of sensitive and specific biomarkers that can rapidly inform understandings of dose-responsiveness, mechanistic specificity, as well as magnitude and duration of pharmacodynamic action in clinical trial settings. Such needs pervade the process of clinical development of NRF2 inducers. Although hundreds—if not thousands—of molecules have been described as inducers and dozens are in clinical development, only four (dimethyl fumarate (DMF), bardoxolone methyl (BARD-Me), oltipraz, and sulforaphane (SFN)) have appeared substantively in the peer-reviewed literature, especially wherein measures of biomarkers have been a component of the clinical protocols. Interestingly, all four of these agents intersect the NRF2 signaling pathway through interactions with cysteine151 in KEAP1 [5]. While this review focuses on biomarkers likely related to NRF2 action, because of their reactivity with thiols all these agents affect multiple targets and pathways in cells. Sole attribution of any response to actions on NRF2 in the context of the biomarkers reported herein has not been firmly established. Nonetheless, there are common themes in the field, and as mechanistic insights become sharpened along with analytic improvements higher confidence in the specificity and sensitivity of some of these biomarkers may evolve.
Summaries of the clinical development and utilization of DMF, BARD-Me, oltipraz, and SFN are provided in the following sections, along with highlights of general evidence for the targeting of NRF2 by these agents based on preclinical studies. Interestingly, the clinical applications of each drug are quite distinct. The distributions of healthy and disease-state study populations enrolled in clinical trials with these four agents are highlighted in Figure 1.
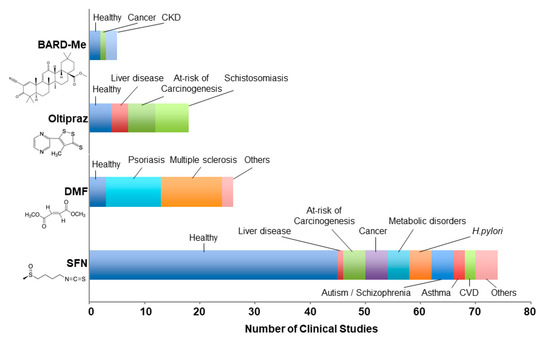
Figure 1.
Study populations examined in peer-reviewed clinical studies of nuclear factor erythroid 2 related factor 2 (NRF2) inducers. Studies for bardoxolone methyl (BARD-Me), oltipraz, dimethyl fumarate (DMF), and sulforaphane (SFN) totaled 5, 18, 26, and 75, respectively, from 1982 to June 2020. Literature searches were conducted on PubMed, Google Scholar, and ClinicalTrials.gov. Publications based on the same clinical trial were aggregated as one study in this graph. The chemical structure for each NRF2 inducer is indicated below each name. CKD, chronic kidney disease; CVD, cardiovascular disease.
2.1. Dimethyl Fumarate (DMF, BG-12, Tecfidera)
Fumaric acid esters are used for the treatment of psoriasis and multiple sclerosis. A mixture of dimethyl fumarate with mono ethylfumarate salts was developed as a drug product (Fumaderm®) and registered in 1994 as an oral agent for the treatment of moderate to severe plaque psoriasis. Dimethyl fumarate alone (Skilarence®) also has EU approval for this indication [13]. Due to the immunomodulatory potential of Fumaderm®, DMF was evaluated in other immune-mediated diseases, which led to testing of DMF in large multicenter phase 2 and 3 trials of relapsing–remitting multiple sclerosis [14]. Oral DMF as a galenical formulation (BG-12, Tecfidera®) was subsequently demonstrated to be an effective agent [15,16] and was approved for use in this disease by the U.S. Food and Drug Administration in 2013 and the European Medicines Agency in 2014 [17]. New analogs featuring better bioavailability and efficacy are under development [10]. DMF has been linked to several treatment-limiting adverse events, including flushing and gastrointestinal complaints, and less frequently to persistent lymphopenia. Such action likely precludes the use of DMF in any disease prevention settings. A summary of clinical trials utilizing fumaric acid esters, all of which include DMF, is listed in Table S1 [15,16,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56].
DMF is almost completely converted to monomethyl fumarate (MMF) by intestinal esterases. Thus, it is likely that DMF serves as a prodrug in the elaboration of its therapeutic actions, with MMF the likely active form. Talalay reported in 1990 [57] that DMF and other fumaric acid esters induced Nrf2 target gene (Nqo1 and Gst) activities in various organs of mice and rats after dietary administration. DMF and MMF contain double bonds, allowing them to act as Michael acceptors to form adducts with thiol groups, such as C151 in Keap1. Much more recently, Linker et al. [58] demonstrated in mice that oral DMF induces the expression of Nrf2 target genes in multiple cell types of the central nervous system. Studies in Nrf2-disrupted mice indicate a loss of protective effect of DMF in a multiple sclerosis model of experimental autoimmune encephalitis and MPTP-induced experimental Parkinson’s disease [58,59,60]. However, Nrf2-independent actions are also being described. For example, DMF and MMF activate the nicotinic receptor hydroxycarboxylic acid receptor 2, which is expressed in immune cells and gut epithelial cells, resulting in NRF2-independent anti-inflammatory responses, in addition to anti-inflammatory actions associated with Nrf2 signaling. There is substantial evidence that DMF and MMF influence multiple aspects of the immune system that contribute to both the therapeutic effects and its major side effects [10,17].
2.2. Bardoxolone-methyl (BARD-Me; CDDO-Me)
Many dozens of synthetic oleanane triterpenoids have been synthesized, guided by efforts to enhance the weak anti-inflammatory activity of the naturally occurring triterpenoid oleanolic acid [61], which in vitro have been shown to: (1) suppress inflammation-like responses and oxidative stress, and therefore to be cytoprotective, especially at low nanomolar doses; (2) induce differentiation; and (3) block cell proliferation and induce apoptosis at higher micromolar doses [62]. Extensive animal data have been described demonstrating efficacy of triterpenoids in the prevention or amelioration of neurodegenerative diseases and in diseases of the eye, lung, cardiovascular system, liver, gastrointestinal tract, and kidney, as well as in cancer and in metabolic and inflammatory or autoimmune disorders [62,63]. Two triterpenoids have been studied extensively in preclinical studies (CDDO-Im; 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole); (CDDO-Me; bardoxolone methyl; methyl 2-cyano-3,12-dioxooleana-1,9(11)dien-28-oate) and are likely to have strongly overlapping, but not entirely concordant, mechanisms of action. As summarized in Table S1, CDDO-Me (BARD-Me) was the featured synthetic oleanane triterpenoid selected by Reata Pharmaceuticals for clinical development [64,65,66,67,68,69,70,71,72,73,74,75].
Over 30 clinical trials with BARD-Me have been registered in ClinicalTrials.gov. The first phase I clinical trial of BARD-Me was conducted in patients with advanced solid tumor and lymphoma to determine the dose-limiting toxicity and the maximum tolerated dose [66]. In this first trial, an increase in estimated glomerular filtration rate (eGFR) was also noted. This observation led to the evaluation of BARD-Me for treatment of patients with chronic kidney disease (CKD) and prompted several follow-up studies, including a phase 2 Bardoxolone Methyl Treatment: Renal Function in CKD/Type 2 Diabetes (BEAM); and phase 3 Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes Mellitus: the Occurrence of Renal Events (BEACON) trials. While BEAM showed a promising increase in estimated glomerular filtration rate (eGFR), BEACON was terminated early due to heart failure events within the first 4 weeks of treatment [64,67]. Bardoxolone-methyl is currently in clinical trials for Alport’s syndrome (A Phase 2/3 Trial of the Efficacy and Safety of Bardoxolone Methyl in Patients With Alport Syndrome-CARDINAL, NCT03019185), IgA nephropathy, type 1 diabetic nephropathy, focal segmental glomerulosclerosis, and autosomal dominant polycystic kidney disease (A Phase 2 Trial of the Safety and Efficacy of Bardoxolone Methyl in Patients With Rare Chronic Kidney Diseases - PHOENIX, NCT03366337). An additional phase 3 study in patients with diabetic kidney disease is also being performed with a primary outcome of time to onset of a 30% decline in eGFR or end-stage renal disease (A phase 3 study of bardoxolone methyl in patients with diabetic kidney disease, AYAME, NCT03550443) [76].
The role of NRF2 is inferred to be a central component of the mechanism of action of the bardoxolone methyl. CDDO-Im [77] and CDDO-Me [78] protect against acute kidney injury in mice, while Nrf2 null mice are sensitized to injury [79]. Similarly, CDDO-Im completely abrogates aflatoxin hepatocarcinogenesis in rats [80], whilst Nrf2 knockout rats show markedly enhanced sensitivity to the hepatotoxic effects of aflatoxin [81]. Interestingly, although CDDO-Im, CDDO-Me, and DMF all activate the Nrf2 pathway, they target distinct genes and signaling pathways, and in the case of DMF result in opposite effects for the prevention of experimental lung cancer in mice [82]. Highlighting the importance of additional mechanisms of action, Ball et al. [83] recently reported that CDDO-Me relieves immunosuppression in the breast tumor microenvironment and unleashes host adaptive antitumor immunity. This may be mediated in part by conversion of breast-tumor-activated macrophages from a tumor-promoting to a tumor-inhibiting activation state [84]. Some anti-inflammatory actions of CDDO-Me are likely mediated through additional actions on nuclear factor-κB (NF-κB) [85].
2.3. Oltipraz
Oltipraz, [4-methyl-5-(2-pyrazinyl)-3H-1,2-dithiole-3-thione], was originally developed by Rhône-Poulenc (subsequently acquired by Sanofi) as a possible treatment for schistosomiasis and was extensively evaluated in clinical trials in the early 1980s. Field trials in Mali, Gabon, Sudan, and other sites, using short courses with durations of 1–5 days with total doses of 1.25–7.5 g, achieved cure rates of greater than 90% [86,87,88,89,90,91]. However, side effects occurred in about 10% of participants, principally related to the digestive system and to fingertip pain, with the latter being amplified by exposure to sunlight. Although all effects were reported as mild, subsided within a few days, and did not require discontinuation of the drug, the concerns regarding photosensitivity led to the abandonment of oltipraz for the treatment of schistosomiasis. This decision was surely influenced by concurrent clinical progress of less expensive, equi-effective, and less problematic drugs for the chemotherapy of schistosomiasis.
While studying mechanisms of antischistosomiasis by oltipraz, Ernest Bueding and colleagues at Johns Hopkins initially noted that giving the drug to mice infected with Schistosoma mansoni caused a dramatic reduction in the glutathione stores of the parasite, while paradoxically markedly elevating glutathione levels in many tissues of the host [92]. Subsequent studies demonstrated that oltipraz and some structurally related 1,2-dithiole-3- thiones were potent inducers of enzymes concerned with the maintenance of reduced glutathione pools, as well as enzymes important to electrophile detoxication in multiple tissues of rats and mice [93]. These results prompted Bueding to predict that oltipraz might have cancer chemopreventive properties [94], and that perhaps at lower doses the side effects would be mitigated.
Largely under the aegis of the drug development program of the Chemoprevention Branch of the National Cancer Institute, oltipraz underwent extensive evaluation for anticarcinogenic efficacy in animal models. As reviewed elsewhere [95,96], oltipraz has shown chemopreventive activity against different classes of carcinogens targeting the trachea, lung, stomach, small intestine, colon, pancreas, liver, urinary bladder, mammary gland, hematopoietic cells, and skin. The most dramatic actions of oltipraz occurred in the colon and liver, where dietary administration resulted in significant reductions in both tumor incidence and multiplicity. Accordingly, subsequent clinical trials of oltipraz (Figure 1; Table S1 [86,87,88,89,90,91,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111] focused on cancer-preventive interventions targeted to the colon and liver, and most recently mitigation of hepatic fibrosis and nonalcoholic fatty liver disease (NAFLD). Only one current trial with oltipraz (NCT04142749: A Multi-center, Randomized, Double-blind, Placebo-controlled, Parallel, Phase II Clinical Trial to Evaluate the Efficacy and Safety of Oltipraz) is listed on ClinicalTrials.gov, indicating that while this agent has provided historical perspectives on biomarker utilization directed towards the NRF2 pathway, it appears to be at a defining point for a possible clinical path forward.
A role for the involvement of Nrf2 in the chemopreventive actions of oltipraz in animals was built on the foundation of induction of Nrf2 target genes and enzymes in multiple rodent tissues [93], amplified by an appreciation for the role of AREs in mediating these responses [112] and capped by studies demonstrating that protective effects of oltipraz against DNA adduct formation and tumorigenesis were abrogated in Nrf2-disrupted mice [113,114]. These observations, in turn, guided the choice of biomarkers selected as intermediate endpoints for clinical studies with oltipraz.
2.4. Sulforaphane (SFN)
Sulforaphane was described in the middle of the last century as an antibiotic, and was isolated from red cabbage and from the western USA rangeland weed hoary cress. Various groups have since synthesized it, however Talalay and Zhang were the first to isolate it from broccoli [115] and to demonstrate its cancer protective properties (against mammary carcinogenesis in rats) [116]. Its biogenic precursor, glucoraphanin, was then found in abundance in broccoli sprouts. Sprout-based preparations were confirmed to be active for prevention in animal carcinogenesis models [117].
The highly reactive isothiocyanate SFN is produced in plants as an inert precursor, the glucosinolate glucoraphanin. Upon disruption of plant tissue integrity, glucoraphanin interacts with myrosinase, which catalyzes the hydrolysis of glucoraphanin to yield SFN. The β-thioglucosidases within the human microbiome also catalyze this bioactivation. Glucoraphanin occurs in all tissues of broccoli plants, though it is most abundant in the aerial portions, and the developing florets and ultimately the seeds are richest in this compound [117]. Studies to examine the pharmacokinetics of SFN in humans began in 1998 [118]. In the subsequent 75 or so studies examining pharmacokinetics, pharmacodynamics, or efficacy, a multitude of formulations have been used. Broccoli-based preparations have consisted typically of either glucoraphanin; SFN; glucoraphanin with added active myrosinase; the raw, cooked, or dried vegetables themselves (either broccoli or broccoli sprouts); or extracts of broccoli seeds or sprouts, which are glucoraphanin-rich, SFN-rich, or both. This diversity of formulations—and thus lack of consistency—has provided uncertainty regarding actual administered doses and confusion in the interpretation of study results. The broccoli (sulforaphane) literature with regards to formulation, dose, and response in preclinical and clinical studies has been reviewed recently [11]. Relatively few studies have evaluated the efficacy of SFN in its various formulations against clinical endpoints of disease. As depicted in Figure 1, more than half of the published studies have involved healthy volunteers. However, in part based on a broad spectrum of beneficial responses observed in animal models, clinical trials focused on neurodevelopmental diseases such as autism and schizophrenia, cardiovascular disease, sickle cell anemia, chronic obstructive pulmonary disease, asthma, Helicobacter pylori infection, and environmental factors contributing to carcinogenesis, as well as diabetes, metabolic syndrome, and related disorders have been undertaken (reviewed in [11,119] and are listed in Table S1 [118,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196]).
