Particulate Matter 2.5 Mediates Cutaneous Cellular Injury by Inducing Mitochondria-Associated Endoplasmic Reticulum Stress: Protective Effects of Ginsenoside Rb1

 ,
,  and
and 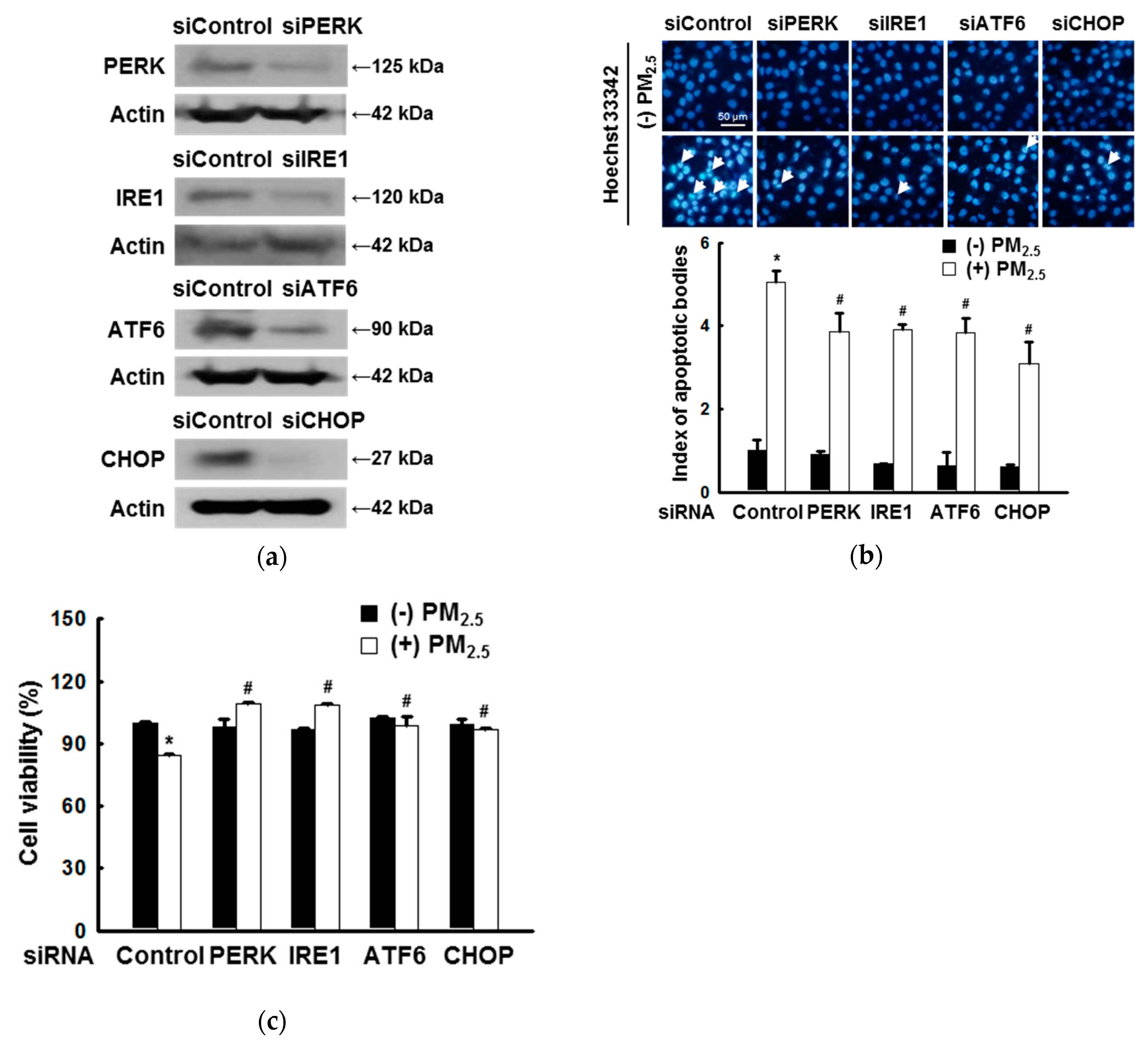{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Transient Transfection with Small Interfering RNA
