Flavonoids Activation of the Transcription Factor Nrf2 as a Hypothesis Approach for the Prevention and Modulation of SARS-CoV-2 Infection Severity


Abstract
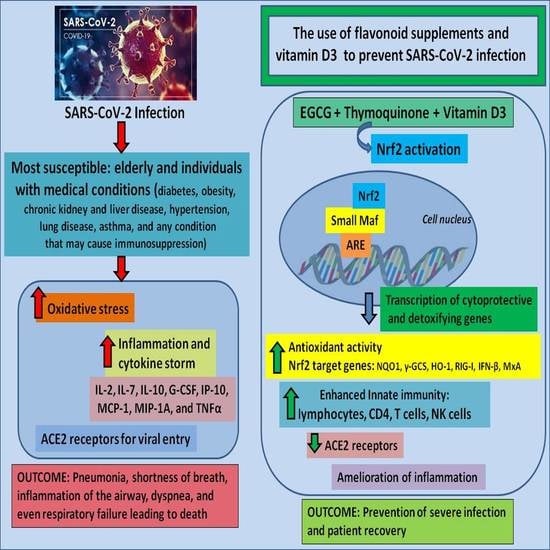
{kind=link}
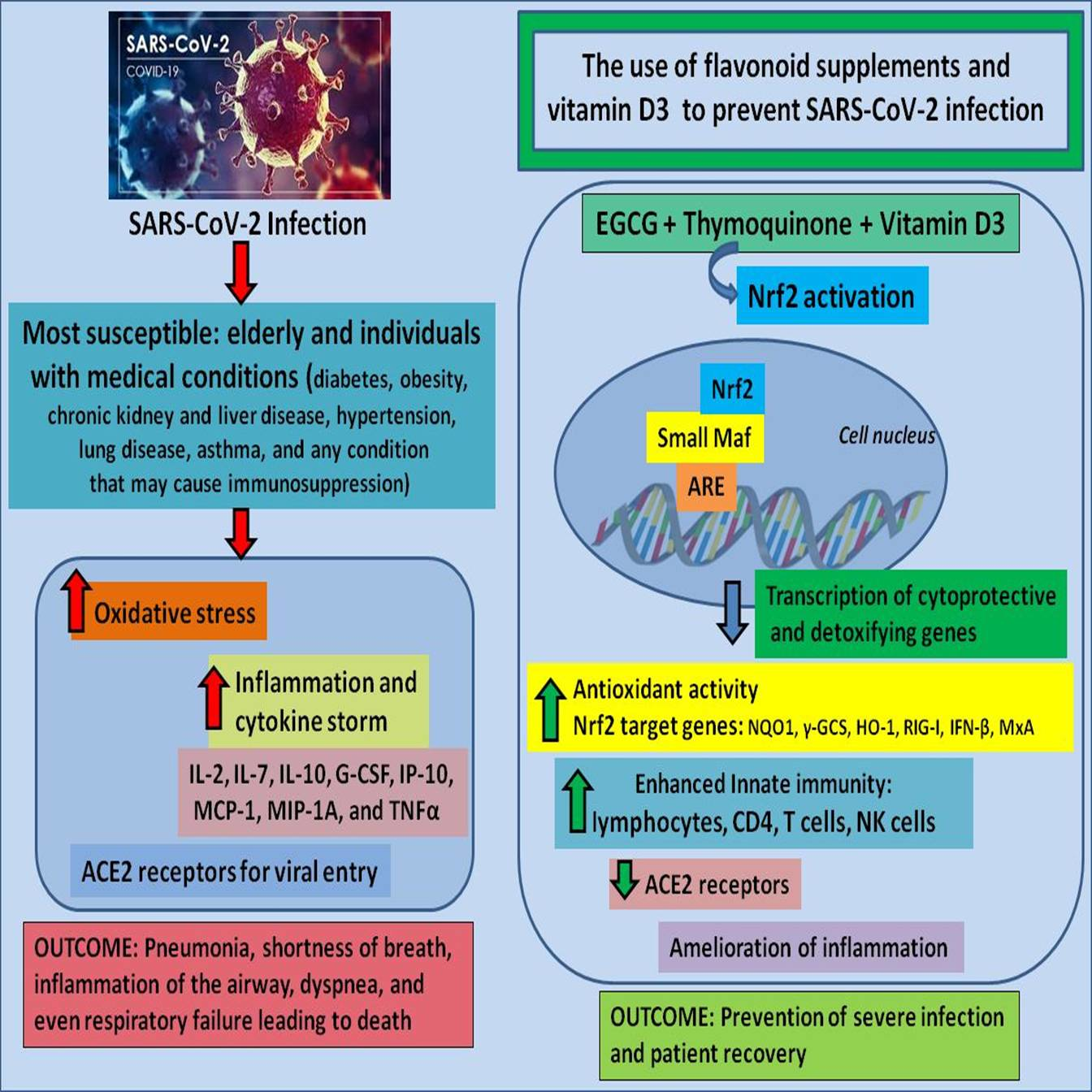
{kind=link}
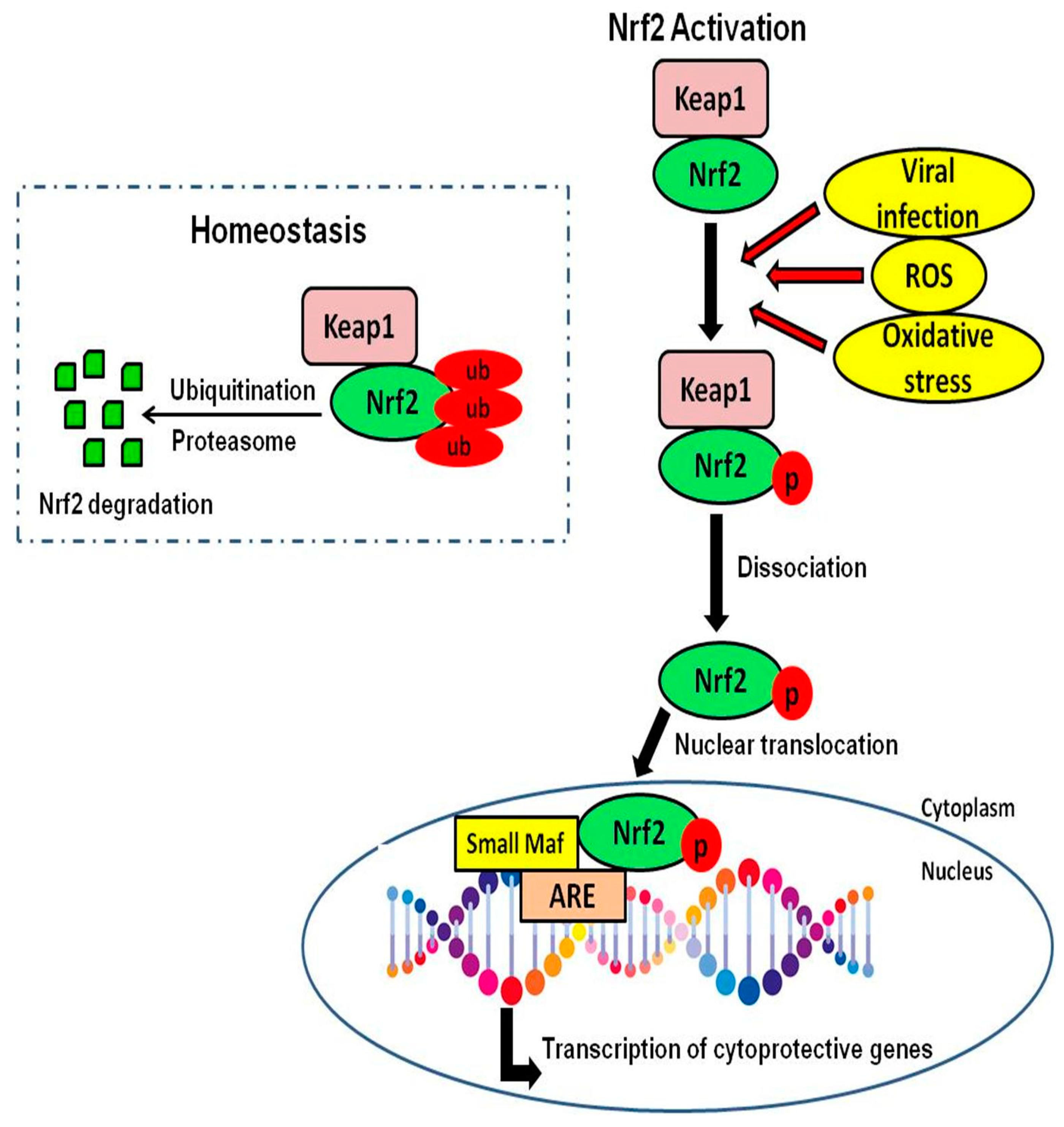
{kind=link}
{kind=link}
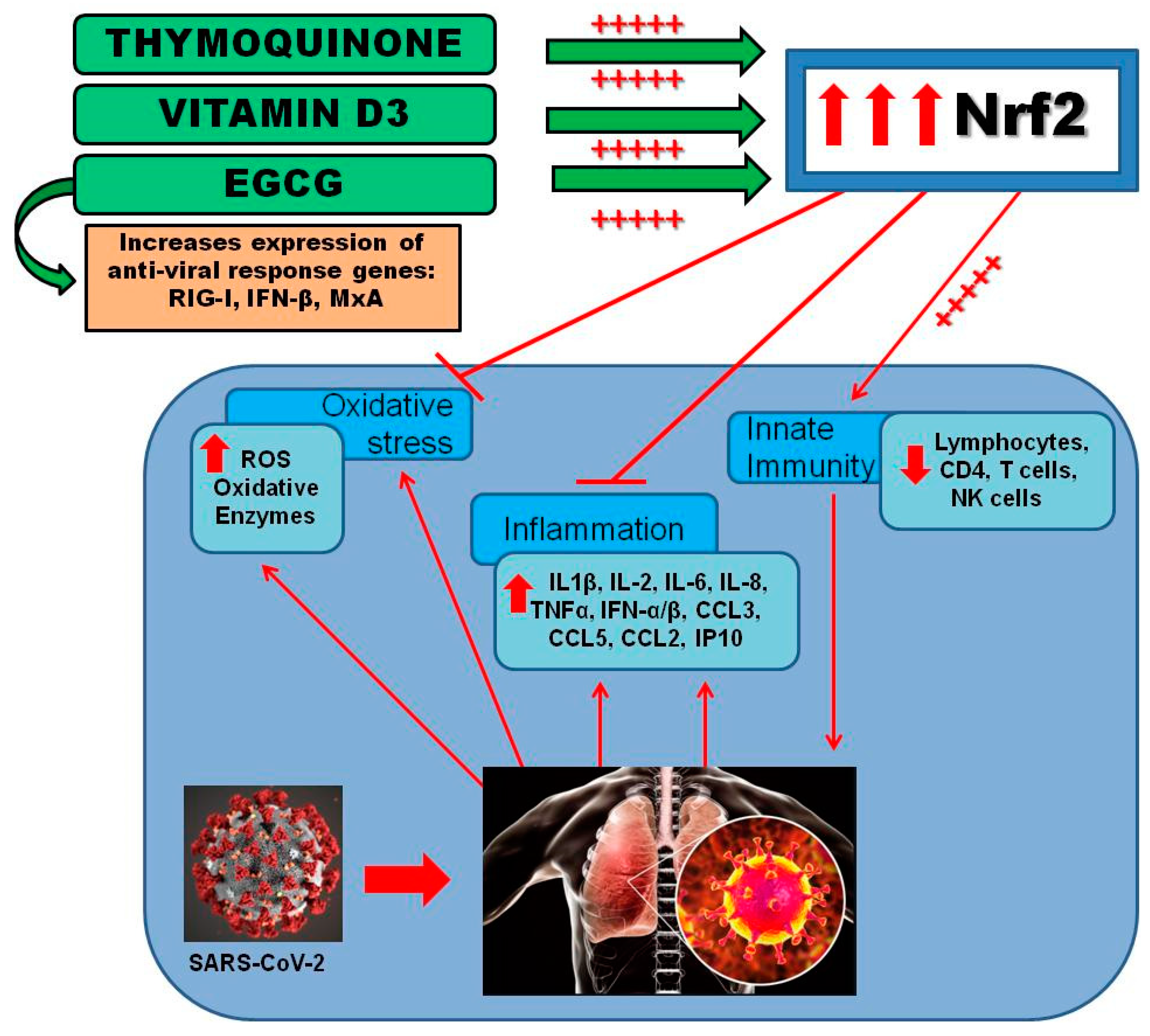{kind=link}
1. Introduction
2. COVID 19 Pathogenesis
3. Nuclear Factor Erythroid-Derived 2-Related Factor 2 (Nrf2)
4. Nrf2 Activation and COVID-19 Infection
5. Viral Infection and Oxidative Stress
6. Nrf2 Activation Downregulates ACE2 Receptor Expression
7. Oxidative Stress-Mediated Hypomethylation Increases Expression of ACE2 Receptor
8. Nrf2 Downregulation, Oxidative Stress, and Proteases Expression
9. Nrf2 Activation Modulates Viral Infection Severity
10. Nrf2 Activation Enhances Innate Immune Response
11. Nrf2 Reduces Oxidative Stress and Inflammation
12. Nrf2 Low Expression Levels in Elderly
13. Nrf2, Inflammation, and Obesity
14. Nrf2 Reduces Oxidative Stress Associated with Hyperglycemia
15. Flavonoids Role as Antivirals and Nrf2 Activators
15.1. Epigallocatechin-3-Gallate
15.2. Thymoquinone
16. Vitamin D3 Supplementation to Enhance Immune Response
17. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACACA | Acetyl-CoA carboxylase alpha |
| ACACB | Acetyl-CoA carboxylase beta |
| ACE2 | Angiotensin-converting enzyme 2 |
| ALI | Acute lung injury |
| ARDS | Acute respiratory distress syndrome |
| ARE | Antioxidant response elements |
| CAT | Catalase |
| CDC | Centers for Disease Control and Prevention |
| CDDO-Me | Bardoxolone-methyl |
| CNS | Central Nervous System |
| COVID-19 | Coronavirus disease 2019 |
| DM | Diabetes mellitus |
| DNMT1 | DNA methyltransferase |
| EGCG | Epigallocatechin-3-gallate |
| EpRE | Electrophilic response element |
| FASN | Fatty acid synthase |
| GCL | Glutamate-cysteine ligase |
| Glut 4 | Glucose transporter type 4 |
| GST | Glutathione S-transferase |
| HAT | Human airway trypsin-like protease |
| HFD | High-fat diet |
| HO-1 | Heme oxygenase 1 |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IFN | Interferons |
| IL-6 | Interleukin-6 |
| Keap1 | Kelch-like ECH-associated protein 1 |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| MxA | Mx GTPases |
| NK | Natural killer |
| NQO1 | NAD(P)H: quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor erythroid-derived 2-related factor 2 |
| PGE2 | Prostaglandin E2 |
| ROS | Reactive oxygen species |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SLPI | Secretory leukocyte proteinase inhibitor |
| SOD | Superoxide dismutase |
| SOD1 | Superoxide dismutase 1 |
| TMPRSS2 | Transmembrane protease serine 2 |
| TQ | Thymoquinone |
| WT | Wild type |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Yuen, K.Y.; Osterhaus, A.D.; Stöhr, K. The severe acute respiratory syndrome. N. Engl. J. Med. 2003, 349, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019–nCoV) In Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Cortegiani, A.; Ingoglia, G.; Ippolito, M.; Giarratano, A.; Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid–19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, S.M.; Yu, X.H.; Tang, S.L.; Tang, C.K. Coronavirus disease 2019 (COVID–19): Current status and future perspectives. Int. J. Antimicrob. Agents 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Yu, P.; Zhu, J.; Zhang, Z.; Han, Y.; Huang, L. A familial cluster of infection associated with the 2019 novel coronavirus indicating potential person-to-person transmission during the incubation period. J. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Tian, S.; Hu, W.; Niu, L.; Liu, H.; Xu, H.; Xiao, S.Y. Pulmonary Pathology of Early–Phase 2019 Novel Coronavirus (COVID–19) Pneumonia in Two Patients With Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Rothe, C.; Schunk, M.; Sothmann, P.; Bretzel, G.; Froeschl, G.; Wallrauch, C.; Zimmer, T.; Thiel, V.; Janke, C.; Guggemos, W.; et al. Transmission of 2019-nCoV Infection from an Asymptomatic Contact in Germany. N. Engl. J. Med. 2020, 382, 970–971. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.; Riley, S.; Anderson, R.M.; Ferguson, N.M. Factors that make an infectious disease outbreak controllable. Proc. Natl. Acad. Sci. USA 2004, 101, 6146–6151. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar] [CrossRef]
- Mahallawi, W.H.; Khabour, O.F.; Zhang, Q.; Makhdoum, H.M.; Suliman, B.A. MERS–CoV infection in humans, is associated with a pro–inflammatory Th1 and Th17 cytokine profile. Cytokine 2018, 104, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Lam, C.W.; Wu, A.K.; Ip, W.K.; Lee, N.L.; Chan, I.H.; Lit, L.C.; Hui, D.S.; Chan, M.H.; Chung, S.S.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- O’Connell, M.A.; Hayes, J.D. The Keap1/Nrf2 pathway in health and disease: From the bench to the clinic. Biochem. Soc. Trans. 2015, 43, 687–689. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed]
- Silva–Palacios, A.; Ostolga-Chavarria, M.; Zazueta, C.; Konigsberg, M. Nrf2: Molecular and epigenetic regulation during aging. Ageing Res. Rev. 2018, 47, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Kerr, F.; Sofola–Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez–Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R. Direct Keap1–Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 2017, 13, e1006593. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Sekhar, K.R.; Freeman, M.L.; Liebler, D.C. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 2005, 280, 31768–31775. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Iida, K.; Kang, M.I.; Kobayashi, A.; Mizukami, M.; Tong, K.I.; McMahon, M.; Hayes, J.D.; Itoh, K.; Yamamoto, M. Evolutionary conserved N–terminal domain of Nrf2 is essential for the Keap1–mediated degradation of the protein by the proteasome. Arch. Biochem. Biophys. 2005, 433, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Nrf2–Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzym. Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef]