Bioassay guided fractionation of acetonitrile extracts of SAGA broccoli led to the isolation of SFN as the major inducer [115]. The bioassay tracked the induction of Nqo1 activity in murine hepatoma cells. Thus, a Nrf2 target gene was at the center of its isolation from broccoli. There are now over 300 publications in which the actions of SFN in mice invoked a role for Nrf2. Studies in which these actions are diminished or abrogated in parallel experiments in Nrf2-disrupted mice provide the strongest lines of evidence for a key role of this transcription factor in its actions. With that said, it is equally evident that other modes of action contribute to the molecular responses to SFN in animals and humans [119,197,198]. Such polypharmacy may well contribute to the efficacy of the agent in disease prevention and mitigation but obfuscates the value of specific pharmacodynamic biomarkers in the clinical development and evaluation of SFN—perhaps even more so, because unlike the situation with triterpenoids (e.g., CDDO-Im and CDDO-Me), where there appears to be a hierarchical activation of targets/pathways with increasing dose [62], it is more likely that the multiple targets of SFN are activated at similar concentrations [198].
2.5. Other Natural Product Inducers
While SFN is generally considered to be the most potent natural product inducer of Nrf2 signaling, there are many plant-derived molecules that activate the pathway. They have been identified principally in cell culture screens. Several comprehensive reviews of this topic have been published recently [193,199]. These molecules can be natural products, natural product-derived, or natural product-inspired. Moreover, they can activate the pathway through different mechanisms—almost all intersecting with Keap1 directly or indirectly. However, relatively few have seen rigorous evaluation in clinical studies or trials. Some natural products of clinical use include curcumin, resveratrol, and flaxseed.
The polyphenol curcumin, which is isolated from Curcuma longa and provides yellow color to the spice turmeric, is a weak inducer of Nrf2 signaling in cell culture [200]. Long used in Ayurveda for the treatment of many conditions, two clinical trials of curcumin have examined NRF2-driven biomarkers. Yang et al. [201] reported that curcumin (500 mg, daily) induced NQO1 levels in lymphocytes and reduced plasma malondialdehyde levels in plasma of T2DM patients. Modulation of inflammatory biomarkers was also observed. Jiménez-Osorio et al. [202] failed to observe any effects of curcumin (320 mg/d for 8 weeks) on the activities of NRF2 target genes (glutathione peroxidase, glutathione reductase, superoxide dismutase (SOD), and catalase) in peripheral blood mononuclear cells (PBMCs) of diabetic proteinuric chronic kidney disease (CKD) patients enrolled in a placebo-controlled, double-blind trial. Similar mixed results were seen in two placebo-controlled, randomized trials with resveratrol, another polyphenol, which in this instance is found in the skins of grapes and berries and is widely marketed as a dietary supplement. Saldanha [203] reported no effect of 500 mg/d on NRF2 expression in peripheral blood mononuclear cells (PBMCs) of non-dialized CKD patients, whilst Seyyedbrahimi et al. [204] observed significant changes in NRF2 and SOD levels with 400 mg twice-daily in T2DM patients. Flaxseed, a dietary botanical supplement with high fiber, lignan phenolics, and omega-3 fatty acids, has anti-inflammatory and antioxidant properties in murine models of acute and chronic lung injury. Ten cystic fibrosis patients and five healthy volunteers consumed 40 g of flaxseed daily for 4 weeks in a pilot study of tolerability and possible pharmacodynamic action. No significant effects were observed on biomarkers of NRF2 signaling or attenuation of oxidative stress. Thus, the translation of these broadly touted natural products towards effective modulation of NRF2 signaling and possible disease mitigation remain largely unfulfilled. This outcome likely reflects the relative lack of potency of these molecules and the myriad of molecular targets that may be modified.
3. Biomarker-Based Clinical Studies and NRF2 Inducers
Prospective clinical studies have revolutionized the development of medicine by providing reliable evidence on the efficacy and safety of novel treatment strategies. In newer paradigms of drug development, biomarkers are often used to guide early clinical development (e.g., phase 0 studies). A biomarker is defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [205]. A simple distinction can be made between clinical biomarkers and mechanism-specific biomarkers. Clinical biomarkers are thought to reflect disease activity and pathophysiology, which are not discussed in any detail in this article. The mechanism-specific biomarkers, which are the main focus of this article, reflect the molecular action of an agent on the pharmacology or pathophysiology, where biomarkers are used to evaluate pharmacokinetic and pharmacodynamic aspects, including target activation [206].
Screening of peer-reviewed clinical studies of NRF2 inducers from 1982 to June 2020 (Table S1) identified 12, 21, 41, and 78 published studies for BARD-Me, oltipraz, DMF, and SFN, respectively. Not all studies utilized mechanistic biomarkers. The most frequently used mechanistic biomarkers are enumerated in Figure 2A, according to their placement in 6 broad categories, which are reflective of NRF2 and other modes of action. To give some context regarding the actual usage of these biomarkers, the distributions of these biomarkers in the clinical studies with each of the 4 agents were further analyzed (Figure 2B). There is a varied landscape with unique distributions for each clinical agent. As the NRF2 signaling pathway is a new and evolving proposed molecular target for clinical application, mechanistically linked assessments within clinical studies would a priori seem to be fundamental. Indeed, several mechanically linked biomarkers have been investigated widely in the clinical studies for SFN and oltipraz, agents that were developed largely in academic settings. Presumably, these biomarkers have been used to guide selection of dose or formulation in advance of efficacy trials. On the other hand, within the clinical studies for BARD-Me and DMF, agents developed by Pharma, clinical biomarkers account for the majority usage in publications. Here, mechanistic biomarker use tends to be confirmatory once some efficacy has been established.
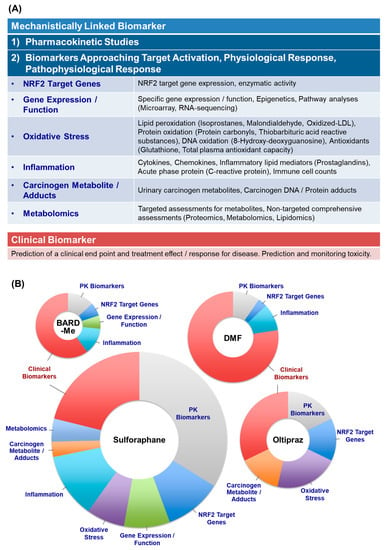
Figure 2.
(A) Classification of biomarkers used in published clinical studies of “NRF2 Inducers”. (B) Distribution of biomarker categories used in the clinical studies. The size of each pie reflects the total number of times a biomarker of each class was used in the clinical studies. Multiple classes of biomarkers were used in some studies. PK, pharmacokinetics.
Although standardizing NRF2 biomarker definitions presents a challenge as there is a significant overlap in biomarker categories, here we breakdown and describe each mechanistically linked biomarker that can be potentially modulated by NRF2 inducers.
3.1. Nrf2 Target Genes
Quickly after its discovery, it was established in mice that Nrf2 regulates the expression of several hundreds of genes, encoding a network of enzymes involved in a broad-based cellular defense system in mice [6,8]. Less extensive cataloging has been undertaken in humans; however, there appears to a largely concordant, albeit likely more limited set of NRF2 target genes [207,208,209]. Thus, tracking changes in the expression or activities of NRF2-targeted enzymes could be one of the most direct biomarkers for monitoring the effects on the downstream pathway of NRF2 signaling. NAD(P)H: quinone oxidoreductase-1 (NQO1) and heme oxygenase-1 (HMOX1) are widely examined NRF2-targeted enzymes exhibiting sensitivity and reliability of their quantification, wherein the mRNA expression levels of these genes have been evaluated in clinical samples, such as in PBMCs and biopsy samples from study subjects.
Reduced glutathione (GSH; the oxidized form of which is glutathione disulfide, GSSG) is the most abundant cellular antioxidant and a major thiol utilized for detoxication of reactive intermediates. NRF2 regulates the expression of enzymes involved in the synthesis and recycling of GSH, such as the catalytic and modulator subunits of glutamate–cysteine ligase (GCLC and GCLM), glutathione reductase (GR), glutathione peroxidase (GPX), and several glutathione S-transferases (GSTs). Moreover, several proteins within the redoxin family, such as thioredoxin (TRX), thioredoxin reductases (TrxRs), peroxiredoxins (Prxs), and sulfiredoxins (SRXNs), are all regulated by NRF2 and provide compartmentalized sensing and signal transduction of regional production of reactive oxygen species. Thus, the redox regulation by NRF2-targeted genes can modulate the level of oxidative stress, which is considered to be one of therapeutic targets by NRF2 inducers. In this article, these genes are considered as NRF2 target gene biomarkers, as distinguished from oxidative stress biomarkers. GCLC, GCLM, GPX, and GST have been used as biomarkers for several clinical studies of NRF2 inducers [104,107,134].
3.2. Gene Expression and Function
In addition to prototypic NRF2 target genes, other genes are also examined as biomarkers, where links to NRF2 signaling are more tenuous or perhaps nonexistent. Mounting reports describe how SFN affects multiple potential downstream pathways, such as epigenetic alterations and heat shock. Epigenetic effects mediated through modulation of histone deacetylase (HDAC) activity have been explored in several clinical studies and may relate to the chemopreventive effects of SFN [129,135,140,159,170]. In a study targeting autism spectrum disorder, heat shock proteins were employed as biomarkers reflecting therapeutic effects by SFN [195]. Comprehensive, unbiased analyses for gene expression (e.g., microarray, RNA-sequencing, and ChIP-seq) have provided powerful assessments for gene expression profiling and transcriptional networks. Several studies, principally focused on SFN, have been performed [128,131,193], revealing characteristic and novel gene expression profiling correlated with intervention.
Rajendran et al. [170] reported that wild-type mice, which are more susceptible to dimethylhydrazine-induced colon carcinogenesis than Nrf2-deficient mice, had higher HDAC levels globally and locally. SFN treatment reduced tumor burden, most notably in the wild-type mice, and reduced HDAC3 expression. Thus, Nrf2 status may influence HDAC levels and signaling by its downstream targets, including p16.
3.3. Oxidative-Stress-Mediated Biomarkers
Oxidative stress has been defined as “an imbalance in pro-oxidants and antioxidants, with associated disruption of redox circuitry and macromolecular damage” [210]. Measuring oxidative stress in the context of clinical trials has a long and somewhat unsuccessful history, especially relating to intervention studies with “traditional” direct antioxidant compounds. In accord with the definition, many oxidative markers found in the body have been proposed, including lipid peroxidation products, such as malondialdehyde (MDA), isoprostane products, oxidized low-density lipoproteins (LDL), hydroperoxides, and 4-hydroxynonenal; protein oxidation products, such as thiobarbituric acid reactive substances (TBARS); carbohydrate oxidation products, such as 3-nitrotyrosine; and nucleic acid oxidation products, such as 8-hydroxy-2-deoxyguanosine (8-OHdG) [211,212]. These biomarkers, based on molecular oxidation products, have also been applied in trials with NRF2 inducers. As alternatives to measures of oxidation products, tests measuring total oxidative status (TOS), total antioxidant capacity (TAC), and oxidative stress index (OSI) have also been used as oxidative stress markers for clinical studies with NRF2 inducers [138,175]. TOS and total antioxidant status (TAS) are usually used to measure the overall oxidation status or antioxidant status of the body, the ratio of which is expressed as the OSI. The TAC measures the amount of free radicals scavenged by a test solution and is used to evaluate the antioxidant capacity of biological samples. GSH and its oxidized form GSSG in blood are global indices of the redox status in the whole organism. In the case of NRF2 inducers, GSH measurements attempt to reflect the balance between oxidant stress and the biosynthesis of GSH, as well as the recycling of oxidized glutathione through genes responding within the NRF2 downstream pathway. More comprehensive means to assess redox dynamics have been developed by monitoring the reversible oxidation of sulfur-containing amino acids and peptides, notably GSH. The redox states of glutathione/glutathione disulfide (GSH/GSSG) and cysteine/cystine (Cys/CySS) are oxidized in association with several known oxidative-stress-related exposures, health conditions, and measures of physiologic function [210]. Applications to NRF2-targeting drugs have not been undertaken to date.
Nrf2 knockout (Nrf2−/−) mice are more susceptible to oxidative-stress-based diseases [213]. Many lines of animal experiments show that Nrf2 activation by pharmacological or genetic approaches significantly reduces cellular damage caused by oxidative stress and suppress the development of several kinds of diseases [214,215]. Because an increased oxidative stress is crucial in the pathogenesis of several kinds of diseases in humans, the notion of targeting oxidative stress with NRF2 inducers has been expanding. As mentioned above, the target genes of NRF2 include genes involved in the regulation of the synthesis and conjugation of GSH. Supported by molecular biology and in vivo experimental studies, assessments of changes in levels of oxidative stress biomarkers could be one important means for optimizing clinical studies of NRF2 inducers (Figure 3).
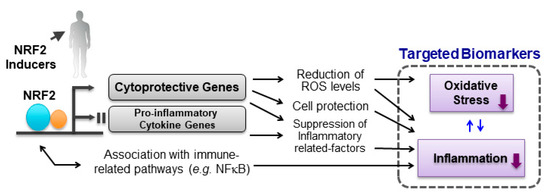
Figure 3.
NRF2 signaling pathway and biomarkers targeting oxidative stress and inflammation. ROS, reactive oxygen species.
3.4. Inflammation-Mediated Biomarkers
The oldest definition of inflammation is by Aulus Cornelius Celsus, who defined the four hallmarks of inflammation: “rubor, et tumor, cum calore, et dolore”, meaning redness, swelling, heat, and pain [216], respectively, which are described as the cardinal signs of inflammation. Surprisingly, the concept of inflammatory biomarkers was established in the first century AD. Studies investigating the molecular basis of inflammation have led to the identification of markers that may also serve as new targets of therapy for inflammation-related diseases. Inflammation and oxidative stress are tightly linked. Although oxidative stress biomarkers mainly approach the formation of effector molecules, such as lipid peroxides and oxidized proteins of DNA, the inflammatory markers measure the response of the organism, for example through the production of inflammatory cytokines and lipid mediators (Figure 2A) [217].
Emerging evidence demonstrates the linkage between NRF2 signaling and inflammatory response. Inflammation and oxidative stress can cause tissue damage with a complex interplay of processes, indicating that the defensive response to oxidative and electrophilic insults by activation of NRF2 can lead to the synergic anti-inflammatory effects. In addition, many lines of experimental results indicate that the activation of NRF2 signaling modifies inflammatory reaction though multiple pathways (Figure 3) [5].