2.4. Western Blotting
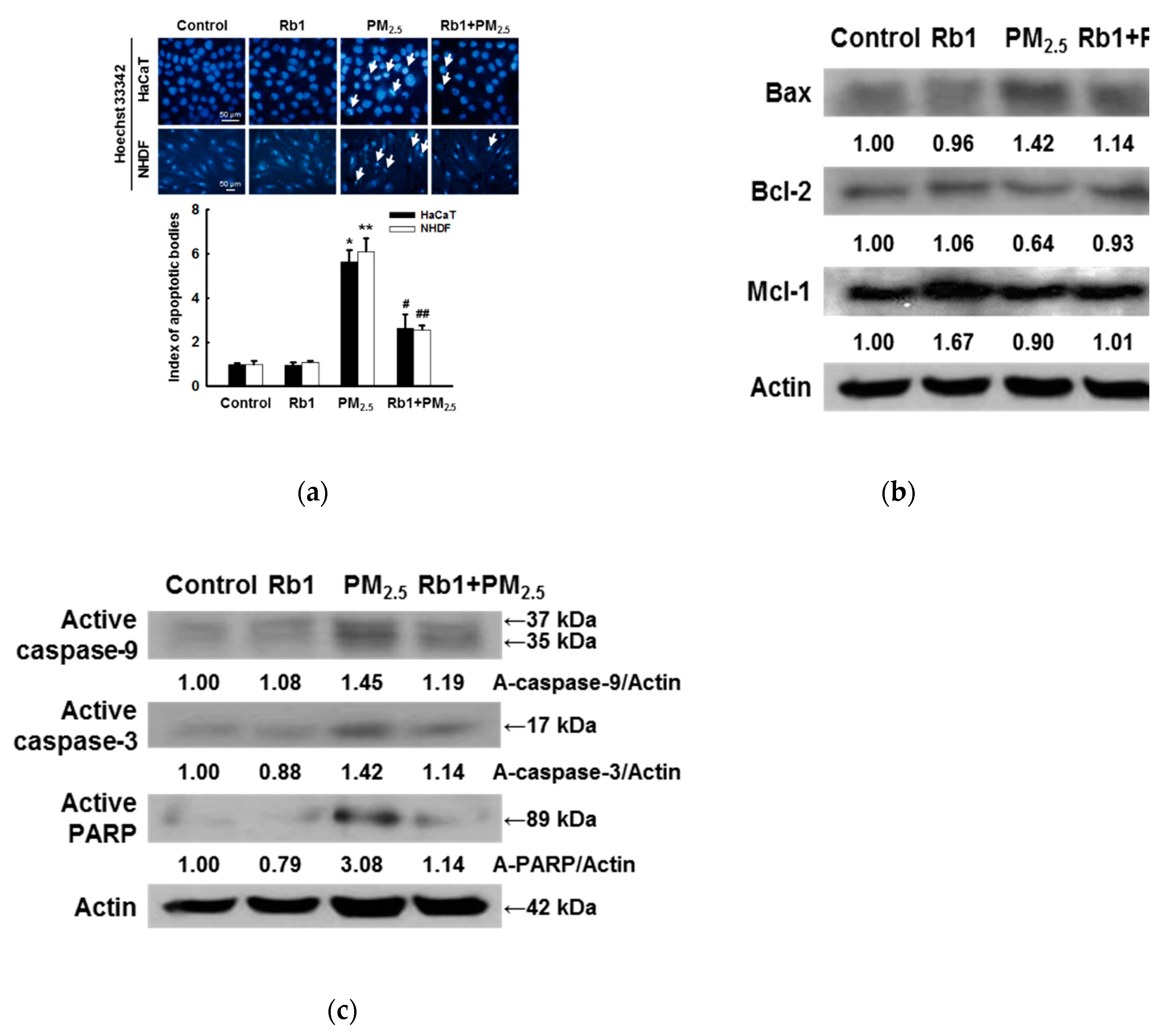
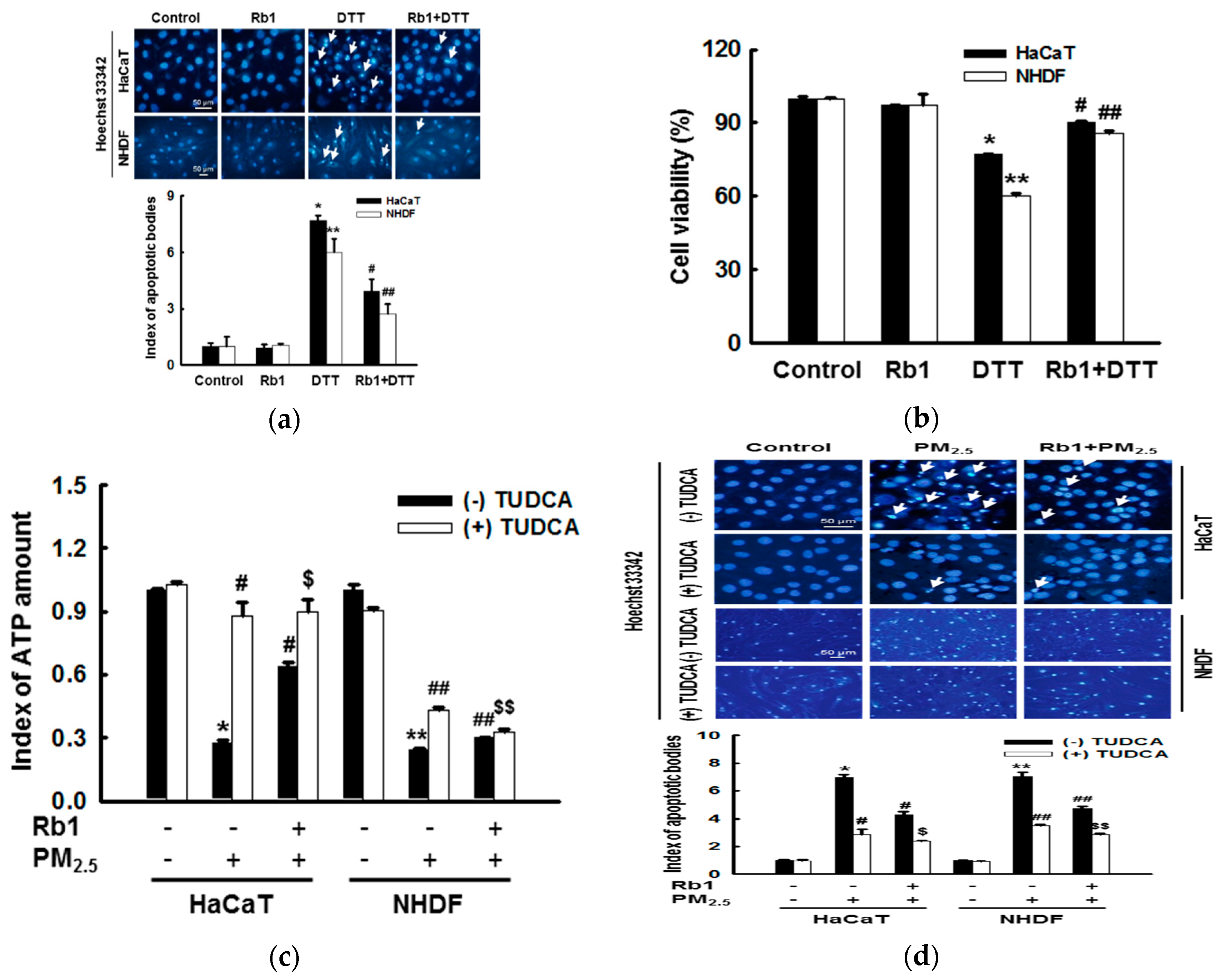
2.5. Detection of Apoptosis
2.6. Cytotoxicity Assay