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as a key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. SUA 2004, 101, 2046–2051. [Google Scholar] [CrossRef]
- Su, C.; Zhang, P.; Song, X.; Shi, Q.; Fu, J.; Xia, X.; Bai, H.; Hu, L.; Xu, D.; Song, E.; et al. Tetrachlorobenzoquinone activates Nrf2 signaling by Keap1 cross-linking and ubiquitin translocation but not Keap1–Cullin3 complex dissociation. Chem. Res. Toxicol. 2015, 28, 765–774. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef]
- Li, J.; Johnson, D.; Calkins, M.; Wright, L.; Svendsen, C.; Johnson, J. Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol. Sci. Off. J. Soc. Toxicol. 2005, 83, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.N.; Mele, J.; Hayes, J.D.; Buffenstein, R. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr. Comp. Biol. 2010, 50, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Kaspar, J.W.; Shen, J.; Jaiswal, A.K. Nrf2 signaling and cell survival. Toxicol. Appl. Pharm. 2010, 244, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-Y.; Reddy, S.P.; DeBiase, A.; Yamamoto, M.; Kleeberger, S.R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 2005, 38, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Das, B.N.; Kim, Y.-W.; Keum, Y.-S. Mechanisms of Nrf2/Keap1–dependent phase II cytoprotective and detoxifying gene expression and potential cellular targets of chemopreventive isothiocyanates. Oxidative Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; So, Y.K.; Han, S.N.; Kim, J.-B. Isoegomaketone upregulates heme oxygenase-1 in RAW264. 7 cells via ROS/p38 MAPK/Nrf2 pathway. Biomol. Ther. 2016, 24, 510. [Google Scholar] [CrossRef] [PubMed]
- Solis, W.A.; Dalton, T.P.; Dieter, M.Z.; Freshwater, S.; Harrer, J.M.; He, L.; Shertzer, H.G.; Nebert, D.W. Glutamate–cysteine ligase modifier subunit: Mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem. Pharmacol. 2002, 63, 1739–1754. [Google Scholar] [CrossRef]
- Battino, M.; Giampieri, F.; Pistollato, F.; Sureda, A.; de Oliveira, M.R.; Pittalà, V.; Fallarino, F.; Nabavi, S.F.; Atanasov, A.G.; Nabavi, S.M. Nrf2 as a regulator of innate immunity: A molecular Swiss army knife! Biotechnol. Adv. 2018, 36, 358–370. [Google Scholar] [CrossRef]
- Biton, S.; Ashkenazi, A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell 2011, 145, 92–103. [Google Scholar] [CrossRef]
- Rocourt, C.R.; Wu, M.; Chen, B.P.; Cheng, W.H. The catalytic subunit of DNA-dependent protein kinase is downstream of ATM and feeds forward oxidative stress in the selenium–induced senescence response. J. Nutr. Biochem. 2013, 24, 781–787. [Google Scholar] [CrossRef]
- Chhunchha, B.; Kubo, E. Sulforaphane–Induced Klf9/Prdx6 Axis Acts as a Molecular Switch to Control Redox Signaling and Determines Fate of Cells. Cells 2019, 8, 1159. [Google Scholar] [CrossRef] [PubMed]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi–Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Therapeutic Modulation of Virus–Induced Oxidative Stress via the Nrf2–Dependent Antioxidative Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 6208067. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS–CoV–2 infected patients. E. Bio. Med. 2020, 55, 102763. [Google Scholar] [CrossRef]
- Lomeli, N.; Bota, D.A.; Davies, K.J.A. Diminished stress resistance and defective adaptive homeostasis in age-related diseases. Clin. Sci. 2017, 131, 2573–2599. [Google Scholar] [CrossRef]
- McCord, J.M.; Hybertson, B.M.; Cota–Gomez, A.; Gao, B. Nrf2 Activator PB125® as a Potential Therapeutic Agent Against COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. Identification of SARS-CoV2-mediated suppression of NRF2 signaling reveals a potent antiviral and anti–inflammatory activity of 4–octyl-itaconate and dimethyl fumarate. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Komaravelli, N.; Casola, A. Respiratory Viral Infections and Subversion of Cellular Antioxidant Defenses. J. Pharm. Pharm. 2014, 5. [Google Scholar] [CrossRef]
- Cuadrado, A.; Pajares, M.; Benito, C.; Jiménez–Villegas, J.; Escoll, M.; Fernández–Ginés, R.; Garcia Yagüe, A.J.; Lastra, D.; Manda, G.; Rojo, A.I.; et al. Can activation of NRF2 be a strategy against COVID–19? Trends Pharmacol. Sci. 2020. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.; Higgins, M.; Hams, E. Itaconate is an anti–inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113. [Google Scholar] [CrossRef]
- Wang, M.M.; Lu, M.; Zhang, C.L.; Wu, X.; Chen, J.X.; Lv, W.W.; Sun, T.; Qiu, H.; Huang, S.H. Oxidative stress modulates the expression of toll-like receptor 3 during respiratory syncytial virus infection in human lung epithelial A549 cells. Mol. Med. Rep. 2018, 18, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Jantzi, P.D.; Esham, D.L.; Spratt, H.; Kurosky, A.; Casola, A.; Garofalo, R.P. Viral–mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am. J. Respir. Crit. Care Med. 2011, 183, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll–like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-Y.; Imani, F.; Miller-DeGraff, L.; Walters, D.; Melendi, G.A.; Yamamoto, M.; Polack, F.P.; Kleeberger, S.R. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 2009, 179, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ren, F.; Hesketh, J.; Shi, X.; Li, J.; Gan, F.; Huang, K. Reactive oxygen species regulate the replication of porcine circovirus type 2 via NF–κB pathway. Virology 2012, 426, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, A.; Anticoli, S.; Nencioni, L.; Sgarbanti, R.; Garaci, E.; Palamara, A.T. Interplay between Hepatitis C Virus and Redox Cell Signaling. Int. J. Mol. Sci. 2013, 14, 4705–4721. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF–E2–related factor 2 (Nrf2), an NF–E2–like basic leucine zipper transcriptional activator that binds to the tandem NF–E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T.D.; Memish, Z.A.; Drosten, C. The continuing 2019–nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264. [Google Scholar] [CrossRef]