Nrf2 activation by the triterpenoid (CDDO-Im) induces a series of antioxidant genes, accompanied by suppressed expression of inflammatory-related genes, such as TNF- α, IL-6, MCP-1 (monocyte chemo attractant protein-1), and MIP2 (macrophage inflammatory protein-2) in LPS-stimulated mouse peritoneal macrophages, as well as LPS-treated mice (in which anti-inflammatory effects did not occur in Nrf2-deficient cells and mice) [218]. In mouse peritoneal macrophages stimulated by prostaglandin, Nrf2 induces Hmox1 and peroxiredoxin 1 (Prx-1) gene expression, which appears to inhibit inflammatory-related gene expression [219]. Interestingly, human PRX-1 is reported to negatively regulate macrophage migration inhibitory factor (MIF) [220], a crucial factor in the regulation of inflammation. Accordingly, MIF activity was measured in human urine samples after SFN administration to investigate the possibility for MIF as a biomarker to assess anti-inflammatory efficacy in interventions [142]. SFN seems to affect multiple downstream pathways associated with anti-inflammatory actions and NRF2 signaling may be but one pivotal pathway. Pretreatment of primary peritoneal macrophages by SFN inhibits inflammatory-related gene expression, including Cox-2 and iNOS, which was attenuated in Nrf2-deficient cells [221]. The suppression of COX-2 expression in PBMCs was indicated in a clinical study, where the treatment response by SFN for autism spectrum disorder patients was examined [195]. The association between Keap1-Nrf2 signaling and the nuclear factor-κB (NF-κB) signaling pathway, one of the essential immune-related pathways, is another potential mechanism underlying anti-inflammatory effects by Nrf2 activation [222,223,224]. Nrf2 and NF-κB individually affect other signaling cascades, in addition to crosstalk between two pathways. The absence of Nrf2 amplifies NF-κB activity, suggesting an inhibitory action of Nrf2 toward NF-κB [225], whereas NF-κB can modulate Nrf2 transcriptional activity [226]. Understanding the molecular links between these two signaling pathways may lead to more informative biomarkers. Intracellular NF-κB signaling molecules have been examined to evaluate responses for inflammatory disease in clinical studies using DMF and BARD-Me [50,66]. Other molecular mechanisms behind Nrf2-mediated anti-inflammatory effects have been described, such as transcriptional suppression of proinflammatory cytokine genes (Il-6 and Il-1β) [227] and the correlation between Nrf2 signaling and NLRP3 inflammasome activity [228].
Leaving aside the linkage of specific molecular pathways indicated above, the general approaches for monitoring anti-inflammatory responses are widely examined as well, using global inflammatory markers such as C-reactive proteins (CRP), cytokines, chemokines, inflammatory lipid mediators, and the immune cell counts.
3.5. Carcinogen Metabolites/DNA Adducts
Initial studies probing the functional consequences of enhanced Nrf2 signaling have focused on toxicological models, in which pharmacologic and genetic means were used to alter pathway flux in mice [6,229]. For example, Nrf2-deficient mice had a significantly higher burden of gastric neoplasia after treatment with benzo[a]pyrene than did wild-type mice [113]. Oltipraz significantly reduced the multiplicity of gastric neoplasia in wild-type mice but had no effect on tumor burden in Nrf2-deficient mice. A similar result was observed when SFN was used as the chemopreventive agent in this model [230]. At the same time, biomarkers based on carcinogen metabolites and excreted DNA adducts were being developed and validated as modifiable, short-term endpoints to assess the efficacy of chemopreventive interventions and for cohort selection in clinical trials [231].
Aflatoxin biomarkers were first used in 1995 as intermediate endpoints in a chemoprevention trial of oltipraz in Qidong, China, a hotspot for the development of liver cancer [100]. Aflatoxin, found in moldy corn, is a potent human hepatocarcinogen. In a placebo-controlled, double-blind study of daily oltipraz, median urinary levels of a detoxication product—aflatoxin-mercapturic acid (a glutathione conjugate derivative)—were elevated six-fold. Increased formation of aflatoxin-mercapturic acid reflects induction of aflatoxin conjugation through the actions of oltipraz on the expression of GSTs and is presumed to be a NRF2-mediated action (Figure 4). When metabolically activated, aflatoxin can also form stable covalent adducts with a lysine residue in serum albumin and the N7 atom of guanine in DNA. In as much as albumin has a circulating half-life of about 3-weeks, albumin adducts provide an integrated estimate of sub-chronic to chronic exposures to toxicants. By contrast, the DNA adduct, which rapidly depurinates from DNA and is excreted in urine, reflects exposures within the past 24 h. In the same trial, aflatoxin-albumin adducts were noted to slowly decline during the active intervention phase with oltipraz and to rebound to placebo control levels after cessation of the intervention [99].
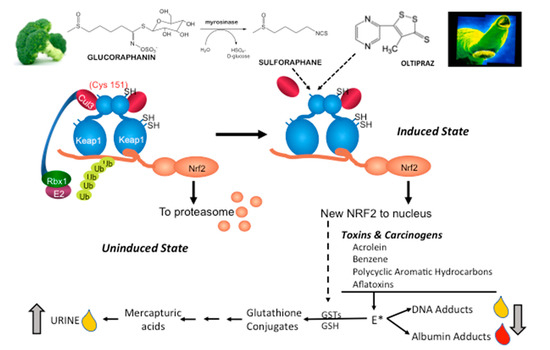
Figure 4.
Induction of KEAP1-NRF2 signaling leads to enhanced detoxication of carcinogens in clinical trials. Air-borne (e.g., acrolein, benzene, polycyclic aromatic hydrocarbons) and food-borne (e.g., aflatoxins) carcinogens are metabolized to reactive electrophiles (E*) by cytochrome P450 and other enzymes. NRF2 target genes such as GSTs can conjugate glutathione (GSH) to E*, leading to formation of nonreactive, water-soluble mercapturic acids. E* can also initiate carcinogenesis by forming promutagenic DNA adducts. Some DNA adducts undergo spontaneous or enzymatic depurination allowing for excretion in urine. E* can also form protein adducts with lysine or cysteine residues in albumin. Mercapturic acids and the adducts can be quantified in clinical samples following ambient exposures using mass spectrometric techniques. Cys, cysteine; SH, sulfhydryl; Ub, ubiquitin; Cul3, cullin 3, Rbx1, ring-box 1.
A broccoli beverage containing defined concentrations of glucoraphanin was evaluated for its ability to alter the metabolic disposition of aflatoxin [125]. In this study, 200 healthy adults drank beverages containing either 400 or <3 μmol glucoraphanin nightly for 2 weeks. Measurement of urinary levels of SFN metabolites indicated striking inter-individual differences in bioavailability, likely reflecting individual differences in the rates of hydrolysis of glucoraphanin to SFN by the intestinal microflora of the study participants. Accounting for this variability, in a secondary analysis a significant inverse association was observed for the uptake and subsequent urinary excretion of SFN and aflatoxin-N7-guanine adducts in individuals receiving broccoli sprout beverage. Changes in the metabolism of phenanthrene-tetraol were also noted in this trial.
Follow-up studies with broccoli-sprout-based interventions in Qidong demonstrated enhanced detoxication of air pollutants over one-week or three-month time frames. Higher urinary excretion levels for benzene and acrolein mercapturic acids were observed [146,154] compared to the placebo beverage. A recent dose-response study with a broccoli sprout beverage indicated that the dynamic range for induction of NRF2-driven detoxication of benzene metabolism to it mercapturic acid was rather limited in participants exposed to ambient air pollutants (~60% elevation) and saturated a relatively low level of SFN dosing [188].
In a study in which smokers were administered phenethyl isothiocyanate (PEITC), the PEITC arm reduced metabolic activation of NNK, one of the most potent lung carcinogens present in cigarettes [232]. Larger increases in rates of excretion of detoxification metabolites (often mercapturic acids) of combustion pollutants such as benzene and aldehydes were observed following PEITC intervention [232]. The mechanisms of action of the two isothiocyanates, SFN and PEITC, are not identical, but do include Nrf2 activation. In a small trial, subjects ingested 250 g each of Brussels sprouts and broccoli per day [233]. At the end of this feeding phase, subjects consumed a cooked meat meal with measured levels of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), a heterocyclic amine carcinogen formed from char-broiling meats) and urine samples were collected. Cruciferous vegetable consumption significantly increased hepatic CYP1A2, as demonstrated by changes in saliva caffeine kinetics. and significantly increased the urinary excretion of N2-hydroxy-PhIP-N2-glucuronide, another possible detoxication outcome through NRF2 induction in humans.
Many—but certainly not all—environmental carcinogens such as benzene, polycyclic aromatic hydrocarbons, and aflatoxins can be detoxified through the molecular pathways induced by agents such as oltipraz and SFN (Figure 4).
3.6. Metabolomics
Widespread application of “omic” technologies is providing precise guidance for selection of therapeutic interventions based on patient biology [234]. Metabolomics is an emerging field of “omics” that characterizes small-molecule metabolites in biological systems. Metabolomic analyses reflect both the steady-state physiological equilibrium of cells or organisms, as well as their dynamic metabolic responses to stimuli, including drugs. The opportunity to use perhaps hundreds of analytes for assessment drug pharmacodynamics, or indeed as descriptors of human health and disease, will provide greater accuracy in unraveling the complexity of human biology. Already, metabolomic profiles obtained prior to, during, or after drug treatment are used to provide insights about the drug mechanism of action and variation of response to treatment [235].
Metabolomics technologies are beginning to be applied to the discovery of biomarkers in clinical studies using NRF2 inducers. Importantly, several untargeted metabolomic studies have been performed [149,183,190]. Bent et al. [182] identified altered urinary metabolites that were correlated with changes in symptoms in patients of autism spectrum disorder by SFN treatment, and which they were clustered into pathways of oxidative stress, amino acid and gut microbiomes, neurotransmitters, hormones, and sphingomyelin metabolism.
Specific metabolite biomarkers are also widely used for monitoring physiological responses and pharmacotherapy in clinical studies. Dyslipidemia is a risk factor for cardiovascular decease and fatty liver. To examine the beneficial effect for dyslipidemia, the lipid profiles in blood samples, such as total cholesterol, triacylglycerol, and lipoproteins (low-density lipoproteins; LDL, high-density lipoproteins; HDL), are investigated in the clinical studies of SFN [122,149,151,161]. It is reported that the activation of Nrf2 represses the expression of key enzymes involved in fatty acid synthesis, with concomitant reduction in the levels of hepatic lipids in mice [236]. Kikuchi et al. indicated that intervention by SFN-rich broccoli sprout extract improved hepatic abnormalities; however, TG, HDL, and LDL-cholesterol were not changed in this study [167]. As these biomarkers are commonly used in clinical diagnostics, they have been allocated as clinical biomarkers in this article (Table S1).
The recent global approach of identifying NRF2 target genes reveals novel gene candidates, including metabolic genes [209,237,238]. In addition to GSH-metabolism-related genes, xenobiotic metabolism, lipid metabolism, glucose metabolism, and several amino acid transporter mediated genes are considered to be NRF2 target genes, which seemingly have expanded the role of NRF2 toward metabolic regulation. Integrated approaches combining metabolomics and genomics could lead to new discoveries of mechanism-based metabolite biomarkers for NRF2 inducers. NRF2 influences heme, iron, and hemoglobin metabolism in human blood cell lines. Furthermore, NRF2 regulates fetal γ-globin gene expression and fetal globin genes [239,240]. Given these findings, NRF2 activation by SFN was introduced to sickle cell disease patients, and mRNA expression of HbF and HbF protein levels was examined [172].
4. Integrated Assessment of Biomarker Outcomes
Outcomes measured in published clinical studies of NRF2 inducers were identified, listed, and categorized under pharmacokinetics or into six classes of mechanism-based biomarkers: NRF2 target genes, gene expression and function, inflammation, oxidative stress, carcinogen metabolites and adducts, and metabolomics, as well as disease-specific and clinical responses (Table S1). Using data derived from the literature summarized in Table S1, a Sankey plot (Figure 5) was developed to indicate the use and outcomes associated with the mechanistic biomarker measures reported for the four clinically used agents. Measured biomarkers were designated “+” for statistically significant or “-“ for statistically nonsignificant changes, no change, or undetectability in the Sankey diagram. In each clinical study, a biomarker category receives only one count, regardless of the number of specific biomarkers measured in that category. As an example, in the 2009 SFN clinical study by Riedl et al. [134], although several NRF2 target genes (HMOX1, NQO1, GSTP1, GSTM1) were measured, the NRF2 target gene biomarker category for SFN still receives only one count for that biomarker category. More detailed presentation of the numbers of individual biomarkers within each classification and their outcomes in the clinical trials is provided in Table 1. Although the Sankey diagram provides a somewhat dire picture of the utility of biomarker measurements for each NRF2 inducer, closer inspection of each specific biomarker presented in Table 1 provides more hopeful insights.
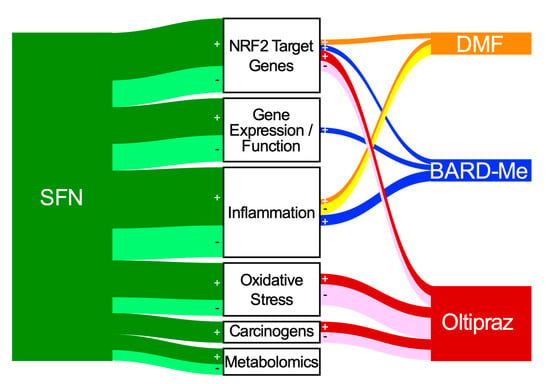
Figure 5.
Sankey diagram of mechanism-based biomarkers measured in clinical trials for SFN (green), DMF (orange), BARD-Me (blue), and oltipraz (red). Darker lines (green, orange, blue, red) marked with “+” are studies in which at least 1 biomarker was reported to exhibit a statistically significant (p < 0.05) change. Lighter lines marked with “-“ indicate nonsignificant (i.e., null) responses for all biomarkers examined within a study. This accounting overemphasizes positive outcomes. Lines emanating from each inducer are connected to different measured biomarker categories. The box height for each NRF2 inducer and the thickness of the flow lines or nodes emanating from each inducer to the biomarker categories are proportional to the number of biomarker category counts.

Table 1.
Significant vs. null (nonsignificant) outcomes of individual biomarker measures.
Overwhelmingly, SFN dominated the use of mechanistically based biomarkers in clinical studies, with 77 specific and distinct biomarkers being measured (Table 1); approximately two-thirds (53) exhibited statistically significant responses. Several were modulated significantly in multiple trials. Oltipraz studies collectively utilized only 18 biomarkers, followed by DMF and BARD-Me at 16 and 11 each, respectively.
NRF2 target gene biomarkers were the only biomarker class to be affected positively by all four agents. Nineteen NRF2 target gene biomarkers were measured for SFN, with 14 (74%) biomarker measurements exhibiting statistical significance. Half of the NRF2 biomarkers measured for oltipraz were statistically significant. Unexpectedly, both BARD-Me and DMF only had one biomarker measurement that was statistically significant. Of note, for the most studied NRF2 target gene, NQO1, transcript levels showed significant induction in 7 out of 13 settings (54%). Of other transcripts with multiple assessments, only γGCS, GCLM, and GSTP transcripts showed 50–100% of measurements as statistically significant. However, the majority of the NRF2 target genes measured exhibited changes that were statistically significant in at least one study.