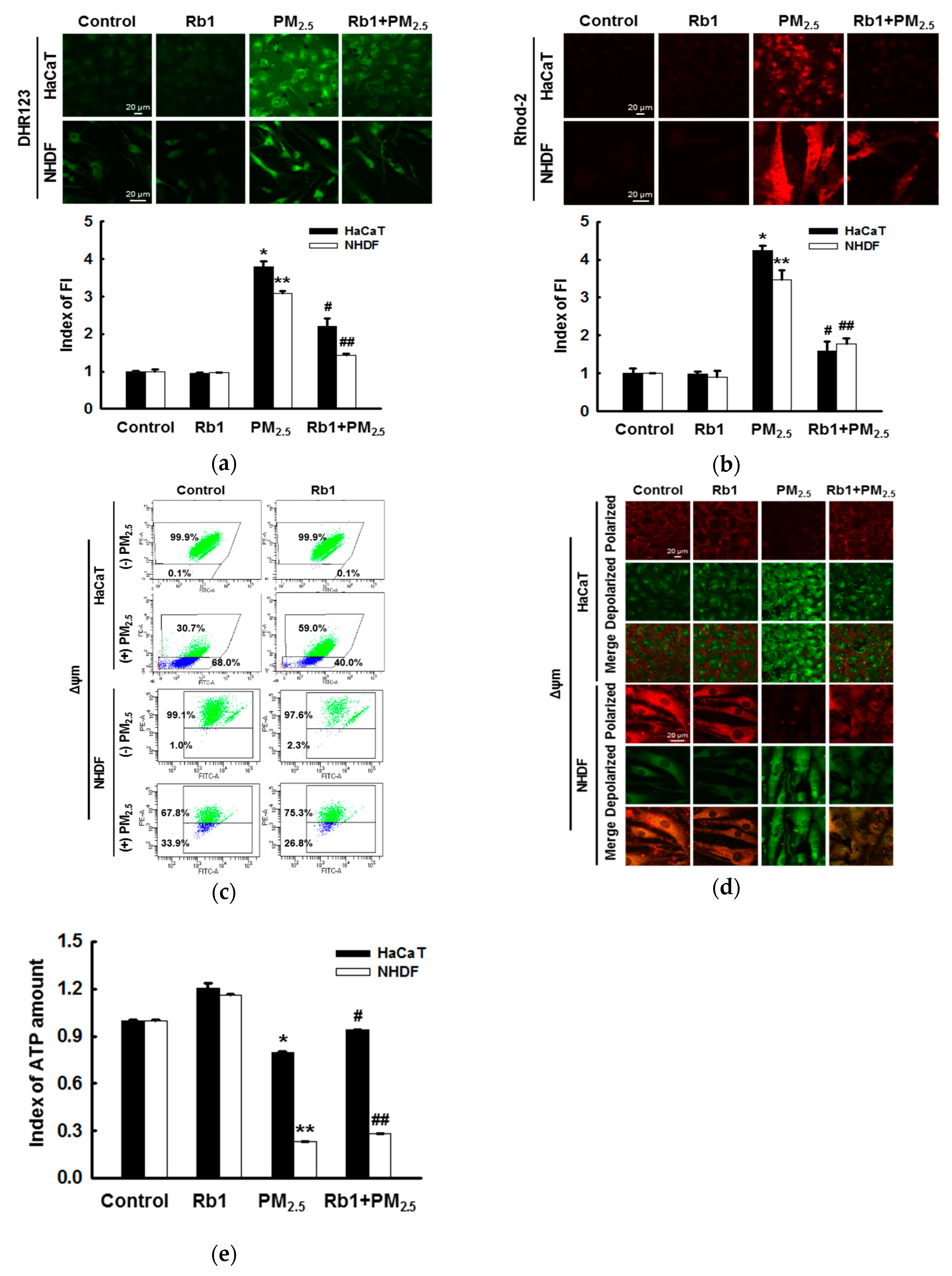
2.7. Detection of ROS
2.8. Single-Cell Gel Electrophoresis Assay (Comet Assay)
2.9. Lipid Peroxidation Assay