- Li, F. Evidence for a common evolutionary origin of coronavirus spike protein receptor–binding subunits. J. Virol. 2012, 86, 2856–2858. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Ghosh, A.; Lo, C.S.; Chenier, I.; Scholey, J.W.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S.D. Nrf2 Deficiency Upregulates Intrarenal Angiotensin-Converting Enzyme–2 and Angiotensin 1–7 Receptor Expression and Attenuates Hypertension and Nephropathy in Diabetic Mice. Endocrinology 2018, 159, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Strickland, F.M.; Patel, D.; Khanna, D.; Somers, E.; Robida, A.M.; Pihalja, M.; Swartz, R.; Marder, W.; Richardson, B. Characterisation of an epigenetically altered CD4+ CD28+ Kir+ T cell subset in autoimmune rheumatic diseases by multiparameter flow cytometry. Lupus Sci. Med. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Gensterblum, E.; Renauer, P.; Coit, P.; Strickland, F.M.; Kilian, N.C.; Miller, S.; Ognenovski, M.; Wren, J.D.; Tsou, P.-S.; Lewis, E.E. CD4+ CD28+ KIR+ CD11ahi T cells correlate with disease activity and are characterized by a pro-inflammatory epigenetic and transcriptional profile in lupus patients. J. Autoimmun. 2018, 86, 19–28. [Google Scholar] [CrossRef]
- Sawalha, A.H.; Zhao, M.; Coit, P.; Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin. Immunol. 2020, 215, 108410. [Google Scholar] [CrossRef]
- Li, Y.; Gorelik, G.; Strickland, F.M.; Richardson, B.C. Oxidative stress, T cell DNA methylation, and lupus. Arthritis Rheumatol. 2014, 66, 1574–1582. [Google Scholar] [CrossRef]
- Gorelik, G.; Fang, J.Y.; Wu, A.; Sawalha, A.H.; Richardson, B. Impaired T cell protein kinase Cδ activation decreases ERK pathway signaling in idiopathic and hydralazine–induced lupus. J. Immunol. 2007, 179, 5553–5563. [Google Scholar] [CrossRef]
- Weeding, E.; Sawalha, A.H. Deoxyribonucleic acid methylation in systemic lupus erythematosus: Implications for future clinical practice. Front. Immunol. 2018, 9, 875. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Taguchi, F. Two-step conformational changes in a coronavirus envelope glycoprotein mediated by receptor binding and proteolysis. J. Virol. 2009, 83, 11133–11141. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Reeves, J.D.; Rennekamp, A.J.; Amberg, S.M.; Piefer, A.J.; Bates, P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS–CoV) spike glycoprotein–mediated viral entry. Proc. Natl. Acad. Sci. USA 2004, 101, 4240–4245. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Kok, K.H. Genomic characterization of the 2019 novel human–pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Yasuoka, S.; Ohnishi, T.; Kawano, S.; Tsuchihashi, S.; Ogawara, M.; Masuda, K.-i.; Yamaoka, K.; Takahashi, M.; Sano, T. Purification, characterization, and localization of a novel trypsin–like protease found in the human airway. Am. J. Respir. Cell Mol. Biol. 1997, 16, 300–308. [Google Scholar] [CrossRef]
- Szabo, R.; Bugge, T.H. Type II transmembrane serine proteases in development and disease. Int. J. Biochem. Cell Biol. 2008, 40, 1297–1316. [Google Scholar] [CrossRef]
- Kesic, M.J.; Simmons, S.O.; Bauer, R.; Jaspers, I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic. Biol. Med. 2011, 51, 444–453. [Google Scholar] [CrossRef]
- Iizuka, T.; Ishii, Y.; Itoh, K.; Kiwamoto, T.; Kimura, T.; Matsuno, Y.; Morishima, Y.; Hegab, A.E.; Homma, S.; Nomura, A. Nrf2–deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 2005, 10, 1113–1125. [Google Scholar] [CrossRef]
- Toapanta, F.R.; Ross, T.M. Impaired immune responses in the lungs of aged mice following influenza infection. Respir. Res. 2009, 10, 112. [Google Scholar] [CrossRef]
- Vasileva, L.V.; Savova, M.S.; Amirova, K.M.; Dinkova–Kostova, A.T.; Georgiev, M.I. Obesity and NRF2–mediated cytoprotection: Where is the missing link? Pharmacol. Res. 2020, 156, 104760. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Lai, C.-L.; Hsieh, S.-H.; Shieh, C.-C.; Huang, L.-M.; Wu-Hsieh, B.A. Influenza A virus induction of oxidative stress and MMP–9 is associated with severe lung pathology in a mouse model. Virus Res. 2013, 178, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Paracha, U.Z.; Fatima, K.; Alqahtani, M.; Chaudhary, A.; Abuzenadah, A.; Damanhouri, G.; Qadri, I. Oxidative stress and hepatitis C virus. Virol. J. 2013, 10, 251. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-Y.; Kleeberger, S.R. Nrf2 protects against airway disorders. Toxicol. Appl. Pharm. 2010, 244, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Coon, A.; Baker, A.F.; Powis, G. Antitumor agent PX–12 inhibits HIF-1α protein levels through an Nrf2/PMF–1–mediated increase in spermidine/spermine acetyl transferase. Cancer Chemother. Pharmacol. 2011, 68, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I. Antioxidant therapeutic advances in COPD. Ther. Adv. Respir. Dis. 2008, 2, 351–374. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Rahbar, R.; Chan, R.W.; Lee, S.M.; Chan, M.C.; Wang, B.X.; Baker, D.P.; Sun, B.; Peiris, J.M.; Nicholls, J.M. Influenza virus non-structural protein 1 (NS1) disrupts interferon signaling. PLoS ONE 2010, 5, e13927. [Google Scholar] [CrossRef]