SFN is the primary inducer to manifest statistically significant measurements for gene expression or function biomarkers (8 of 11), although there have been almost no attempts with the other drugs (one positive gene, cyclin D1, in a large set of cancer genes with BARD-Me). Limited assessments of signaling pathways (e.g., TGF-β, EGFR, and insulin) have been undertaken. Modulation of epigenetic marks through inhibition of HDAC activity and subsequent effects of cell proliferation and tumor development may be an important mode of action that is possibly linked to NRF2.
Only SFN and oltipraz trials elicited statistically significant changes of oxidative stress biomarkers. Most oxidative stress biomarkers (9 out of 12 biomarkers) measured in SFN clinical studies were positive; oltipraz studies were less forthcoming (2 of 6). More than half of the oxidative biomarkers exhibiting significant changes utilized either GSH, 8-OHdG, or MDA levels.
Although trials with all four inducers assessed inflammation biomarkers, SFN showed the most positive responses (19), followed by BARD-Me (3), DMF (1), and oltipraz (0). Upon closer inspection of the specific inflammation biomarkers measured, lipid mediators including PGD2, tetranor-PGEM, 11β-PGF2α, and 11-dehydro-TXB2; select chemokines and cytokines; NF-κB pathway markers; and proinflammatory aggregate transcripts appear to be useful biomarkers in these settings.
Multiple clinical trials using either SNF or oltipraz have successfully used the measurements of carcinogen metabolites or adducts to define a pharmacodynamic action. Outcomes dependent on the carcinogens targeted detoxication of air pollutants (benzene, aldehydes) and aflatoxin, which were consistently enhanced, whilst that of benzo[a]pyrene was not.
Metabolomic studies are an emerging approach to developing mechanism-based biomarkers. Within these four agents, only SFN trials have utilized these technologies, albeit with some success (3 out of 5).
5. Conclusions
5.1. Critical Path for Biomarkers in NRF2 Drug Development?
The clinical development pathways for the four “NRF2 inducers” have been distinct. DMF appears to have been appropriated for treatment of multiple sclerosis in the absence of precise mechanistic understanding of its action in humans beyond the appreciation of its immunomodulatory effects. Biomarkers were of limited use in guiding its regulatory approvals. BARD-Me was selected by Reata Pharmaceuticals as a lead compound from a large series of triterpenoids known to potently activate Nrf2 signaling in cells and animal models. However, biomarker studies in humans have again seemingly played a minor role in its promising clinical development (other than eGFR). Oltipraz was a drug originally developed by Rhône-Poulenc for the chemotherapy of schistosomiasis that was repurposed, largely through the support and guidance of the Chemoprevention Branch of the National Cancer Institute, for evaluation as a cancer chemopreventive agent. Biomarkers were central to these early studies, although issues of drug availability and difficulties related to synthesis (cost) and toxicities short-circuited its development. SFN arose out of extramurally funded academic labs, where small biomarker-driven studies have led to a large series of incremental improvements in formulation, dose selection, and identification of possible cohorts for clinical use. To date, no clinical application shows a clear path to its therapeutic or prophylactic registration. Thus, depending upon the agent, biomarkers have either been critical for their discovery and translational development or ancillary to the process, as can be inferred from the data in Figure 2B.
5.2. Metrics of Success and Confounders
As highlighted in Figure 5 and Table 1, a large range of biomarkers have been employed with somewhat limited success in clinical trials involving inducers of the NRF2 pathway. This uneven outcome arises despite many animal studies supporting roles for the pathways and processes that are measured. However, the approach herein of relying on “statistical significance” as the arbiter of a successful outcome oversimplifies the complexity of the challenge. These biomarkers have been studied in different settings with different populations (healthy and diseased), different treatment regimens (dose, schedule, and time of biomarker measurement), and different formulations, even within one agent, as well as differences in the sources of specimens interrogated (PBMCs vs. target cells). Additionally, there is a lack of consensus regarding methodologies and the extent of validation, standardization, and reproducibility of biomarker measurements in these clinical studies. There is also a three-decade time period in across which these trials have been conducted, masking underlying changes in analytic technologies applied to biomarker measures. Finally, it is evident that these compounds can affect NRF2-independent responses and pathways in humans. With this diversity of study populations, analytical methodologies and study designs, and unclear target specificity, it is difficult to reach overarching evaluations of the performance of each biomarker or the degree of association with the NRF2 pathway currently used in the clinical studies. While cognizant of these limitations, there are some comments that can be made regarding both foundational observations and gaps within the clinical studies of the current four agents thought to modulate NRF2 signaling.
5.3. Lessons from Dose-Response
Examination of dose-responses of biomarkers provides insights into associations between putative mechanisms of action and clinical outcomes. In prevention studies especially, they can help define minimally effective doses, with disease prevention being a setting in which any side-effects are unacceptable. However, to date there have been very few studies that have examined dose-response in the actions of these agents on NRF2-related biomarker levels in humans. Moreover, some of the studies that attempted such analyses resulted in no modulation of biomarkers at any dose. O’Dwyer et al. [98] did observe increases in NQO1 and γGCS transcripts in PBMCs and colon mucosa with doses of 125 to 250 mg/m2 of oltipraz. No additional increases were observed for 500 and 1000 mg/m2 doses. Only the lowest doses increased GST and NQO1 activity in PBMCs and colon mucosa; surprisingly, the higher doses were not different from baseline. Riedl et al. [134] reported graded increases in transcript levels of GSTM, GSTP, HMOX1, and NQO1 in nasal lavage samples from participants receiving 3 daily doses of 25 to 200g of a broccoli sprout homogenate to provide SFN. Maximal increases were of the magnitude of 100%. Ushida et al. [241] also reported modest dose-dependent increases in serum GST and NQO1 activity after administering volunteers low doses of 30 or 60 mg of glucoraphanin-based tablets. Lastly, Doss et al. [172] fed participants with 50, 100, or 150 g of a SFN-rich broccoli sprout homogenate and reported significant induction pre- to post-treatment for HMOX1 mRNA at 150 g, for HBG1 at 100 g, and no significant change at any dose for NQO1. Thus, dose-response data remains elusive to this point, and possibly suggests greater pharmacodynamic action at lower rather than highest doses—a so-called “∩”-shaped curve. In accord with this possibility, Chen et al. [188] reported that the enhanced excretion of benzene mercapturic acids appeared to be saturated at modest doses of a glucoraphanin + SFN-rich beverage. Given that these biomarkers likely represent direct effects on NRF2 signaling, it appears that the dynamic range for pharmacological activation of the pathway might be more limited in humans than in mice.
5.4. Take-Home Messages
Despite the variability in the biomarker responses, there are a few overarching conclusions that may be drawn regarding the approaches taken to assess the pharmacodynamic action of NRF2 inducers through the analysis of biomarkers in clinical trials.
- NRF2 Target Genes: Given the preclinical evidence that all 4 of the agents can activate Nrf2 signaling, it is comforting that all four increased activities or transcript levels of classic NRF2 target genes in clinical trials. NQO1 was most studied and showed reasonable consistency across trials. Worryingly, in most studies the induction of NQO1 transcripts exhibited a limited dynamic range (~ < 2-fold). In an oltipraz study, concordance between expression in surrogate (e.g., PBMCs) and target tissues (e.g., colonic mucosa) was reported. Limited studies suggest possible merit for PCA or clustering analyses to characterize induction “signatures” that may be more revealing than single candidate genes;
- Gene Expression/Function: Most studies here have focused on pathways affecting cancer development and progression. The most promising outcomes (with SFN) have centered on modulation of epigenetic regulators such as histone deacetylase (HDAC) and histone acetyltransferase activities. One cancer-related gene expression panel was largely unaltered in a BARD-Me study and the other two agents were not evaluated in this context;
- Oxidative Stress: Many of the workhorse biomarkers of oxidative stress have been applied to clinical studies with SFN and oltipraz, but not the other two drugs. Oxidized DNA products along with DNA strand breaks have shown protective responses in some of the interventions. Studies using the oxidation products of lipids and proteins have been more variable in their responses, although MDA looks promising. The more integrated measures of TAC, TOS, and OSI have not been revealing in limited studies. Cellular GSH levels have been measured frequently and show repeated, albeit still inconsistent, modulation by intervention;
- Inflammation: In the aggregate over 35 individual inflammation biomarkers have been measured, while barely half evoked a significant response in any study; very few have been evaluated in multiple studies. The context for selection of candidate biomarkers is rarely presented in these studies. The NRF2 target gene Il-6 [227] shows some responsiveness, while other cytokines such as IL-1, IL-8 IL-13, TNFα, and IFNγ have been null in multiple studies. Lipid mediators including PGD2, tetranor-PGEM, 11β-PGF2α, and 11-dehydro-TXB2 offer some promise. Subgroup analyses of responders only within a DMF trial exhibited significant reductions in intracellular NF-κB signaling molecules [50];
- Carcinogen Metabolism/Adducts: Monitoring detoxication metabolites following interventions in study populations provides strong links to canonical NRF2 mechanisms of action. Multiple studies in settings of unavoidable exposures to air pollution and dietary carcinogens highlight successful interventions with oltipraz and SFN. However, such studies require sophisticated mass spectrometry methodologies for metabolite, DNA adduct, and protein adduct quantification. Moreover, interception of all classes of carcinogens and toxins is not achievable. Perhaps phase 0 “microdosing” trials with small, safe amounts of heavy-isotope-labeled substrates can provide an effective means to prioritize tractable exposures [242]. As with all reviewed biomarkers, extrapolation from biomarker change to extent of risk reduction has not been realized;
- Metabolomics: Targeted and nontargeted metabolomics are beginning to be applied successfully as biomarkers in clinical trials of NRF2 inducers, albeit exclusively to date with SFN. Recent studies in mice have shown the power of these tools to define the impact of modulation of Nrf2 signaling on cancer cell metabolism [243] and the maintenance of health in space flight [244]. Combinations of omics approaches are likely to provide more integrated pictures of the actions of targeted NRF2 activation on early, intermediate, and later events on the pathways of disease prevention and mitigation.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-3921/9/8/716/s1: Table S1: Summary of literature reporting clinical studies with dimethyl fumarate, bardoxolone methyl, oltipraz, and sulforaphane. Publications including the use of pharmacodynamic biomarkers are highlighted.
Author Contributions
Conceptualization and supervision, T.W.K. and Y.Y.; review, discussion, and analysis of data, T.W.K., Y.Y., T.N.G.-S., and M.L.M.; data organization, M.L.M.; original draft preparation and writing, T.W.K., Y.Y., T.N.G.-S., and M.L.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health (R35 CA197222, to T.W.K.) the Washington State Andy Hill CARE Fund (to T.W.K.), and the Japan Society for the Promotion of Science Overseas Research Fellowships (JSPS 201860777 (to Y.Y)).
Acknowledgments
The authors thank Nobunao Wakabayashi for valuable discussions. We thank our many colleagues who have contributed to our clinical studies on oltipraz and sulforaphane, and the participants in the clinical studies noted in this review.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. A Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Stewart, D.; Hu, B.; Li, N.; Cook, J.; Nel, A.; Alam, J. Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Delta(12,14)-prostaglandin J(2) is mediated by the stress response elements and transcription factor Nrf2. Antioxid. Redox Signal. 2002, 4, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Yates, M.S.; Kensler, T.W. Chemopreventive promise of targeting the Nrf2 pathway. Drug News Perspect. 2007, 20, 109–117. [Google Scholar] [CrossRef]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of NRF2: A review of their potential for clinical development. Oxid Med. Cell Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or sulforaphane: Is it the source or dose that matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Chapman, E. The role of natural products in revealing NRF2 function. Nat. Prod. Rep. 2020, 37, 797–826. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Dimethyl fumarate: A review in moderate to severe plaque psoriasis. Drugs 2018, 78, 123–130. [Google Scholar] [CrossRef]