2.10. Protein Carbonylation Assay
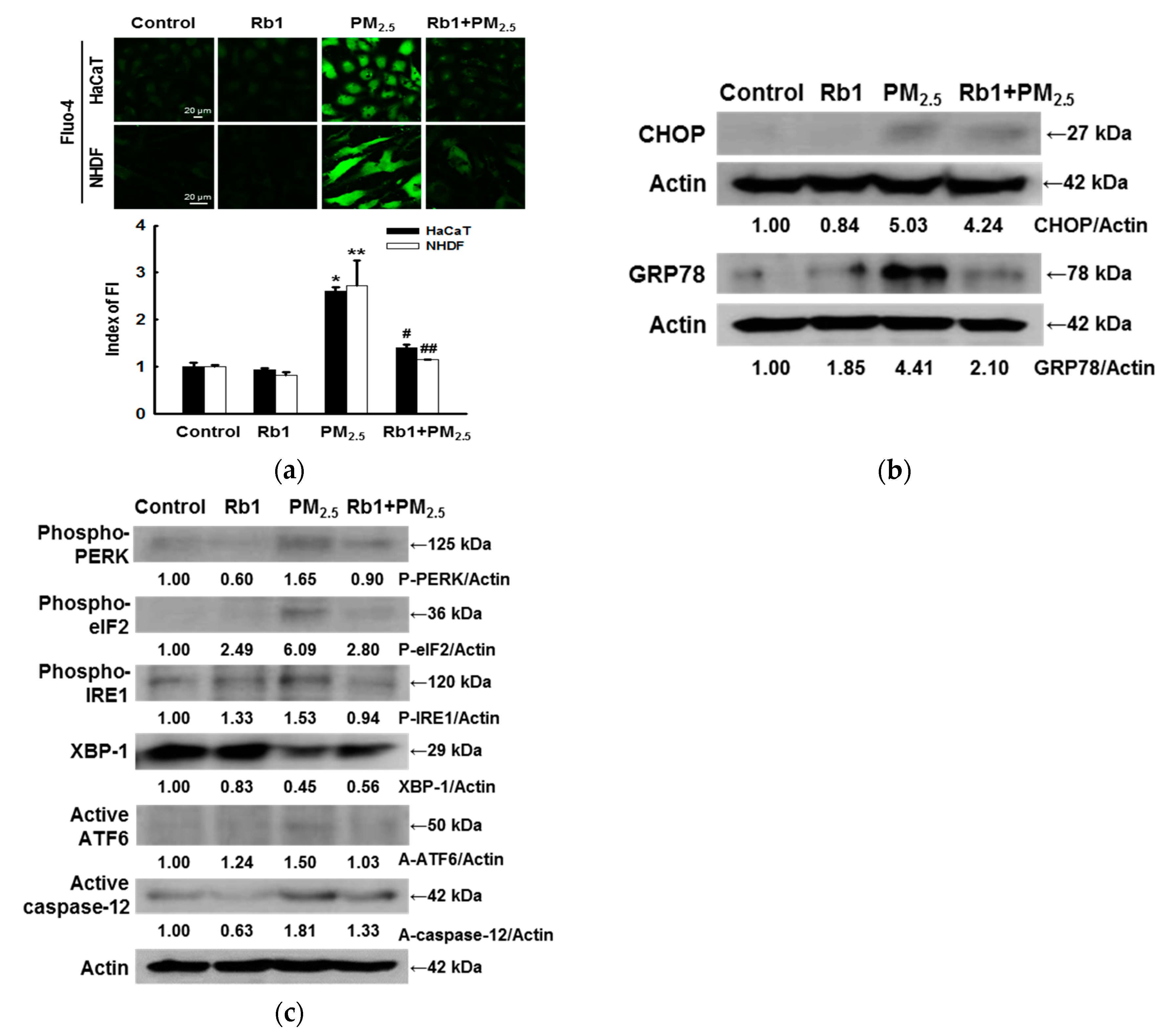
2.11. Quantification of Ca2+ Levels
2.12. Mitochondrial Membrane Potential Measurement