- Chen, S.; Short, J.A.; Young, D.F.; Killip, M.J.; Schneider, M.; Goodbourn, S.; Randall, R.E. Heterocellular induction of interferon by negative-sense RNA viruses. Virology 2010, 407, 247–255. [Google Scholar] [CrossRef]
- Haller, O.; Kochs, G. Interferon-induced mx proteins: Dynamin–like GTPases with antiviral activity. Traffic 2002, 3, 710–717. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Elliott, D.E.; Siddique, S.S.; Weinstock, J.V. Innate immunity in disease. Clin. Gastroenterol. Hepatol. 2014, 12, 749–755. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Juncadella, I.J.; Kadl, A.; Sharma, A.K.; Shim, Y.M.; Hochreiter–Hufford, A.; Borish, L.; Ravichandran, K.S. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 2013, 493, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Thimmulappa, R.K.; Cano, M.; Fujihara, M.; Izumi–Nagai, K.; Kong, X.; Sporn, M.B.; Kensler, T.W.; Biswal, S.; Handa, J.T. Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radic. Biol. Med. 2009, 47, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 2006, 116, 984–995. [Google Scholar] [CrossRef]
- Gomez, J.C.; Dang, H.; Martin, J.R. Nrf2 Modulates Host Defense during Streptococcus pneumoniae Pneumonia in Mice. J. Immunol. 2016, 197, 2864–2879. [Google Scholar] [CrossRef] [PubMed]
- Ennis, F.; Beare, A.; Riley, D.; Schild, G.; Meager, A.; Yi-Hua, Q.; Schwarz, G.; Rook, A. Interferon induction and increased natural killer–cell activity in influenza infections in man. Lancet 1981, 318, 891–893. [Google Scholar] [CrossRef]
- Jonjić, S.; Babić, M.; Polić, B.; Krmpotić, A. Immune evasion of natural killer cells by viruses. Curr. Opin. Immunol. 2008, 20, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Rajasekaran, K.; Nanbakhsh, A.; Gorski, J.; Thakar, M.S.; Malarkannan, S. IL–27 promotes NK cell effector functions via Maf–Nrf2 pathway during influenza infection. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Smyth, M.J. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J. Immunol. 2006, 176, 1517–1524. [Google Scholar] [CrossRef]
- De Colvenaer, V.; Taveirne, S.; Delforche, M.; De Smedt, M.; Vandekerckhove, B.; Taghon, T.; Boon, L.; Plum, J.; Leclercq, G. CD27-deficient mice show normal NK–cell differentiation but impaired function upon stimulation. Immunol. Cell Biol. 2011, 89, 803–811. [Google Scholar] [CrossRef]
- Baird, L.; Swift, S.; Llères, D.; Dinkova–Kostova, A.T. Monitoring Keap1–Nrf2 interactions in single live cells. Biotechnol. Adv. 2014, 32, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and done? Targeting the NRF2 pathway with sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2–antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Suzuki, H.; Itoh, K.; Yamamoto, M.; Sugiyama, Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem. Biophys. Res. Commun. 2003, 310, 824–829. [Google Scholar] [CrossRef]
- Sasaki, K.; Hatta, S.; Wada, K.; Ueda, N.; Yoshimura, T.; Endo, T.; Sakata, M.; Tanaka, T.; Haga, M. Effects of extract of Ginkgo biloba leaves and its constituents on carcinogen–metabolizing enzyme activities and glutathione levels in mouse liver. Life Sci. 2002, 70, 1657–1667. [Google Scholar] [CrossRef]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1–Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2–related factor–2–dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef]
- Braun, S.; Hanselmann, C.; Gassmann, M.G.; auf dem Keller, U.; Born–Berclaz, C.; Chan, K.; Kan, Y.W.; Werner, S. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol. Cell. Biol. 2002, 22, 5492–5505. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Grottelli, S.; Gatticchi, L.; Mierla, A.L.; Minelli, A. α–Tocopheryl succinate pre–treatment attenuates quinone toxicity in prostate cancer PC3 cells. Gene 2014, 539, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2–regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Tucci, A.; Galli, F.; Grottelli, S.; Mierla, A.L.; Pilolli, F.; Minelli, A. Inhibition of NF–κB nuclear translocation via HO–1 activation underlies α-tocopheryl succinate toxicity. J. Nutr. Biochem. 2012, 23, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres–Becker, I. Transcription factors NRF2 and NF–κB are coordinated effectors of the Rho family, GTP–binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Huang, C.; Zhang, G.; Liu, D.; Li, P.; Lu, C.; Li, J. Epidemiological characteristics of novel coronavirus pneumonia in Henan, China. J. Tuberc. Respir. Dis. 2020, 43, E027. [Google Scholar]
- Wan, S.; Yi, Q.; Fan, S.; Lv, J.; Zhang, X.; Guo, L.; Lang, C.; Xiao, Q.; Xiao, K.; Yi, Z.; et al. Characteristics of lymphocyte subsets and cytokines in peripheral blood of 123 hospitalized patients with 2019 novel coronavirus pneumonia (NCP). Med. Rxiv. 2020. [Google Scholar] [CrossRef]
- Franceschi, C.; Bondage, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, X. An Update on Inflamm–Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Bailey–Downs, L.; Sosnowska, D.; Gautam, T.; Koncz, P.; Losonczy, G.; Ballabh, P.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Vascular oxidative stress in aging: A homeostatic failure due to dysregulation of NRF2–mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H363–H372. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Zhang, R.; Guo, Y.; Jiang, Y.; Huang, Y.; Jiang, H.; Li, C. Nrf2 activity is lost in the spinal cord and its astrocytes of aged mice. Vitr. Cell. Dev. Biol. Anim. 2009, 45, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; deBeer, J.; Tarnopolsky, M.A. Dysfunctional Nrf2–Keap1 redox signaling in skeletal muscle of the sedentary old. Free Radic. Biol. Med. 2010, 49, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Gounder, S.S.; Kannan, S.; Devadoss, D.; Miller, C.J.; Whitehead, K.J.; Odelberg, S.J.; Firpo, M.A.; Paine, R., 3rd; Hoidal, J.R.; Abel, E.D.; et al. Impaired transcriptional activity of Nrf2 in age–related myocardial oxidative stress is reversible by moderate exercise training. PLoS ONE 2012, 7, e45697. [Google Scholar] [CrossRef]