- Meissner, M.; Valesky, E.M.; Kippenberger, S.; Kaufmann, R. Dimethyl fumarate-only an anti-psoriatic medication? J. Dtsch. Dermatol. Ges. 2012, 10, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef]
- Mrowietz, U.; Morrison, P.J.; Suhrkamp, I.; Kumanova, M.; Clement, B. The pharmacokinetics of fumaric acid esters reveal their in vivo effects. Trends Pharmacol. Sci. 2018, 39, 1–12. [Google Scholar] [CrossRef]
- Nieboer, C.; de Hoop, D.; Langendijk, P.N.; van Loenen, A.C.; Gubbels, J. Fumaric acid therapy in psoriasis: A double-blind comparison between fumaric acid compound therapy and monotherapy with dimethylfumaric acid ester. Dermatologica 1990, 181, 33–37. [Google Scholar] [CrossRef]
- Altmeyer, P.J.; Matthes, U.; Pawlak, F.; Hoffmann, K.; Frosch, P.J.; Ruppert, P.; Wassilew, S.W.; Horn, T.; Kreysel, H.W.; Lutz, G.; et al. Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J. Am. Acad. Dermatol. 1994, 30, 977–981. [Google Scholar] [CrossRef]
- Mrowietz, U.; Christophers, E.; Altmeyer, P. Treatment of psoriasis with fumaric acid esters: Results of a prospective multicentre study. German Multicentre Study. Br. J. Dermatol. 1998, 138, 456–460. [Google Scholar] [CrossRef]
- Friedrich, M.; Sterry, W.; Klein, A.; Ruckert, R.; Docke, W.D.; Asadullah, K. Addition of pentoxifylline could reduce the side effects of fumaric acid esters in the treatment of psoriasis. Acta Derm. Venereol. 2001, 81, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, H.; Altmeyer, P.; Kaufmann, R.; Ring, J.; Christophers, E.; Pavel, S.; Ziegler, J. Topical calcipotriol plus oral fumaric acid is more effective and faster acting than oral fumaric acid monotherapy in the treatment of severe chronic plaque psoriasis vulgaris. Dermatology 2002, 205, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Stander, H.; Stadelmann, A.; Luger, T.; Traupe, H. Efficacy of fumaric acid ester monotherapy in psoriasis pustulosa palmoplantaris. Br. J. Dermatol. 2003, 149, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Carboni, I.; De Felice, C.; De Simoni, I.; Soda, R.; Chimenti, S. Fumaric acid esters in the treatment of psoriasis: An Italian experience. J. Dermatolog. Treat. 2004, 15, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Venten, I.; Hess, N.; Hirschmuller, A.; Altmeyer, P.; Brockmeyer, N. Treatment of therapy-resistant Alopecia areata with fumaric acid esters. Eur. J. Med. Res. 2006, 11, 300–305. [Google Scholar] [PubMed]
- Kappos, L.; Gold, R.; Miller, D.H.; Macmanus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Yang, M.; et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008, 372, 1463–1472. [Google Scholar] [CrossRef]
- MacManus, D.G.; Miller, D.H.; Kappos, L.; Gold, R.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Eraksoy, M.; et al. BG-12 reduces evolution of new enhancing lesions to T1-hypointense lesions in patients with multiple sclerosis. J. Neurol. 2011, 258, 449–456. [Google Scholar] [CrossRef]
- Kappos, L.; Gold, R.; Miller, D.H.; MacManus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Eraksoy, M.; et al. Effect of BG-12 on contrast-enhanced lesions in patients with relapsing--remitting multiple sclerosis: Subgroup analyses from the phase 2b study. Mult. Scler. 2012, 18, 314–321. [Google Scholar] [CrossRef]
- Sheikh, S.I.; Nestorov, I.; Russell, H.; O’Gorman, J.; Huang, R.; Milne, G.L.; Scannevin, R.H.; Novas, M.; Dawson, K.T. Tolerability and pharmacokinetics of delayed-release dimethyl fumarate administered with and without aspirin in healthy volunteers. Clin. Ther. 2013, 35, 1582–1594. [Google Scholar] [CrossRef]
- Bar-Or, A.; Gold, R.; Kappos, L.; Arnold, D.L.; Giovannoni, G.; Selmaj, K.; O’Gorman, J.; Stephan, M.; Dawson, K.T. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: Subgroup analyses of the DEFINE study. J. Neurol. 2013, 260, 2297–2305. [Google Scholar] [CrossRef]
- Hutchinson, M.; Fox, R.J.; Miller, D.H.; Phillips, J.T.; Kita, M.; Havrdova, E.; O’Gorman, J.; Zhang, R.; Novas, M.; Viglietta, V.; et al. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: Subgroup analyses of the CONFIRM study. J. Neurol. 2013, 260, 2286–2296. [Google Scholar] [CrossRef]
- Arnold, D.L.; Gold, R.; Kappos, L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Yang, M.; Zhang, R.; Stephan, M.; Sheikh, S.I.; et al. Magnetization transfer ratio in the delayed-release dimethyl fumarate DEFINE study. J. Neurol. 2014, 261, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.L.; Gold, R.; Kappos, L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Yang, M.; Zhang, R.; Stephan, M.; Sheikh, S.I.; et al. Effects of delayed-release dimethyl fumarate on MRI measures in the Phase 3 DEFINE study. J. Neurol. 2014, 261, 1794–1802. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Gold, R.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Sarda, S.P.; Agarwal, S.; Zhang, A.; Sheikh, S.I.; et al. Quality of life outcomes with BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: The Define study. Mult. Scler. 2014, 20, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Kita, M.; Fox, R.J.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Sarda, S.P.; Agarwal, S.; Kong, J.; Zhang, A.; Viglietta, V.; et al. Effects of BG-12 (dimethyl fumarate) on health-related quality of life in patients with relapsing-remitting multiple sclerosis: Findings from the CONFIRM study. Mult. Scler. 2014, 20, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Fox, R.J.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Wheeler-Kingshott, C.A.; Tozer, D.J.; MacManus, D.G.; Yousry, T.A.; et al. Effects of delayed-release dimethyl fumarate on MRI measures in the phase 3 CONFIRM study. Neurology 2015, 84, 1145–1152. [Google Scholar] [CrossRef]
- Kappos, L.; Giovannoni, G.; Gold, R.; Phillips, J.T.; Arnold, D.L.; Hotermans, C.; Zhang, A.; Viglietta, V.; Fox, R.J.; DEFINE and CONFIRM study investigators. Time course of clinical and neuroradiological effects of delayed-release dimethyl fumarate in multiple sclerosis. Eur. J. Neurol. 2015, 22, 664–671. [Google Scholar] [CrossRef]
- Freedman, M.S.; Montalban, X.; Miller, A.E.; Dive-Pouletty, C.; Hass, S.; Thangavelu, K.; Leist, T.P. Comparing outcomes from clinical studies of oral disease-modifying therapies (dimethyl fumarate, fingolimod, and teriflunomide) in relapsing MS: Assessing absolute differences using a number needed to treat analysis. Mult. Scler. Relat. Disord. 2016, 10, 204–212. [Google Scholar] [CrossRef][Green Version]
- Lijnen, R.; Otters, E.; Balak, D.; Thio, B. Long-term safety and effectiveness of high-dose dimethylfumarate in the treatment of moderate to severe psoriasis: A prospective single-blinded follow-up study. J. Dermatolog. Treat. 2016, 27, 31–36. [Google Scholar] [CrossRef]
- Zhu, B.; Nestorov, I.; Zhao, G.; Meka, V.; Leahy, M.; Kam, J.; Sheikh, S.I. Evaluation of potential drug-drug interaction between delayed-release dimethyl fumarate and a commonly used oral contraceptive (norgestimate/ethinyl estradiol) in healthy women. Clin. Pharmacol. Drug Dev. 2017, 6, 604–613. [Google Scholar] [CrossRef]
- Fernandez, O.; Giovannoni, G.; Fox, R.J.; Gold, R.; Phillips, J.T.; Potts, J.; Okwuokenye, M.; Marantz, J.L. Efficacy and safety of delayed-release dimethyl fumarate for relapsing-remitting multiple sclerosis in prior interferon users: An integrated nalysis of DEFINE and CONFIRM. Clin. Ther. 2017, 39, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Cutter, G.; Fox, R.J.; Xiao, J.; Lewin, J.B.; Edwards, M.R. Comparative effectiveness of delayed-release dimethyl fumarate versus glatiramer acetate in multiple sclerosis patients: Results of a matching-adjusted indirect comparison. J. Comp. Eff. Res. 2017, 6, 313–323. [Google Scholar] [CrossRef]
- Havrdova, E.; Giovannoni, G.; Gold, R.; Fox, R.J.; Kappos, L.; Phillips, J.T.; Okwuokenye, M.; Marantz, J.L. Effect of delayed-release dimethyl fumarate on no evidence of disease activity in relapsing-remitting multiple sclerosis: Integrated analysis of the phase III DEFINE and CONFIRM studies. Eur. J. Neurol. 2017, 24, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the Phase 3 DEFINE and CONFIRM studies. Mult. Scler. 2017, 23, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Weisenseel, P.; Reich, K.; Griemberg, W.; Merten, K.; Groschel, C.; Gomez, N.N.; Taipale, K.; Brau, B.; Zschocke, I. Efficacy and safety of fumaric acid esters in combination with phototherapy in patients with moderate-to-severe plaque psoriasis (FAST). J. Dtsch. Dermatol. Ges. 2017, 15, 180–186. [Google Scholar] [CrossRef]
- Mrowietz, U.; Szepietowski, J.C.; Loewe, R.; van de Kerkhof, P.; Lamarca, R.; Ocker, W.G.; Tebbs, V.M.; Pau-Charles, I. Efficacy and safety of LAS41008 (dimethyl fumarate) in adults with moderate-to-severe chronic plaque psoriasis: A randomized, double-blind, Fumaderm((R))-and placebo-controlled trial (BRIDGE). Br. J. Dermatol. 2017, 176, 615–623. [Google Scholar] [CrossRef]
- Gold, R.; Arnold, D.L.; Bar-Or, A.; Hutchinson, M.; Kappos, L.; Havrdova, E.; MacManus, D.G.; Yousry, T.A.; Pozzilli, C.; Selmaj, K.; et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult. Scler. 2017, 23, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Koulinska, I.; Riester, K.; Chalkias, S.; Edwards, M.R. Effect of bismuth subsalicylate on gastrointestinal tolerability in healthy volunteers receiving oral delayed-release dimethyl fumarate: PREVENT, a randomized, multicenter, double-blind, placebo-controlled study. Clin. Ther. 2018, 40, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Ochi, H.; Niino, M.; Onizuka, Y.; Hiramatsu, K.; Hase, M.; Yun, J.; Matta, A.; Torii, S. 72-week safety and tolerability of dimethyl fumarate in Japanese patients with relapsing-remitting multiple sclerosis: Analysis of the randomised, double blind, placebo-controlled, phase III APEX study and its open-label extension. Adv. Ther. 2018, 35, 1598–1611. [Google Scholar] [CrossRef]
- Braley, T.J.; Huber, A.K.; Segal, B.M.; Kaplish, N.; Saban, R.; Washnock-Schmid, J.M.; Chervin, R.D. A randomized, subject and rater-blinded, placebo-controlled trial of dimethyl fumarate for obstructive sleep apnea. Sleep 2018, 41. [Google Scholar] [CrossRef]
- Prosperini, L.; Lucchini, M.; Haggiag, S.; Bellantonio, P.; Bianco, A.; Buscarinu, M.C.; Buttari, F.; Centonze, D.; Cortese, A.; De Giglio, L.; et al. Fingolimod vs. dimethyl fumarate in multiple sclerosis: A real-world propensity score-matched study. Neurology 2018, 91, e153–e161. [Google Scholar] [CrossRef] [PubMed]
- Alroughani, R.; Das, R.; Penner, N.; Pultz, J.; Taylor, C.; Eraly, S. Safety and efficacy of delayed-release dimethyl fumarate in pediatric patients with relapsing multiple sclerosis (FOCUS). Pediatr. Neurol. 2018, 83, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Cohan, S.L.; Moses, H.; Calkwood, J.; Tornatore, C.; LaGanke, C.; Smoot, K.E.; Meka, V.; Okwuokenye, M.; Hotermans, C.; Mendoza, J.P.; et al. Clinical outcomes in patients with relapsing-remitting multiple sclerosis who switch from natalizumab to delayed-release dimethyl fumarate: A multicenter retrospective observational study (STRATEGY). Mult. Scler. Relat. Disord. 2018, 22, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Sator, P.; Loewe, R.; Zamani, O.; Holzer, G.; Wolf, P.; Mlynek, A.; Berger, T.; Richter, L.; Schuller, E. Dimethyl fumarate is efficacious in severe plaque psoriasis: Post hoc analysis from the BRIDGE trial in Austria. Wien. Klin. Wochenschr. 2019, 131, 485–492. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef]
- Saida, T.; Yamamura, T.; Kondo, T.; Yun, J.; Yang, M.; Li, J.; Mahadavan, L.; Zhu, B.; Sheikh, S.I. A randomized placebo-controlled trial of delayed-release dimethyl fumarate in patients with relapsing-remitting multiple sclerosis from East Asia and other countries. BMC Neurol. 2019, 19, 5. [Google Scholar] [CrossRef]
- Spencer, C.A.; Groudine, M. Molecular analysis of the c-myc transcription elongation block. Implications for the generation of Burkitt’s lymphoma. Ann. N. Y. Acad. Sci. 1990, 599, 12–28. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef]
- Ahuja, M.; Ammal Kaidery, N.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 signaling mechanisms of fumaric acid esters and their role in neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced experimental Parkinson’s-like disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T.; Sporn, M.B. Synthetic oleanane triterpenoids: Multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef] [PubMed]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and anticancer activity of CDDO and CDDO-Me, two derivatives of natural triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Krauth, M.; Huff, J.W.; Ferguson, D.A.; Ruiz, S.; Meyer, C.J.; Warnock, D.G. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am. J. Nephrol. 2011, 33, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef]
- Lambers Heerspink, H.J.; Chertow, G.M.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Goldsberry, A.; Krauth, M.; Linde, P.; McMurray, J.J.; Meyer, C.J.; et al. Baseline characteristics in the Bardoxolone methyl EvAluation in patients with Chronic kidney disease and type 2 diabetes mellitus: The Occurrence of renal eveNts (BEACON) trial. Nephrol. Dial. Transplant. 2013, 28, 2841–2850. [Google Scholar] [CrossRef]
- Teuscher, N.S.; Kelley, R.J.; Dumas, E.O.; Klein, C.E.; Awni, W.M.; Meyer, C.J. A food effect study and dose proportionality study to assess the pharmacokinetics and safety of bardoxolone methyl in healthy volunteers. Clin. Pharmacol. Drug Dev. 2014, 3, 314–320. [Google Scholar] [CrossRef]
- Chin, M.P.; Reisman, S.A.; Bakris, G.L.; O’Grady, M.; Linde, P.G.; McCullough, P.A.; Packham, D.; Vaziri, N.D.; Ward, K.W.; Warnock, D.G.; et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 2014, 39, 499–508. [Google Scholar] [CrossRef]
- Chin, M.P.; Wrolstad, D.; Bakris, G.L.; Chertow, G.M.; de Zeeuw, D.; Goldsberry, A.; Linde, P.G.; McCullough, P.A.; McMurray, J.J.; Wittes, J.; et al. Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J. Card. Fail. 2014, 20, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Appel, G.B.; Block, G.A.; Chin, M.P.; Coyne, D.W.; Goldsberry, A.; Kalantar-Zadeh, K.; Meyer, C.J.; Molitch, M.E.; Pergola, P.E.; et al. Effects of bardoxolone methyl on body weight, waist circumference and glycemic control in obese patients with type 2 diabetes mellitus and stage 4 chronic kidney disease. J. Diabetes Complicat. 2018, 32, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Bakris, G.L.; Block, G.A.; Chertow, G.M.; Goldsberry, A.; Inker, L.A.; Heerspink, H.J.L.; O’Grady, M.; Pergola, P.E.; Wanner, C.; et al. Bardoxolone methyl improves kidney function in patients with chronic kidney disease stage 4 and type 2 diabetes: Post-hoc analyses from bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes study. Am. J. Nephrol. 2018, 47, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Rizk, D.V.; Silva, A.L.; Pergola, P.E.; Toto, R.; Warnock, D.G.; Chin, M.P.; Goldsberry, A.; O’Grady, M.; Meyer, C.J.; McCullough, P.A. Effects of bardoxolone methyl on magnesium in patients with type 2 diabetes mellitus and chronic kidney disease. Cardiorenal Med. 2019, 9, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Rich, S.; Goldsberry, A.; Grady, M.O.A.; Meyer, C.J. Effects of bardoxolone methyl on QT interval in healthy volunteers. Cardiorenal Med. 2019, 9, 326–333. [Google Scholar] [CrossRef]
- Yamawaki, K.; Kanda, H.; Shimazaki, R. Nrf2 activator for the treatment of kidney diseases. Toxicol. Appl. Pharmacol. 2018, 360, 30–37. [Google Scholar] [CrossRef]
- Liu, M.; Reddy, N.M.; Higbee, E.M.; Potteti, H.R.; Noel, S.; Racusen, L.; Kensler, T.W.; Sporn, M.B.; Reddy, S.P.; Rabb, H. The Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys from ischemia-reperfusion injury in mice. Kidney Int. 2014, 85, 134–141. [Google Scholar] [CrossRef]
- Wu, J.; Liu, X.; Fan, J.; Chen, W.; Wang, J.; Zeng, Y.; Feng, X.; Yu, X.; Yang, X. Bardoxolone methyl (BARD) ameliorates aristolochic acid (AA)-induced acute kidney injury through Nrf2 pathway. Toxicology 2014, 318, 22–31. [Google Scholar] [CrossRef]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef]
- Johnson, N.M.; Egner, P.A.; Baxter, V.K.; Sporn, M.B.; Wible, R.S.; Sutter, T.R.; Groopman, J.D.; Kensler, T.W.; Roebuck, B.D. Complete protection against aflatoxin B(1)-induced liver cancer with a triterpenoid: DNA adduct dosimetry, molecular signature, and genotoxicity threshold. Cancer Prev. Res. 2014, 7, 658–665. [Google Scholar] [CrossRef]
- Taguchi, K.; Takaku, M.; Egner, P.A.; Morita, M.; Kaneko, T.; Mashimo, T.; Kensler, T.W.; Yamamoto, M. Generation of a new model rat: Nrf2 knockout rats are sensitive to aflatoxin B1 toxicity. Toxicol. Sci. 2016, 152, 40–52. [Google Scholar] [CrossRef] [PubMed]
- To, C.; Ringelberg, C.S.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Sporn, M.B.; Liby, K.T. Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis 2015, 36, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.S.; Bhandari, R.; Torres, G.M.; Martyanov, V.; ElTanbouly, M.A.; Archambault, K.; Whitfield, M.L.; Liby, K.T.; Pioli, P.A. CDDO-Me alters the tumor microenvironment in estrogen receptor negative breast cancer. Sci. Rep. 2020, 10, 6560. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.S.; Shipman, E.P.; Kim, H.; Liby, K.T.; Pioli, P.A. CDDO-Me redirects activation of breast tumor associated macrophages. PLoS ONE 2016, 11, e0149600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar] [CrossRef]
- Bella, H.; Rahim, A.G.; Mustafa, M.D.; Ahmed, M.A.; Wasfi, S.; Bennett, J.L. Oltipraz—Antischistosomal efficacy in Sudanese infected with Schistosoma mansoni. Am. J. Trop. Med. Hyg. 1982, 31, 775–778. [Google Scholar] [CrossRef]
- Burchard, G.D.; Kern, P.; Baltes, R.; Dietrich, M. Comparative trial of oltipraz versus praziquantel in the treatment of urinary schistosomiasis in the Gabon. Tropenmed. Parasitol. 1984, 35, 91–94. [Google Scholar]
- Kern, P.; Burchard, G.D.; Dietrich, M. Comparative study of oltipraz versus praziquantel for treatment of schistosomiasis with intestinal manifestation in the Gabon (Schistosoma intercalatum and S. haematobium). Tropenmed. Parasitol. 1984, 35, 95–99. [Google Scholar]
- el Igail, A.B.; el Tayeb, M.; Kardaman, M.W.; Daffalla, A.A.; Dixon, H.G.; Fenwick, A. Dose-finding trial using Oltipraz to treat schoolchildren infected with Schistosoma mansoni in Gezira, Sudan. J. Trop. Med. Hyg. 1985, 88, 101–104. [Google Scholar]
- Kardaman, M.W.; Fenwick, A.; el Igail, A.B.; el Tayeb, M.; Bennett, J.L.; Daffalla, A.A. Field trials with Oltipraz against Schistosoma mansoni in the Gezira Irrigated Area, Sudan. J. Trop. Med. Hyg. 1985, 88, 95–100. [Google Scholar]
- el Tayeb, M.; Daffalla, A.A.; Kardaman, M.W.; See, R.; Fenwick, A. Praziquantel and oltipraz: The treatment of schoolchildren infected with Schistosoma mansoni and/or Schistosoma haematobium in Gezira, Sudan. Ann. Trop. Med. Parasitol. 1988, 82, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Bueding, E.; Dolan, P.; Leroy, J.P. The antischistosomal activity of oltipraz. Res. Commun. Chem. Pathol. Pharmacol. 1982, 37, 293–303. [Google Scholar] [PubMed]
- Ansher, S.S.; Dolan, P.; Bueding, E. Biochemical effects of dithiolthiones. Food Chem. Toxicol. 1986, 24, 405–415. [Google Scholar] [CrossRef]
- Ansher, S.S.; Dolan, P.; Bueding, E. Chemoprotective effects of two dithiolthiones and of butylhydroxyanisole against carbon tetrachloride and acetaminophen toxicity. Hepatology 1983, 3, 932–935. [Google Scholar] [CrossRef]
- Kelloff, G.J.; Boone, C.W.; Steele, V.E.; Fay, J.R.; Lubet, R.A.; Crowell, J.A.; Sigman, C.C. Mechanistic considerations in chemopreventive drug development. J. Cell Biochem. Suppl. 1994, 20, 1–24. [Google Scholar] [CrossRef]
- Kensler, T.W.; Groopman, J.D.; Sutter, T.R.; Curphey, T.J.; Roebuck, B.D. Development of cancer chemopreventive agents: Oltipraz as a paradigm. Chem. Res. Toxicol. 1999, 12, 113–126. [Google Scholar] [CrossRef]
- Ali, H.M.; Homeida, M.M.; Sulaiman, S.M.; Bennett, J.L. Diet-controlled blood levels of oltipraz in healthy male subjects. J. Antimicrob. Chemother. 1984, 13, 465–470. [Google Scholar] [CrossRef]
- O’Dwyer, P.J.; Szarka, C.E.; Yao, K.S.; Halbherr, T.C.; Pfeiffer, G.R.; Green, F.; Gallo, J.M.; Brennan, J.; Frucht, H.; Goosenberg, E.B.; et al. Modulation of gene expression in subjects at risk for colorectal cancer by the chemopreventive dithiolethione oltipraz. J. Clin. Investig. 1996, 98, 1210–1217. [Google Scholar] [CrossRef]
- Kensler, T.W.; He, X.; Otieno, M.; Egner, P.A.; Jacobson, L.P.; Chen, B.; Wang, J.S.; Zhu, Y.R.; Zhang, B.C.; Wang, J.B.; et al. Oltipraz chemoprevention trial in Qidong, People’s Republic of China: Modulation of serum aflatoxin albumin adduct biomarkers. Cancer Epidemiol. Biomark. Prev. 1998, 7, 127–134. [Google Scholar]
- Wang, J.S.; Shen, X.; He, X.; Zhu, Y.R.; Zhang, B.C.; Wang, J.B.; Qian, G.S.; Kuang, S.Y.; Zarba, A.; Egner, P.A.; et al. Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China. J. Natl. Cancer Inst. 1999, 91, 347–354. [Google Scholar] [CrossRef]
- Benson, A.B., III; Olopade, O.I.; Ratain, M.J.; Rademaker, A.; Mobarhan, S.; Stucky-Marshall, L.; French, S.; Dolan, M.E. Chronic daily low dose of 4-methyl-5-(2-pyrazinyl)-1,2-dithiole-3-thione (Oltipraz) in patients with previously resected colon polyps and first degree female relatives of breast cancer patients. Clin. Cancer Res. 2000, 6, 3870–3877. [Google Scholar] [PubMed]
- O’Dwyer, P.J.; Szarka, C.; Brennan, J.M.; Laub, P.B.; Gallo, J.M. Pharmacokinetics of the chemopreventive agent oltipraz and of its metabolite M3 in human subjects after a single oral dose. Clin. Cancer Res. 2000, 6, 4692–4696. [Google Scholar]
- Camoirano, A.; Bagnasco, M.; Bennicelli, C.; Cartiglia, C.; Wang, J.B.; Zhang, B.C.; Zhu, Y.R.; Qian, G.S.; Egner, P.A.; Jacobson, L.P.; et al. Oltipraz chemoprevention trial in Qidong, People’s Republic of China: Results of urine genotoxicity assays as related to smoking habits. Cancer Epidemiol. Biomark. Prev. 2001, 10, 775–783. [Google Scholar]
- Pendyala, L.; Schwartz, G.; Bolanowska-Higdon, W.; Hitt, S.; Zdanowicz, J.; Murphy, M.; Lawrence, D.; Creaven, P.J. Phase I/pharmacodynamic study of N-acetylcysteine/oltipraz in smokers: Early termination due to excessive toxicity. Cancer Epidemiol. Biomark. Prev. 2001, 10, 269–272. [Google Scholar]
- Dimitrov, N.V.; Leece, C.M.; Tompkins, E.R.; Seymour, E.; Bennink, M.; Gardiner, J.; Crowell, J.; Hawk, E.; Nashawaty, M.; Bennett, J.L. Oltipraz concentrations in plasma, buccal mucosa cells, and lipids: Pharmacological studies. Cancer Epidemiol. Biomark. Prev. 2001, 10, 201–207. [Google Scholar]
- Szarka, C.E.; Yao, K.S.; Pfeiffer, G.R.; Balshem, A.M.; Litwin, S.; Frucht, H.; Goosenberg, E.B.; Engstrom, P.F.; Clapper, M.L.; O’Dwyer, P.J. Chronic dosing of oltipraz in people at increased risk for colorectal cancer. Cancer Detect. Prev. 2001, 25, 352–361. [Google Scholar]
- Kelley, M.J.; Glaser, E.M.; Herndon, J.E., II; Becker, F.; Bhagat, R.; Zhang, Y.J.; Santella, R.M.; Carmella, S.G.; Hecht, S.S.; Gallot, L.; et al. Safety and efficacy of weekly oral oltipraz in chronic smokers. Cancer Epidemiol. Biomark. Prev. 2005, 14, 892–899. [Google Scholar] [CrossRef]
- Glintborg, B.; Weimann, A.; Kensler, T.W.; Poulsen, H.E. Oltipraz chemoprevention trial in Qidong, People’s Republic of China: Unaltered oxidative biomarkers. Free Radic. Biol. Med. 2006, 41, 1010–1014. [Google Scholar] [CrossRef]
- Kim, S.G.; Kim, Y.M.; Choi, Y.H.; Lee, M.G.; Choi, J.Y.; Han, J.Y.; Cho, S.H.; Jang, J.W.; Um, S.H.; Chon, C.Y.; et al. Pharmacokinetics of oltipraz and its major metabolite (RM) in patients with liver fibrosis or cirrhosis: Relationship with suppression of circulating TGF-beta1. Clin. Pharmacol Ther. 2010, 88, 360–368. [Google Scholar] [CrossRef]
- Kim, S.G.; Kim, Y.M.; Choi, J.Y.; Han, J.Y.; Jang, J.W.; Cho, S.H.; Um, S.H.; Chon, C.Y.; Lee, D.H.; Jang, J.J.; et al. Oltipraz therapy in patients with liver fibrosis or cirrhosis: A randomized, double-blind, placebo-controlled phase II trial. J. Pharm. Pharmacol. 2011, 63, 627–635. [Google Scholar] [CrossRef]
- Kim, W.; Kim, B.G.; Lee, J.S.; Lee, C.K.; Yeon, J.E.; Chang, M.S.; Kim, J.H.; Kim, H.; Yi, S.; Lee, J.; et al. Randomised clinical trial: The efficacy and safety of oltipraz, a liver X receptor alpha-inhibitory dithiolethione in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 45, 1073–1083. [Google Scholar] [CrossRef]
- Egner, P.A.; Kensler, T.W.; Prestera, T.; Talalay, P.; Libby, A.H.; Joyner, H.H.; Curphey, T.J. Regulation of phase 2 enzyme induction by oltipraz and other dithiolethiones. Carcinogenesis 1994, 15, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Interactive effects of Nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis 2003, 24, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. [Google Scholar] [CrossRef]
- Fahey, J.W.; Zhang, Y.; Talalay, P. Broccoli sprouts: An exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc. Natl. Acad. Sci. USA 1997, 94, 10367–10372. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol. Biomark. Prev. 1998, 7, 1091–1100. [Google Scholar]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and done? Targeting the NRF2 pathway with sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef]
- Conaway, C.C.; Getahun, S.M.; Liebes, L.L.; Pusateri, D.J.; Topham, D.K.; Botero-Omary, M.; Chung, F.L. Disposition of glucosinolates and sulforaphane in humans after ingestion of steamed and fresh broccoli. Nutr. Cancer 2000, 38, 168–178. [Google Scholar] [CrossRef]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta 2002, 316, 43–53. [Google Scholar] [CrossRef]
- Murashima, M.; Watanabe, S.; Zhuo, X.G.; Uehara, M.; Kurashige, A. Phase 1 study of multiple biomarkers for metabolism and oxidative stress after one-week intake of broccoli sprouts. Biofactors 2004, 22, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Galan, M.V.; Kishan, A.A.; Silverman, A.L. Oral broccoli sprouts for the treatment of Helicobacter pylori infection: A preliminary report. Dig. Dis. Sci. 2004, 49, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Gasper, A.V.; Al-Janobi, A.; Smith, J.A.; Bacon, J.R.; Fortun, P.; Atherton, C.; Taylor, M.A.; Hawkey, C.J.; Barrett, D.A.; Mithen, R.F. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am. J. Clin. Nutr. 2005, 82, 1283–1291. [Google Scholar] [CrossRef]
- Kensler, T.W.; Chen, J.G.; Egner, P.A.; Fahey, J.W.; Jacobson, L.P.; Stephenson, K.K.; Ye, L.; Coady, J.L.; Wang, J.B.; Wu, Y.; et al. Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People’s Republic of China. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2605–2613. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Fahey, J.W.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Stephenson, K.K.; Wade, K.L.; Ye, L.; Talalay, P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: A clinical phase I study. Nutr. Cancer 2006, 55, 53–62. [Google Scholar] [CrossRef]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.S.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef]
- Gasper, A.V.; Traka, M.; Bacon, J.R.; Smith, J.A.; Taylor, M.A.; Hawkey, C.J.; Barrett, D.A.; Mithen, R.F. Consuming broccoli does not induce genes associated with xenobiotic metabolism and cell cycle control in human gastric mucosa. J. Nutr. 2007, 137, 1718–1724. [Google Scholar] [CrossRef]