- Alarcon–Aguilar, A.; Luna–Lopez, A.; Ventura–Gallegos, J.L.; Lazzarini, R.; Galvan–Arzate, S.; Gonzalez–Puertos, V.Y.; Moran, J.; Santamaria, A.; Konigsberg, M. Primary cultured astrocytes from old rats are capable to activate the Nrf2 response against MPP+ toxicity after tBHQ pretreatment. Neurobiol. Aging 2014, 35, 1901–1912. [Google Scholar] [CrossRef]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15–deoxy–Delta(12,14)–prostaglandin j(2). Mol. Cell. Biol. 2004, 24, 36–45. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Shah, S.; MacEwan, D.J. TNF mediates the sustained activation of Nrf2 in human monocytes. J. Immunol. 2011, 187, 702–707. [Google Scholar] [CrossRef]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear erythroid factor 2–mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef]
- Jodar, L.; Mercken, E.M.; Ariza, J.; Younts, C.; Gonzalez–Reyes, J.A.; Alcain, F.J.; Buron, I.; de Cabo, R.; Villalba, J.M. Genetic deletion of Nrf2 promotes immortalization and decreases life span of murine embryonic fibroblasts. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66, 247–256. [Google Scholar] [CrossRef]
- Zhang, Y.; Unnikrishnan, A.; Deepa, S.S.; Liu, Y.; Li, Y.; Ikeno, Y.; Sosnowska, D.; Van Remmen, H.; Richardson, A. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1(-/)(-) mice is correlated to increased cellular senescence. Redox Biol. 2017, 11, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; Mancuso, E.; Spiga, R.; Giuliani, A.; Matacchione, G.; Lazzarini, R.; Marcheselli, F.; Recchioni, R.; Testa, R.; et al. Short–term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. 2018, 15, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Corenblum, M.J.; Ray, S.; Remley, Q.W.; Long, M.; Harder, B.; Zhang, D.D.; Barnes, C.A.; Madhavan, L. Reduced Nrf2 expression mediates the decline in neural stem cell function during a critical middle-age period. Aging Cell 2016, 15, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2017, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Ziros, P.G.; Sykiotis, G.P.; Zaravinos, A.; Psyrogiannis, A.I.; Kyriazopoulou, V.E.; Spandidos, D.A.; Habeos, I.G. Nrf2 activation diminishes during adipocyte differentiation of ST2 cells. Int. J. Mol. Med. 2011, 28, 823–828. [Google Scholar]
- Hou, Y.; Xue, P.; Bai, Y.; Liu, D.; Woods, C.G.; Yarborough, K.; Fu, J.; Zhang, Q.; Sun, G.; Collins, S. Nuclear factor erythroid-derived factor 2–related factor 2 regulates transcription of CCAAT/enhancer-binding protein β during adipogenesis. Free Radic. Biol. Med. 2012, 52, 462–472. [Google Scholar] [CrossRef]
- Xu, J.; Kulkarni, S.R.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin–deficient mice. Diabetes 2012, 61, 3208–3218. [Google Scholar] [CrossRef]
- Xu, J.; Donepudi, A.C.; More, V.R.; Kulkarni, S.R.; Li, L.; Guo, L.; Yan, B.; Chatterjee, T.; Weintraub, N.; Slitt, A.L. Deficiency in N rf2 transcription factor decreases adipose tissue mass and hepatic lipid accumulation in leptin–deficient mice. Obesity 2015, 23, 335–344. [Google Scholar] [CrossRef]
- Yu, Z.; Shao, W.; Chiang, Y.; Foltz, W.; Zhang, Z.; Ling, W.; Fantus, I.G.; Jin, T. Oltipraz upregulates the nuclear factor (erythroid–derived 2)–like 2 [corrected] (NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high–fat diet in C57BL/6J mice. Diabetologia 2011, 54, 922–934. [Google Scholar] [CrossRef]
- Shin, S.M.; Kim, S.G. Inhibition of arachidonic acid and iron–induced mitochondrial dysfunction and apoptosis by oltipraz and novel 1,2-dithiole–3–thione congeners. Mol. Pharmacol. 2009, 75, 242–253. [Google Scholar] [CrossRef]
- Shin, S.; Wakabayashi, N.; Misra, V.; Biswal, S.; Lee, G.H.; Agoston, E.S.; Yamamoto, M.; Kensler, T.W. NRF2 modulates aryl hydrocarbon receptor signaling: Influence on adipogenesis. Mol. Cell. Biol. 2007, 27, 7188–7197. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B.; Ludwig, D.S. Obesity, and impaired metabolic health in patients with COVID–19. Nat. Rev. Endocrinol. 2020, 16, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Tian, M.; Spirohn, K.; Boutros, M.; Bohmann, D. Keap1–independent regulation of Nrf2 activity by protein acetylation and a BET bromodomain protein. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- El-Bab, M.F.; Zaki, N.S.; Mojaddidi, M.A.; Al-Barry, M.; El-Beshbishy, H.A. Diabetic retinopathy is associated with oxidative stress and mitigation of gene expression of antioxidant enzymes. Int. J. Gen. Med. 2013, 6, 799. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vanessa Fiorentino, T.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia–induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Block, K. Nox as a target for diabetic complications. Clin. Sci. 2013, 125, 361–382. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit–Dahm, K. Diabetes and kidney disease: Role of oxidative stress. Antioxid. Redox Signal 2016, 25, 657–684. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I.H., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress, and therapeutic strategies. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 2709–2729. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Jong, C.J.; Mozaffari, M. Role of oxidative stress in diabetes–mediated vascular dysfunction: Unifying hypothesis of diabetes revisited. Vasc. Pharmacol. 2012, 57, 139–149. [Google Scholar] [CrossRef] [PubMed]