- Myzak, M.C.; Tong, P.; Dashwood, W.M.; Dashwood, R.H.; Ho, E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp. Biol. Med. 2007, 232, 227–234. [Google Scholar]
- Rungapamestry, V.; Duncan, A.J.; Fuller, Z.; Ratcliffe, B. Effect of meal composition and cooking duration on the fate of sulforaphane following consumption of broccoli by healthy human subjects. Br. J. Nutr. 2007, 97, 644–652. [Google Scholar] [CrossRef]
- Traka, M.; Gasper, A.V.; Melchini, A.; Bacon, J.R.; Needs, P.W.; Frost, V.; Chantry, A.; Jones, A.M.; Ortori, C.A.; Barrett, D.A.; et al. Broccoli consumption interacts with GSTM1 to perturb oncogenic signalling pathways in the prostate. PLoS ONE 2008, 3, e2568. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M.; Klopping-Ketelaars, I.W.; van den Berg, R.; Vaes, W.H. Bioavailability and kinetics of sulforaphane in humans after consumption of cooked versus raw broccoli. J. Agric. Food Chem. 2008, 56, 10505–10509. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, N.; Coldham, N.; Gielbert, A.; Sauer, M.J.; Ioannides, C. Repeated intake of broccoli does not lead to higher plasma levels of sulforaphane in human volunteers. Cancer Lett. 2009, 284, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Riedl, M.A.; Saxon, A.; Diaz-Sanchez, D. Oral sulforaphane increases Phase II antioxidant enzymes in the human upper airway. Clin. Immunol. 2009, 130, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Riso, P.; Martini, D.; Visioli, F.; Martinetti, A.; Porrini, M. Effect of broccoli intake on markers related to oxidative stress and cancer risk in healthy smokers and nonsmokers. Nutr. Cancer 2009, 61, 232–237. [Google Scholar] [CrossRef]
- Yanaka, A.; Fahey, J.W.; Fukumoto, A.; Nakayama, M.; Inoue, S.; Zhang, S.; Tauchi, M.; Suzuki, H.; Hyodo, I.; Yamamoto, M. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori-infected mice and humans. Cancer Prev. Res. 2009, 2, 353–360. [Google Scholar] [CrossRef]
- Christiansen, B.; Bellostas Muguerza, N.; Petersen, A.M.; Kveiborg, B.; Madsen, C.R.; Thomas, H.; Ihlemann, N.; Sorensen, J.C.; Kober, L.; Sorensen, H.; et al. Ingestion of broccoli sprouts does not improve endothelial function in humans with hypertension. PLoS ONE 2010, 5, e12461. [Google Scholar] [CrossRef]
- Bahadoran, Z.; Mirmiran, P.; Hosseinpanah, F.; Hedayati, M.; Hosseinpour-Niazi, S.; Azizi, F. Broccoli sprouts reduce oxidative stress in type 2 diabetes: A randomized double-blind clinical trial. Eur. J. Clin. Nutr. 2011, 65, 972–977. [Google Scholar] [CrossRef]
- Hauder, J.; Winkler, S.; Bub, A.; Rufer, C.E.; Pignitter, M.; Somoza, V. LC-MS/MS quantification of sulforaphane and indole-3-carbinol metabolites in human plasma and urine after dietary intake of selenium-fortified broccoli. J. Agric. Food Chem. 2011, 59, 8047–8057. [Google Scholar] [CrossRef]
- Clarke, J.D.; Riedl, K.; Bella, D.; Schwartz, S.J.; Stevens, J.F.; Ho, E. Comparison of isothiocyanate metabolite levels and histone deacetylase activity in human subjects consuming broccoli sprouts or broccoli supplement. J. Agric. Food Chem. 2011, 59, 10955–10963. [Google Scholar] [CrossRef]
- Egner, P.A.; Chen, J.G.; Wang, J.B.; Wu, Y.; Sun, Y.; Lu, J.H.; Zhu, J.; Zhang, Y.H.; Chen, Y.S.; Friesen, M.D.; et al. Bioavailability of sulforaphane from two broccoli sprout beverages: Results of a short-term, cross-over clinical trial in Qidong, China. Cancer Prev. Res. 2011, 4, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Healy, Z.R.; Liu, H.; Holtzclaw, W.D.; Talalay, P. Inactivation of tautomerase activity of macrophage migration inhibitory factor by sulforaphane: A potential biomarker for anti-inflammatory intervention. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Bahadoran, Z.; Tohidi, M.; Nazeri, P.; Mehran, M.; Azizi, F.; Mirmiran, P. Effect of broccoli sprouts on insulin resistance in type 2 diabetic patients: A randomized double-blind clinical trial. Int. J. Food Sci. Nutr. 2012, 63, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.M.; Teran-Garcia, M.; Jeffery, E.H. Enhancing sulforaphane absorption and excretion in healthy men through the combined consumption of fresh broccoli sprouts and a glucoraphanin-rich powder. Br. J. Nutr. 2012, 107, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Wehage, S.L.; Holtzclaw, W.D.; Kensler, T.W.; Egner, P.A.; Shapiro, T.A.; Talalay, P. Protection of humans by plant glucosinolates: Efficiency of conversion of glucosinolates to isothiocyanates by the gastrointestinal microflora. Cancer Prev. Res. 2012, 5, 603–611. [Google Scholar] [CrossRef]
- Kensler, T.W.; Ng, D.; Carmella, S.G.; Chen, M.; Jacobson, L.P.; Munoz, A.; Egner, P.A.; Chen, J.G.; Qian, G.S.; Chen, T.Y.; et al. Modulation of the metabolism of airborne pollutants by glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages in Qidong, China. Carcinogenesis 2012, 33, 101–107. [Google Scholar] [CrossRef]
- Mirmiran, P.; Bahadoran, Z.; Hosseinpanah, F.; Keyzad, A.; Azizi, F. Effects of broccoli sprout with high sulforaphane concentration on inflammatory markers in type 2 diabetic patients: A randomized double-blind placebo-controlled clinical trial. J. Funct. Foods 2012, 4, 837–841. [Google Scholar] [CrossRef]
- Saha, S.; Hollands, W.; Teucher, B.; Needs, P.W.; Narbad, A.; Ortori, C.A.; Barrett, D.A.; Rossiter, J.T.; Mithen, R.F.; Kroon, P.A. Isothiocyanate concentrations and interconversion of sulforaphane to erucin in human subjects after consumption of commercial frozen broccoli compared to fresh broccoli. Mol. Nutr. Food Res. 2012, 56, 1906–1916. [Google Scholar] [CrossRef]
- Armah, C.N.; Traka, M.H.; Dainty, J.R.; Defernez, M.; Janssens, A.; Leung, W.; Doleman, J.F.; Potter, J.F.; Mithen, R.F. A diet rich in high-glucoraphanin broccoli interacts with genotype to reduce discordance in plasma metabolite profiles by modulating mitochondrial function. Am. J. Clin. Nutr. 2013, 98, 712–722. [Google Scholar] [CrossRef]
- Meyer, M.; Kesic, M.J.; Clarke, J.; Ho, E.; Simmen, R.C.; Diaz-Sanchez, D.; Noah, T.L.; Jaspers, I. Sulforaphane induces SLPI secretion in the nasal mucosa. Respir. Med. 2013, 107, 472–475. [Google Scholar] [CrossRef]
- Poulton, E.J.; Levy, L.; Lampe, J.W.; Shen, D.D.; Tracy, J.; Shuhart, M.C.; Thummel, K.E.; Eaton, D.L. Sulforaphane is not an effective antagonist of the human pregnane X-receptor in vivo. Toxicol. Appl. Pharmacol. 2013, 266, 122–131. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bahadoran, Z.; Mirmiran, P.; Yeganeh, M.Z.; Hosseinpanah, F.; Zojaji, H.; Azizi, F. Complementary and alternative medicinal effects of broccoli sprouts powder on Helicobacter pylori eradication rate in type 2 diabetic patients: A randomized clinical trial. J. Funct. Foods 2014, 7, 390–397. [Google Scholar] [CrossRef]
- Baier, S.R.; Zbasnik, R.; Schlegel, V.; Zempleni, J. Off-target effects of sulforaphane include the derepression of long terminal repeats through histone acetylation events. J. Nutr. Biochem. 2014, 25, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Chen, J.G.; Zarth, A.T.; Ng, D.K.; Wang, J.B.; Kensler, K.H.; Jacobson, L.P.; Munoz, A.; Johnson, J.L.; Groopman, J.D.; et al. Rapid and sustainable detoxication of airborne pollutants by broccoli sprout beverage: Results of a randomized clinical trial in China. Cancer Prev. Res. 2014, 7, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Heber, D.; Li, Z.; Garcia-Lloret, M.; Wong, A.M.; Lee, T.Y.; Thames, G.; Krak, M.; Zhang, Y.; Nel, A. Sulforaphane-rich broccoli sprout extract attenuates nasal allergic response to diesel exhaust particles. Food Funct. 2014, 5, 35–41. [Google Scholar] [CrossRef]
- Noah, T.L.; Zhang, H.; Zhou, H.; Glista-Baker, E.; Muller, L.; Bauer, R.N.; Meyer, M.; Murphy, P.C.; Jones, S.; Letang, B.; et al. Effect of broccoli sprouts on nasal response to live attenuated influenza virus in smokers: A randomized, double-blind study. PLoS ONE 2014, 9, e98671. [Google Scholar] [CrossRef]
- Oliviero, T.; Verkerk, R.; Vermeulen, M.; Dekker, M. In vivo formation and bioavailability of isothiocyanates from glucosinolates in broccoli as affected by processing conditions. Mol. Nutr. Food Res. 2014, 58, 1447–1456. [Google Scholar] [CrossRef]
- Singh, K.; Connors, S.L.; Macklin, E.A.; Smith, K.D.; Fahey, J.W.; Talalay, P.; Zimmerman, A.W. Sulforaphane treatment of autism spectrum disorder (ASD). Proc. Natl. Acad. Sci. USA 2014, 111, 15550–15555. [Google Scholar] [CrossRef]
- Atwell, L.L.; Hsu, A.; Wong, C.P.; Stevens, J.F.; Bella, D.; Yu, T.W.; Pereira, C.B.; Lohr, C.V.; Christensen, J.M.; Dashwood, R.H.; et al. Absorption and chemopreventive targets of sulforaphane in humans following consumption of broccoli sprouts or a myrosinase-treated broccoli sprout extract. Mol. Nutr. Food Res. 2015, 59, 424–433. [Google Scholar] [CrossRef]
- Alumkal, J.J.; Slottke, R.; Schwartzman, J.; Cherala, G.; Munar, M.; Graff, J.N.; Beer, T.M.; Ryan, C.W.; Koop, D.R.; Gibbs, A.; et al. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Investig. New Drugs 2015, 33, 480–489. [Google Scholar] [CrossRef]
- Armah, C.N.; Derdemezis, C.; Traka, M.H.; Dainty, J.R.; Doleman, J.F.; Saha, S.; Leung, W.; Potter, J.F.; Lovegrove, J.A.; Mithen, R.F. Diet rich in high glucoraphanin broccoli reduces plasma LDL cholesterol: Evidence from randomised controlled trials. Mol. Nutr. Food Res. 2015, 59, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Atwell, L.L.; Zhang, Z.; Mori, M.; Farris, P.; Vetto, J.T.; Naik, A.M.; Oh, K.Y.; Thuillier, P.; Ho, E.; Shannon, J. Sulforaphane bioavailability and chemopreventive activity in women scheduled for breast biopsy. Cancer Prev. Res. 2015, 8, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Reynolds, C.; Brooker, A.; Talalay, P.; Fahey, J.W. Sulforaphane improves the bronchoprotective response in asthmatics through Nrf2-mediated gene pathways. Respir. Res. 2015, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Jang, J.Y.; Kim, Y.H.; Kim, J.W.; Shim, J.J. The effects of broccoli sprout extract containing sulforaphane on lipid peroxidation and Helicobacter pylori infection in the gastric mucosa. Gut Liver 2015, 9, 486–493. [Google Scholar] [CrossRef]
- Cipolla, B.G.; Mandron, E.; Lefort, J.M.; Coadou, Y.; Della Negra, E.; Corbel, L.; Le Scodan, R.; Azzouzi, A.R.; Mottet, N. Effect of sulforaphane in men with biochemical recurrence after radical prostatectomy. Cancer Prev. Res. 2015, 8, 712–719. [Google Scholar] [CrossRef]
- Fahey, J.W.; Holtzclaw, W.D.; Wehage, S.L.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Sulforaphane bioavailability from glucoraphanin-rich broccoli: Control by active endogenous myrosinase. PLoS ONE 2015, 10, e0140963. [Google Scholar] [CrossRef]
- Kikuchi, M.; Ushida, Y.; Shiozawa, H.; Umeda, R.; Tsuruya, K.; Aoki, Y.; Suganuma, H.; Nishizaki, Y. Sulforaphane-rich broccoli sprout extract improves hepatic abnormalities in male subjects. World J. Gastroenterol. 2015, 21, 12457–12467. [Google Scholar] [CrossRef]
- Medina, S.; Dominguez-Perles, R.; Moreno, D.A.; Garcia-Viguera, C.; Ferreres, F.; Gil, J.I.; Gil-Izquierdo, A. The intake of broccoli sprouts modulates the inflammatory and vascular prostanoids but not the oxidative stress-related isoprostanes in healthy humans. Food Chem. 2015, 173, 1187–1194. [Google Scholar] [CrossRef]
- Shiina, A.; Kanahara, N.; Sasaki, T.; Oda, Y.; Hashimoto, T.; Hasegawa, T.; Yoshida, T.; Iyo, M.; Hashimoto, K. An open study of sulforaphane-rich broccoli sprout extract in patients with schizophrenia. Clin. Psychopharmacol. Neurosci. 2015, 13, 62–67. [Google Scholar] [CrossRef]
- Rajendran, P.; Dashwood, W.M.; Li, L.; Kang, Y.; Kim, E.; Johnson, G.; Fischer, K.A.; Lohr, C.V.; Williams, D.E.; Ho, E.; et al. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clin. Epigenetics 2015, 7, 102. [Google Scholar] [CrossRef]
- Bauman, J.E.; Zang, Y.; Sen, M.; Li, C.; Wang, L.; Egner, P.A.; Fahey, J.W.; Normolle, D.P.; Grandis, J.R.; Kensler, T.W.; et al. Prevention of carcinogen-induced oral cancer by sulforaphane. Cancer Prev. Res. 2016, 9, 547–557. [Google Scholar] [CrossRef]
- Doss, J.F.; Jonassaint, J.C.; Garrett, M.E.; Ashley-Koch, A.E.; Telen, M.J.; Chi, J.T. Phase 1 study of a sulforaphane-containing broccoli sprout homogenate for sickle cell disease. PLoS ONE 2016, 11, e0152895. [Google Scholar] [CrossRef] [PubMed]
- Duran, C.G.; Burbank, A.J.; Mills, K.H.; Duckworth, H.R.; Aleman, M.M.; Kesic, M.J.; Peden, D.B.; Pan, Y.; Zhou, H.; Hernandez, M.L. A proof-of-concept clinical study examining the NRF2 activator sulforaphane against neutrophilic airway inflammation. Respir. Res. 2016, 17, 89. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Meyer, M.; Bauer, R.N.; Zhou, H.; Zhang, H.; Jones, S.; Robinette, C.; Noah, T.L.; Jaspers, I. Effect of broccoli sprouts and live attenuated influenza virus on peripheral blood natural killer cells: A randomized, double-blind study. PLoS ONE 2016, 11, e0147742. [Google Scholar] [CrossRef] [PubMed]
- Sudini, K.; Diette, G.B.; Breysse, P.N.; McCormack, M.C.; Bull, D.; Biswal, S.; Zhai, S.; Brereton, N.; Peng, R.D.; Matsui, E.C. A randomized controlled trial of the effect of broccoli sprouts on antioxidant gene expression and airway inflammation in asthmatics. J. Allergy Clin. Immunol. Pract. 2016, 4, 932–940. [Google Scholar] [CrossRef]
- Wise, R.A.; Holbrook, J.T.; Criner, G.; Sethi, S.; Rayapudi, S.; Sudini, K.R.; Sugar, E.A.; Burke, A.; Thimmulappa, R.; Singh, A.; et al. Lack of effect of oral sulforaphane administration on Nrf2 expression in COPD: A randomized, double-blind, placebo controlled trial. PLoS ONE 2016, 11, e0163716. [Google Scholar] [CrossRef]
- Axelsson, A.S.; Tubbs, E.; Mecham, B.; Chacko, S.; Nenonen, H.A.; Tang, Y.; Fahey, J.W.; Derry, J.M.J.; Wollheim, C.B.; Wierup, N.; et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Davidson, R.; Gardner, S.; Jupp, O.; Bullough, A.; Butters, S.; Watts, L.; Donell, S.; Traka, M.; Saha, S.; Mithen, R.; et al. Isothiocyanates are detected in human synovial fluid following broccoli consumption and can affect the tissues of the knee joint. Sci. Rep. 2017, 7, 3398. [Google Scholar] [CrossRef]
- Fahey, J.W.; Wade, K.L.; Wehage, S.L.; Holtzclaw, W.D.; Liu, H.; Talalay, P.; Fuchs, E.; Stephenson, K.K. Stabilized sulforaphane for clinical use: Phytochemical delivery efficiency. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Nucifora, L.G.; Koga, M.; Shaffer, L.S.; Higgs, C.; Tanaka, T.; Wang, A.M.; Coughlin, J.M.; Barker, P.B.; Fahey, J.W.; et al. Sulforaphane augments glutathione and influences brain metabolites in human subjects: A clinical pilot study. Mol. Neuropsychiatry 2018, 3, 214–222. [Google Scholar] [CrossRef]
- Tahata, S.; Singh, S.V.; Lin, Y.; Hahm, E.R.; Beumer, J.H.; Christner, S.M.; Rao, U.N.; Sander, C.; Tarhini, A.A.; Tawbi, H.; et al. Evaluation of biodistribution of sulforaphane after administration of oral broccoli sprout extract in melanoma patients with multiple atypical nevi. Cancer Prev. Res. 2018, 11, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Bent, S.; Lawton, B.; Warren, T.; Widjaja, F.; Dang, K.; Fahey, J.W.; Cornblatt, B.; Kinchen, J.M.; Delucchi, K.; Hendren, R.L. Identification of urinary metabolites that correlate with clinical improvements in children with autism treated with sulforaphane from broccoli. Mol. Autism 2018, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Housley, L.; Magana, A.A.; Hsu, A.; Beaver, L.M.; Wong, C.P.; Stevens, J.F.; Choi, J.; Jiang, Y.; Bella, D.; Williams, D.E.; et al. Untargeted metabolomic screen reveals changes in human plasma metabolite profiles following consumption of fresh broccoli sprouts. Mol. Nutr. Food Res. 2018, 62, e1700665. [Google Scholar] [CrossRef] [PubMed]
- Okunade, O.; Niranjan, K.; Ghawi, S.K.; Kuhnle, G.; Methven, L. Supplementation of the diet by exogenous myrosinase via mustard seeds to increase the bioavailability of sulforaphane in healthy human subjects after the consumption of cooked broccoli. Mol. Nutr. Food Res. 2018, 62, e1700980. [Google Scholar] [CrossRef]
- Sivapalan, T.; Melchini, A.; Saha, S.; Needs, P.W.; Traka, M.H.; Tapp, H.; Dainty, J.R.; Mithen, R.F. Bioavailability of glucoraphanin and sulforaphane from high-glucoraphanin broccoli. Mol. Nutr. Food Res. 2018, 62, e1700911. [Google Scholar] [CrossRef]
- Charron, C.S.; Vinyard, B.T.; Ross, S.A.; Seifried, H.E.; Jeffery, E.H.; Novotny, J.A. Absorption and metabolism of isothiocyanates formed from broccoli glucosinolates: Effects of BMI and daily consumption in a randomised clinical trial. Br. J. Nutr. 2018, 120, 1370–1379. [Google Scholar] [CrossRef]
- Lopez-Chillon, M.T.; Carazo-Diaz, C.; Prieto-Merino, D.; Zafrilla, P.; Moreno, D.A.; Villano, D. Effects of long-term consumption of broccoli sprouts on inflammatory markers in overweight subjects. Clin. Nutr. 2019, 38, 745–752. [Google Scholar] [CrossRef]
- Chen, J.G.; Johnson, J.; Egner, P.; Ng, D.; Zhu, J.; Wang, J.B.; Xue, X.F.; Sun, Y.; Zhang, Y.H.; Lu, L.L.; et al. Dose-dependent detoxication of the airborne pollutant benzene in a randomized trial of broccoli sprout beverage in Qidong, China. Am. J. Clin. Nutr. 2019, 110, 675–684. [Google Scholar] [CrossRef]
- Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Panjwani, A.A.; Liu, H.; Cornblatt, G.; Cornblatt, B.S.; Ownby, S.L.; Fuchs, E.; Holtzclaw, W.D.; et al. Bioavailability of sulforaphane following ingestion of glucoraphanin-rich broccoli sprout and seed extracts with active myrosinase: A pilot study of the effects of proton pump inhibitor administration. Nutrients 2019, 11, 1489. [Google Scholar] [CrossRef]
- Traka, M.H.; Melchini, A.; Coode-Bate, J.; Al Kadhi, O.; Saha, S.; Defernez, M.; Troncoso-Rey, P.; Kibblewhite, H.; O’Neill, C.M.; Bernuzzi, F.; et al. Transcriptional changes in prostate of men on active surveillance after a 12-mo glucoraphanin-rich broccoli intervention-results from the Effect of Sulforaphane on prostate CAncer PrEvention (ESCAPE) randomized controlled trial. Am. J. Clin. Nutr. 2019, 109, 1133–1144. [Google Scholar] [CrossRef]
- Palliyaguru, D.L.; Salvatore, S.R.; Schopfer, F.J.; Cheng, X.; Zhou, J.; Kensler, T.W.; Wendell, S.G. Evaluation of 2-thiothiazolidine-4-carboxylic acid, a common metabolite of isothiocyanates, as a potential biomarker of cruciferous vegetable intake. Mol. Nutr. Food Res. 2019, 63, e1801029. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Ziros, P.G.; Chen, J.G.; Groopman, J.D.; Kensler, T.W.; Sykiotis, G.P. Broccoli sprout beverage is safe for thyroid hormonal and autoimmune status: Results of a 12-week randomized trial. Food Chem. Toxicol. 2019, 126, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Garzotto, M.; Davis, E.W., II; Mori, M.; Stoller, W.A.; Farris, P.E.; Wong, C.P.; Beaver, L.M.; Thomas, G.V.; Williams, D.E.; et al. Sulforaphane bioavailability and chemopreventive activity in men presenting for biopsy of the prostate gland: A randomized controlled trial. Nutr. Cancer 2020, 72, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Langston-Cox, A.; Anderson, D.; Creek, D.J.; Palmer, K.; Wallace, E.M.; Marshall, S.A. Measuring sulforaphane and its metabolites in human plasma: A high throughput method. Molecules 2020, 25, 829. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zimmerman, A.W.; Singh, K.; Connors, S.L.; Diggins, E.; Stephenson, K.K.; Dinkova-Kostova, A.T.; Fahey, J.W. Biomarker exploration in human peripheral blood mononuclear cells for monitoring sulforaphane treatment responses in autism spectrum disorder. Sci. Rep. 2020, 10, 5822. [Google Scholar] [CrossRef]
- Sun, J.; Charron, C.S.; Novotny, J.A.; Peng, B.; Yu, L.; Chen, P. Profiling glucosinolate metabolites in human urine and plasma after broccoli consumption using non-targeted and targeted metabolomic analyses. Food Chem. 2020, 309, 125660. [Google Scholar] [CrossRef]
- Dashwood, R.H.; Ho, E. Dietary histone deacetylase inhibitors: From cells to mice to man. Semin. Cancer Biol. 2007, 17, 363–369. [Google Scholar] [CrossRef]
- Zhang, Y. The molecular basis that unifies the metabolism, cellular uptake and chemopreventive activities of dietary isothiocyanates. Carcinogenesis 2012, 33, 2–9. [Google Scholar] [CrossRef]
- Paunkov, A.; Chartoumpekis, D.V.; Ziros, P.G.; Sykiotis, G.P. A bibliometric review of the Keap1/Nrf2 pathway and its related antioxidant compounds. Antioxidants 2019, 8, 353. [Google Scholar] [CrossRef]
- Shen, T.; Jiang, T.; Long, M.; Chen, J.; Ren, D.M.; Wong, P.K.; Chapman, E.; Zhou, B.; Zhang, D.D. A curcumin derivative that inhibits vinyl carbamate-induced lung carcinogenesis via activation of the Nrf2 protective response. Antioxid. Redox Signal. 2015, 23, 651–664. [Google Scholar] [CrossRef]
- Yang, H.; Xu, W.; Zhou, Z.; Liu, J.; Li, X.; Chen, L.; Weng, J.; Yu, Z. Curcumin attenuates urinary excretion of albumin in type II diabetic patients with enhancing nuclear factor erythroid-derived 2-like 2 (Nrf2) system and repressing inflammatory signaling efficacies. Exp. Clin. Endocrinol. Diabetes 2015, 123, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Osorio, A.S.; Garcia-Nino, W.R.; Gonzalez-Reyes, S.; Alvarez-Mejia, A.E.; Guerra-Leon, S.; Salazar-Segovia, J.; Falcon, I.; Montes de Oca-Solano, H.; Madero, M.; Pedraza-Chaverri, J. The effect of dietary supplementation with curcumin on redox status and Nrf2 activation in patients with nondiabetic or diabetic proteinuric chronic kidney disease: A pilot study. J. Ren. Nutr. 2016, 26, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, J.F.; Leal, V.O.; Rizzetto, F.; Grimmer, G.H.; Ribeiro-Alves, M.; Daleprane, J.B.; Carraro-Eduardo, J.C.; Mafra, D. Effects of resveratrol supplementation in Nrf2 and NF-kappaB expressions in nondialyzed chronic kidney disease patients: A randomized, double-blind, placebo-controlled, crossover clinical trial. J. Ren. Nutr. 2016, 26, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Seyyedebrahimi, S.; Khodabandehloo, H.; Nasli Esfahani, E.; Meshkani, R. The effects of resveratrol on markers of oxidative stress in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled clinical trial. Acta Diabetol. 2018, 55, 341–353. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working, G. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Cohen, A.F.; Burggraaf, J.; van Gerven, J.M.; Moerland, M.; Groeneveld, G.J. The use of biomarkers in human pharmacology (Phase I) studies. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 55–74. [Google Scholar] [CrossRef]
- Devling, T.W.; Lindsay, C.D.; McLellan, L.I.; McMahon, M.; Hayes, J.D. Utility of siRNA against Keap1 as a strategy to stimulate a cancer chemopreventive phenotype. Proc. Natl. Acad. Sci. USA 2005, 102, 7280–7285A. [Google Scholar] [CrossRef]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef]
- Goodman, M.; Bostick, R.M.; Kucuk, O.; Jones, D.P. Clinical trials of antioxidants as cancer prevention agents: Past, present, and future. Free Radic. Biol. Med. 2011, 51, 1068–1084. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and clinical significance of biomarkers of oxidative stress in humans. Oxid Med. Cell Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- Czerska, M.; Mikolajewska, K.; Zielinski, M.; Gromadzinska, J.; Wasowicz, W. Today’s oxidative stress markers. Med. Pr. 2015, 66, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Wakabayashi, N.; Misra, V.; Nilles, T.; Biswal, S.; Trush, M.A.; Kensler, T.W. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch. Biochem. Biophys. 2006, 454, 7–15. [Google Scholar] [CrossRef]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar] [CrossRef]
- Celsus, A.C. De medicina. Self-published 25.
- Ghezzi, P.; Floridi, L.; Boraschi, D.; Cuadrado, A.; Manda, G.; Levic, S.; D’Acquisto, F.; Hamilton, A.; Athersuch, T.J.; Selley, L. Oxidative stress and inflammation induced by environmental and psychological stressors: A biomarker perspective. Antioxid. Redox Signal. 2018, 28, 852–872. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2). Mol. Cell Biol. 2004, 24, 36–45. [Google Scholar] [CrossRef]
- Jung, H.; Kim, T.; Chae, H.Z.; Kim, K.T.; Ha, H. Regulation of macrophage migration inhibitory factor and thiol-specific antioxidant protein PAG by direct interaction. J. Biol. Chem. 2001, 276, 15504–15510. [Google Scholar] [CrossRef]
- Lin, W.; Wu, R.T.; Wu, T.; Khor, T.O.; Wang, H.; Kong, A.N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Brigelius-Flohe, R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid. Redox Signal. 2005, 7, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Tsai, P.Y.; Ka, S.M.; Chang, J.M.; Chen, H.C.; Shui, H.A.; Li, C.Y.; Hua, K.F.; Chang, W.L.; Huang, J.J.; Yang, S.S.; et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Biol. Med. 2011, 51, 744–754. [Google Scholar] [CrossRef]
- Taguchi, K.; Kensler, T.W. Nrf2 in liver toxicology. Arch. Pharm. Res. 2020, 43, 337–349. [Google Scholar] [CrossRef]
- Fahey, J.W.; Haristoy, X.; Dolan, P.M.; Kensler, T.W.; Scholtus, I.; Stephenson, K.K.; Talalay, P.; Lozniewski, A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7610–7615. [Google Scholar] [CrossRef]
- Kensler, T.W.; Groopman, J.D. Carcinogen-DNA and protein adducts: Biomarkers for cohort selection and modifiable endpoints in chemoprevention trials. J. Cell Biochem. Suppl. 1996, 25, 85–91. [Google Scholar] [CrossRef]
- Yuan, J.M.; Stepanov, I.; Murphy, S.E.; Wang, R.; Allen, S.; Jensen, J.; Strayer, L.; Adams-Haduch, J.; Upadhyaya, P.; Le, C.; et al. Clinical trial of 2-phenethyl isothiocyanate as an inhibitor of metabolic activation of a tobacco-specific lung carcinogen in cigarette smokers. Cancer Prev. Res. 2016, 9, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.G.; Young, P.J.; Agus, C.; Knize, M.G.; Boobis, A.R.; Gooderham, N.J.; Lake, B.G. Cruciferous vegetable consumption alters the metabolism of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) in humans. Carcinogenesis 2004, 25, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Tolstikov, V.; Moser, A.J.; Sarangarajan, R.; Narain, N.R.; Kiebish, M.A. Current status of metabolomic biomarker discovery: Impact of study design and demographic characteristics. Metabolites 2020, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Schmidt, M.A.; Kaddurah-Daouk, R. Current concepts in pharmacometabolomics, biomarker discovery, and precision medicine. Metabolites 2020, 10, 129. [Google Scholar] [CrossRef]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef]
- Macari, E.R.; Lowrey, C.H. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood 2011, 117, 5987–5997. [Google Scholar] [CrossRef]
- Campbell, M.R.; Karaca, M.; Adamski, K.N.; Chorley, B.N.; Wang, X.; Bell, D.A. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid Med. Cell Longev. 2013, 2013, 120305. [Google Scholar] [CrossRef]
- Ushida, Y.; Suganuma, H.; Yanaka, A. Low-dose of the sulforaphane precursor glucoraphanin as a dietary supplement induces chemoprotective enzymes in humans. Food Nutr. Sci. 2015, 6, 1603–1612. [Google Scholar] [CrossRef]
- Kensler, T.W.; Groopman, J.D. Is it time to advance the chemoprevention of environmental carcinogenesis with microdosing trials? Cancer Prev. Res. 2009, 2, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Uruno, A.; Yumoto, A.; Taguchi, K.; Suzuki, M.; Harada, N.; Ryoke, R.; Naganuma, E.; Osanai, N.; Goto, A.; et al. Space travel of mice displays contribution of Nrf2 to maintenance of homeostasis. Commun. Biol. 2020, in press. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).