- De Haan, J.B. Nrf2 activators as attractive therapeutics for diabetic nephropathy. Diabetes 2011, 60, 2683–2684. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Reisman, S.A.; Yeager, R.L.; Goedken, M.J.; Klaassen, C.D. Nuclear factor erythroid 2-related factor 2 deletion impairs glucose tolerance and exacerbates hyperglycemia in type 1 diabetic mice. J. Pharmacol. Exp. Ther. 2010, 333, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Bhakkiyalakshmi, E.; Sireesh, D.; Rajaguru, P.; Paulmurugan, R.; Ramkumar, K.M. The emerging role of redox-sensitive Nrf2–Keap1 pathway in diabetes. Pharmacol. Res. 2015, 91, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.H.; Schachner, D.; Werner, E.R.; Dirsch, V.M. Active NF–E2–related factor (Nrf2) contributes to keep endothelial NO synthase (eNOS) in the coupled state role of reactive oxygen species (ROS), eNOS, and heme oxygenase (Ho–1) levels. J. Biol. Chem. 2009, 284, 31579–31586. [Google Scholar] [CrossRef]
- Uruno, A.; Yagishita, Y.; Yamamoto, M. The Keap1–Nrf2 system and diabetes mellitus. Arch. Biochem. Biophys. 2015, 566, 76–84. [Google Scholar] [CrossRef]
- Beyer, T.A.; Xu, W.; Teupser, D.; Auf dem Keller, U.; Bugnon, P.; Hildt, E.; Thiery, J.; Kan, Y.W.; Werner, S. Impaired liver regeneration in Nrf2 knockout mice: Role of ROS–mediated insulin/IGF–1 resistance. EMBO J. 2008, 27, 212–223. [Google Scholar] [CrossRef]
- Camer, D.; Yu, Y.; Szabo, A.; Dinh, C.H.; Wang, H.; Cheng, L.; Huang, X.-F. Bardoxolone methyl prevents insulin resistance and the development of hepatic steatosis in mice fed a high–fat diet. Mol. Cell. Endocrinol. 2015, 412, 36–43. [Google Scholar] [CrossRef]
- Jung, K.-A.; Kwak, M.-K. The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules 2010, 15, 7266–7291. [Google Scholar] [CrossRef]
- Zakaryan, H.; Arabyan, E.; Oo, A.; Zandi, K. Flavonoids: Promising natural compounds against viral infections. Arch. Virol. 2017, 162, 2539–2551. [Google Scholar] [CrossRef]
- Chiang, L.; Chiang, W.; Liu, M.; Lin, C. In vitro antiviral activities of Caesalpinia pulcherrima and its related flavonoids. J. Antimicrob. Chemother. 2003, 52, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Lyu, S.-Y.; Rhim, J.-Y.; Park, W.-B. Antiherpetic activities of flavonoids against herpes simplex virus type 1 (HSV–1) and type 2 (HSV–2) in vitro. Arch. Pharmacal. Res. 2005, 28, 1293–1301. [Google Scholar] [CrossRef]
- Evers, D.L.; Chao, C.-F.; Wang, X.; Zhang, Z.; Huong, S.-M.; Huang, E.-S. Human cytomegalovirus–inhibitory flavonoids: Studies on antiviral activity and mechanism of action. Antivir. Res. 2005, 68, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.B.; Jeong, H.J.; Kim, J.H.; Kim, Y.M.; Park, J.Y.; Kim, D.; Nguyen, T.T.; Park, S.J.; Chang, J.S.; Park, K.H.; et al. Biflavonoids from Torreya nucifera displaying SARS–CoV 3CL(pro) inhibition. Bioorg. Med. Chem. 2010, 18, 7940–7947. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Woo, H.J.; Kang, H.K.; Nguyen, V.D.; Kim, Y.M.; Kim, D.W.; Ahn, S.A.; Xia, Y.; Kim, D. Flavonoid–mediated inhibition of SARS coronavirus 3C-like protease expressed in Pichia pastoris. Biotechnol. Lett. 2012, 34, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Barve, A.; Khor, T.-O.; Shen, G.-x.; Lin, W.; Chan, J.Y.; Cai, L.; Kong, A.-N. Regulation of Nrf2–and AP–1–mediated gene expression by epigallocatechin–3–gallate and sulforaphane in prostate of Nrf2–knockout or C57BL/6J mice and PC-3 AP-1 human prostate cancer cells. Acta Pharmacol. Sin. 2010, 31, 1223–1240. [Google Scholar] [CrossRef]
- Shinkai, Y.; Sumi, D.; Fukami, I.; Ishii, T.; Kumagai, Y. Sulforaphane, an activator of Nrf2, suppresses cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. FEBS Lett. 2006, 580, 1771–1774. [Google Scholar] [CrossRef]
- Narotzki, B.; Reznick, A.Z.; Aizenbud, D.; Levy, Y. Green tea: A promising natural product in oral health. Arch. Oral Biol. 2012, 57, 429–435. [Google Scholar] [CrossRef]
- Chacko, S.M.; Thambi, P.T.; Kuttan, R.; Nishigaki, I. Beneficial effects of green tea: A literature review. Chin. Med. 2010, 5, 13. [Google Scholar] [CrossRef]
- Yang, C.S.; Wang, H. Cancer Preventive Activities of Tea Catechins. Molecules 2016, 21, 1679. [Google Scholar] [CrossRef]
- Xu, J.; Xu, Z.; Zheng, W. A Review of the Antiviral Role of Green Tea Catechins. Molecules 2017, 22, 1337. [Google Scholar] [CrossRef]
- Mandel, S.A.; Youdim, M.B. In the rush for green gold: Can green tea delay age-progressive brain neurodegeneration? Recent Pat. CNS Drug Discov. 2012, 7, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D.; Kennett, M.J.; Sang, S.; Reuhl, K.R.; Ju, J.; Yang, C.S. Hepatotoxicity of high oral dose (-)-epigallocatechin–3–gallate in mice. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2010, 48, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L. Hepatotoxicity associated with supplements containing Chinese green tea (Camellia sinensis). Ann. Intern. Med. 2006, 144, 68–71. [Google Scholar] [CrossRef]
- Federico, A.; Tiso, A.; Loguercio, C. A case of hepatotoxicity caused by green tea. Free Radic. Biol. Med. 2007, 43, 474. [Google Scholar] [CrossRef]
- Kumar, N.B.; Pow-Sang, J.; Spiess, P.E.; Park, J.; Salup, R.; Williams, C.R.; Parnes, H.; Schell, M.J. Randomized, placebo–controlled trial evaluating the safety of one–year administration of green tea catechins. Oncotarget 2016, 7, 70794–70802. [Google Scholar] [CrossRef]
- Pavlovic, J.; Zürcher, T.; Haller, O.; Staeheli, P. Resistance to influenza virus and vesicular stomatitis virus conferred by expression of human MxA protein. J. Virol. 1990, 64, 3370–3375. [Google Scholar] [CrossRef]
- Schlee, M.; Hartmann, G. The chase for the RIG-I ligand—recent advances. Mol. Ther. 2010, 18, 1254–1262. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG–I has an essential function in double–stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Na, H.-K.; Kim, E.-H.; Jung, J.-H.; Lee, H.-H.; Hyun, J.-W.; Surh, Y.-J. Epigallocatechin gallate induces Nrf2–mediated antioxidant enzyme expression via activation of PI3K and ERK in human mammary epithelial cells. Arch. Biochem. Biophys. 2008, 476, 171–177. [Google Scholar] [CrossRef]
- Guerrero-Beltrán, C.E.; Calderón-Oliver, M.; Pedraza-Chaverri, J.; Chirino, Y.I. Protective effect of sulforaphane against oxidative stress: Recent advances. Exp. Toxicol. Pathol. 2012, 64, 503–508. [Google Scholar] [CrossRef]
- Wan, S.B.; Landis-Piwowar, K.R.; Kuhn, D.J.; Chen, D.; Dou, Q.P.; Chan, T.H. Structure–activity study of epi–gallocatechin gallate (EGCG) analogs as proteasome inhibitors. Bioorganic Med. Chem. 2005, 13, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Chen, Y.; Li, R.C. Oral absorption, and bioavailability of tea catechins. Planta Med. 2000, 66, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Hsu, M.; Hsieh, C.; Lin, J.; Lai, P.; Wung, B. Upregulation of heme oxygenase-1 by Epigallocatechin-3–gallate via the phosphatidylinositol 3–kinase/Akt and ERK pathways. Life Sci. 2006, 78, 2889–2897. [Google Scholar] [CrossRef] [PubMed]
- Ali, B.H.; Blunden, G. Pharmacological and toxicological properties of Nigella sativa. Phytother. Res. 2003, 17, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Entok, E.; Ustuner, M.C.; Ozbayer, C.; Tekin, N.; Akyuz, F.; Yangi, B.; Kurt, H.; Degirmenci, I.; Gunes, H.V. Anti–inflammatuar and anti–oxidative effects of Nigella sativa L.: 18FDG-PET imaging of inflammation. Mol. Biol. Rep. 2014, 41, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- Molla, S.; Azad, A.; Hasib, A.; Hossain, M.; Ahammed, S.; Rana, S.; Islam, M. A review on antiviral effects of nigella sativa L. Pharmacol. OnLine 2019, 2, 47–53. [Google Scholar]
- Tavakkoli, A.; Mahdian, V.; Razavi, B.M.; Hosseinzadeh, H. Review on Clinical Trials of Black Seed (Nigella sativa) and Its Active Constituent, Thymoquinone. J. Pharmacopunct. 2017, 20, 179–193. [Google Scholar] [CrossRef]
- Amini, M.; Fallah Huseini, H.; Mohtashami, R.; Sadeqhi, Z.; Ghamarchehre, M. Hypolipidemic Effects of Nigella sativa L. Seeds Oil in Healthy Volunteers: A Randomized, Double–Blind, Placebo–Controlled Clinical Trial. J. Med. Plants 2011, 4, 133–138. [Google Scholar]
- Fallah Huseini, H.; Amini, M.; Mohtashami, R.; Ghamarchehre, M.E.; Sadeqhi, Z.; Kianbakht, S.; Fallah Huseini, A. Blood pressure lowering effect of Nigella sativa L. seed oil in healthy volunteers: A randomized, double–blind, placebo-controlled clinical trial. Phytother Res. 2013, 27, 1849–1853. [Google Scholar] [CrossRef]
- Bamosa, A.O.; Kaatabi, H.; Lebdaa, F.M.; Elq, A.; Al-Sultanb, A. Effect of Nigella sativa seeds on the glycemic control of patients with type 2 diabetes mellitus. Indian J. Physiol Pharm. 2010, 54, 344–354. [Google Scholar]
- Qidwai, W.; Hamza, H.B.; Qureshi, R.; Gilani, A. Effectiveness, safety, and tolerability of powdered Nigella sativa (kalonji) seed in capsules on serum lipid levels, blood sugar, blood pressure, and body weight in adults: Results of a randomized, double-blind controlled trial. J. Altern. Complementary Med. 2009, 15, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Padhye, S.; Azmi, A.; Wang, Z.; Philip, P.A.; Kucuk, O.; Sarkar, F.H.; Mohammad, R.M. Review on molecular and therapeutic potential of thymoquinone in cancer. Nutr. Cancer 2010, 62, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Badary, O.A.; Gamal El-Din, A.M. Inhibitory effects of thymoquinone against 20–methylcholanthrene-induced fibrosarcoma tumorigenesis. Cancer Detect. Prev. 2001, 25, 362–368. [Google Scholar] [PubMed]
- Elbarbry, F.; Ragheb, A.; Marfleet, T.; Shoker, A. Modulation of hepatic drug metabolizing enzymes by dietary doses of thymoquinone in female New Zealand White rabbits. Phytother. Res. 2012, 26, 1726–1730. [Google Scholar] [CrossRef]
- Kanter, M.; Demir, H.; Karakaya, C.; Ozbek, H. Gastroprotective activity of Nigella sativa L oil and its constituent, thymoquinone against acute alcohol–induced gastric mucosal injury in rats. World J. Gastroenterol 2005, 11, 6662–6666. [Google Scholar] [CrossRef]
- Prawan, A.; Kundu, J.K.; Surh, Y.J. Molecular basis of heme oxygenase-1 induction: Implications for chemoprevention and chemoprotection. Antioxid. Redox Signal. 2005, 7, 1688–1703. [Google Scholar] [CrossRef]
- Kundu, J.; Kim, D.H.; Kundu, J.K.; Chun, K.S. Thymoquinone induces heme oxygenase–1 expression in HaCaT cells via Nrf2/ARE activation: Akt and AMPKα as upstream targets. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2014, 65, 18–26. [Google Scholar] [CrossRef]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt–dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Velagapudi, R.; Kumar, A.; Bhatia, H.S.; El-Bakoush, A.; Lepiarz, I.; Fiebich, B.L.; Olajide, O.A. Inhibition of neuroinflammation by thymoquinone requires activation of Nrf2/ARE signalling. Int. Immunopharmacol. 2017, 48, 17–29. [Google Scholar] [CrossRef]
- Samarghandian, S.; Shoshtari, M.E.; Sargolzaei, J.; Hossinimoghadam, H.; Farahzad, J.A. Anti-tumor activity of safranal against neuroblastoma cells. Pharmacogn. Mag. 2014, 10, S419–S424. [Google Scholar] [CrossRef] [PubMed]
- Desforges, M.; Le Coupanec, A.; Dubeau, P.; Bourgouin, A.; Lajoie, L.; Dubé, M.; Talbot, P.J. Human Coronaviruses and Other Respiratory Viruses: Underestimated Opportunistic Pathogens of the Central Nervous System? Viruses 2020, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Netland, J.; Meyerholz, D.K.; Moore, S.; Cassell, M.; Perlman, S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 2008, 82, 7264–7275. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS–CoV2 may play a role in the respiratory failure of COVID–19 patients. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Banks, W.A. Role of the immune system in HIV–associated neuroinflammation and neurocognitive implications. Brain. Behav. Immun. 2015, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; McKim, D.B.; Sheridan, J.F.; Godbout, J.P. Monocyte trafficking to the brain with stress and inflammation. Front. Neurosci. 2015, 9. [Google Scholar]
- Desforges, M.; Miletti, T.C.; Gagnon, M.; Talbot, P.J. Activation of human monocytes after infection by human coronavirus 229E. Virus Res. 2007, 130, 228–240. [Google Scholar] [CrossRef]
- Arbour, N.; Day, R.; Newcombe, J.; Talbot, P.J. Neuroinvasion by human respiratory coronaviruses. J. Virol. 2000, 74, 8913–8921. [Google Scholar] [CrossRef]
- Troyer, E.A.; Kohn, J.N.; Hong, S. Are we facing a crashing wave of neuropsychiatric sequelae of COVID–19? Neuropsychiatric symptoms and potential immunologic mechanisms. Brain Behav. Immun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, D.; Bland, R.; Walker, E.A.; Bradwell, A.R.; Howie, A.J.; Hewison, M.; Stewart, P.M. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in the human kidney. J. Am. Soc. Nephrol. JASN 1999, 10, 2465–2473. [Google Scholar]
- Gunville, C.F.; Mourani, P.M.; Ginde, A.A. The role of vitamin D in prevention and treatment of infection. Inflamm. Allergy Drug Targets 2013, 12, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Hansdottir, S.; Monick, M.M.; Lovan, N.; Powers, L.; Gerke, A.; Hunninghake, G.W. Vitamin D decreases respiratory syncytial virus induction of NF–kappaB–linked chemokines and cytokines in airway epithelium while maintaining the antiviral state. J. Immunol. 2010, 184, 965–974. [Google Scholar] [CrossRef]
- Hansdottir, S.; Monick, M.M.; Hinde, S.L.; Lovan, N.; Look, D.C.; Hunninghake, G.W. Respiratory epithelial cells convert inactive vitamin D to its active form: Potential effects on host defense. J. Immunol. 2008, 181, 7090–7099. [Google Scholar] [CrossRef]
- Aranow, C. Vitamin D and the immune system. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2011, 59, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Laaksi, I.; Ruohola, J.P.; Tuohimaa, P.; Auvinen, A.; Haataja, R.; Pihlajamäki, H.; Ylikomi, T. An association of serum vitamin D concentrations <40 nmol/L with acute respiratory tract infection in young Finnish men. Am. J. Clin. Nutr. 2007, 86, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Sabetta, J.R.; DePetrillo, P.; Cipriani, R.J.; Smardin, J.; Burns, L.A.; Landry, M.L. Serum 25–hydroxyvitamin d and the incidence of acute viral respiratory tract infections in healthy adults. PLoS ONE 2010, 5, e11088. [Google Scholar] [CrossRef] [PubMed]
- Matthews, L.R.; Ahmed, Y.; Wilson, K.L.; Griggs, D.D.; Danner, O.K. Worsening severity of vitamin D deficiency is associated with increased length of stay, surgical intensive care unit cost, and mortality rate in surgical intensive care unit patients. Am. J. Surg. 2012, 204, 37–43. [Google Scholar] [CrossRef]
- Braun, A.; Chang, D.; Mahadevappa, K.; Gibbons, F.K.; Liu, Y.; Giovannucci, E.; Christopher, K.B. Association of low serum 25-hydroxyvitamin D levels and mortality in the critically ill. Crit. Care Med. 2011, 39, 671. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.M.; Wischmeyer, P.E.; Queensland, K.M.; Sillau, S.H.; Sufit, A.J.; Heyland, D.K. Relationship of vitamin D deficiency to clinical outcomes in critically ill patients. J. Parenter. Enter. Nutr. 2012, 36, 713–720. [Google Scholar] [CrossRef]
- Chen, L.; Yang, R.; Qiao, W.; Zhang, W.; Chen, J.; Mao, L.; Goltzman, D.; Miao, D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2–antioxidant signaling and inactivation of p16/p53–senescence signaling. Aging Cell 2019, 18, e12951. [Google Scholar] [CrossRef]
- Mendes, M.M.; Hart, K.H.; Botelho, P.B.; Lanham–New, S.A. Vitamin D status in the tropics: Is sunlight exposure the main determinant? Nutr. Bull. 2018, 43, 428–434. [Google Scholar] [CrossRef]
- Maeda, S.S.; Kunii, I.S.; Hayashi, L.F.; Lazaretti–Castro, M. Increases in summer serum 25–hydroxyvitamin D (25OHD) concentrations in elderly subjects in São Paulo, Brazil vary with age, gender, and ethnicity. BMC Endocr. Disord. 2010, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Levis, S.; Gomez, A.; Jimenez, C.; Veras, L.; Ma, F.; Lai, S.; Hollis, B.; Roos, B.A. Vitamin d deficiency and seasonal variation in an adult South Florida population. J. Clin. Endocrinol. Metab. 2005, 90, 1557–1562. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendonca, P.; Soliman, K.F.A. Flavonoids Activation of the Transcription Factor Nrf2 as a Hypothesis Approach for the Prevention and Modulation of SARS-CoV-2 Infection Severity. Antioxidants 2020, 9, 659. https://doi.org/10.3390/antiox9080659
Mendonca P, Soliman KFA. Flavonoids Activation of the Transcription Factor Nrf2 as a Hypothesis Approach for the Prevention and Modulation of SARS-CoV-2 Infection Severity. Antioxidants. 2020; 9(8):659. https://doi.org/10.3390/antiox9080659
Chicago/Turabian StyleMendonca, Patricia, and Karam F. A. Soliman. 2020. "Flavonoids Activation of the Transcription Factor Nrf2 as a Hypothesis Approach for the Prevention and Modulation of SARS-CoV-2 Infection Severity" Antioxidants 9, no. 8: 659. https://doi.org/10.3390/antiox9080659
APA StyleMendonca, P., & Soliman, K. F. A. (2020). Flavonoids Activation of the Transcription Factor Nrf2 as a Hypothesis Approach for the Prevention and Modulation of SARS-CoV-2 Infection Severity. Antioxidants, 9(8), 659. https://doi.org/10.3390/antiox9080659