2.13. Detection of ATP Levels
2.14. Statistical Analysis
3. Results
3.1. ER Stress Mediates Apoptosis after Exposure to PM2.5
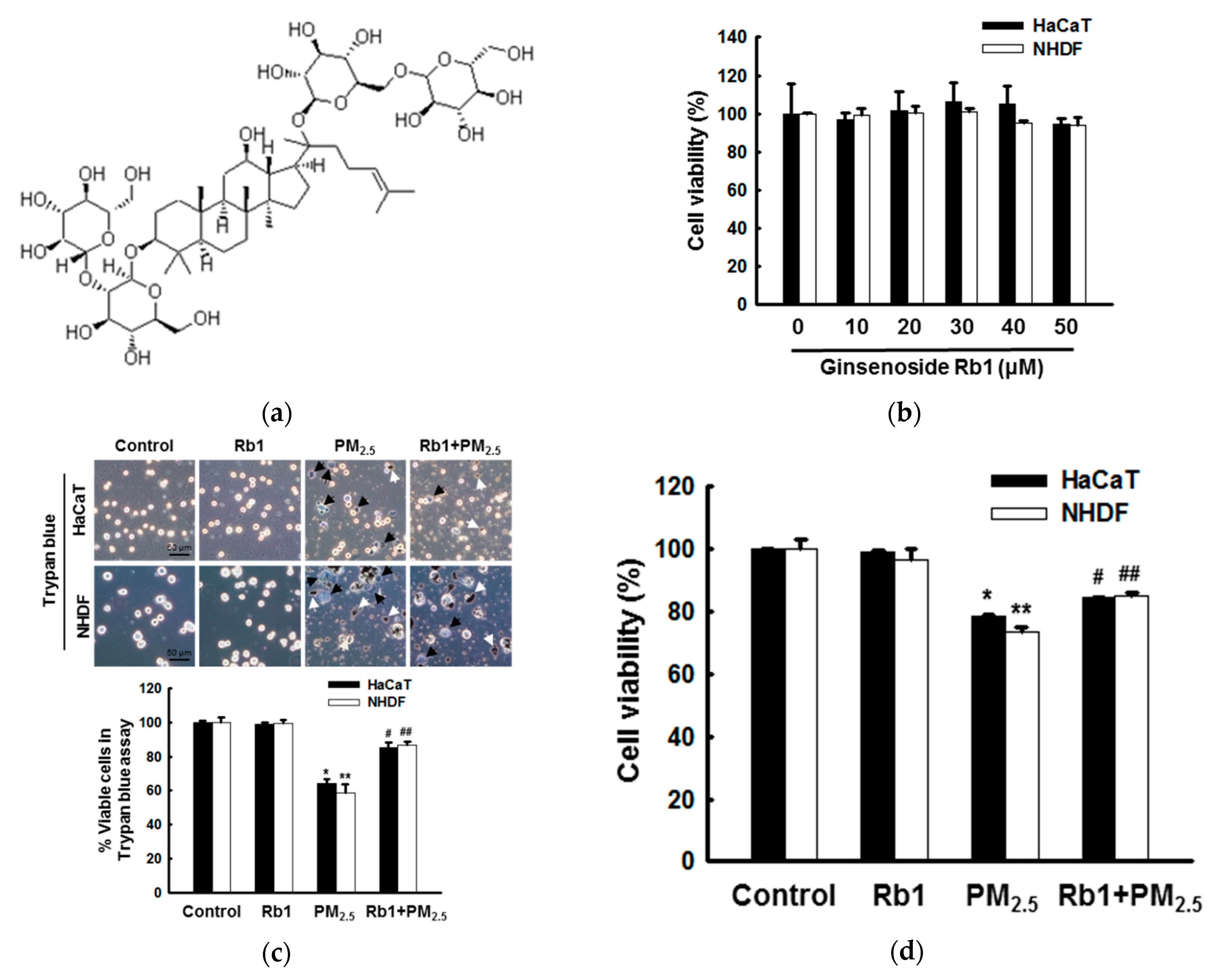
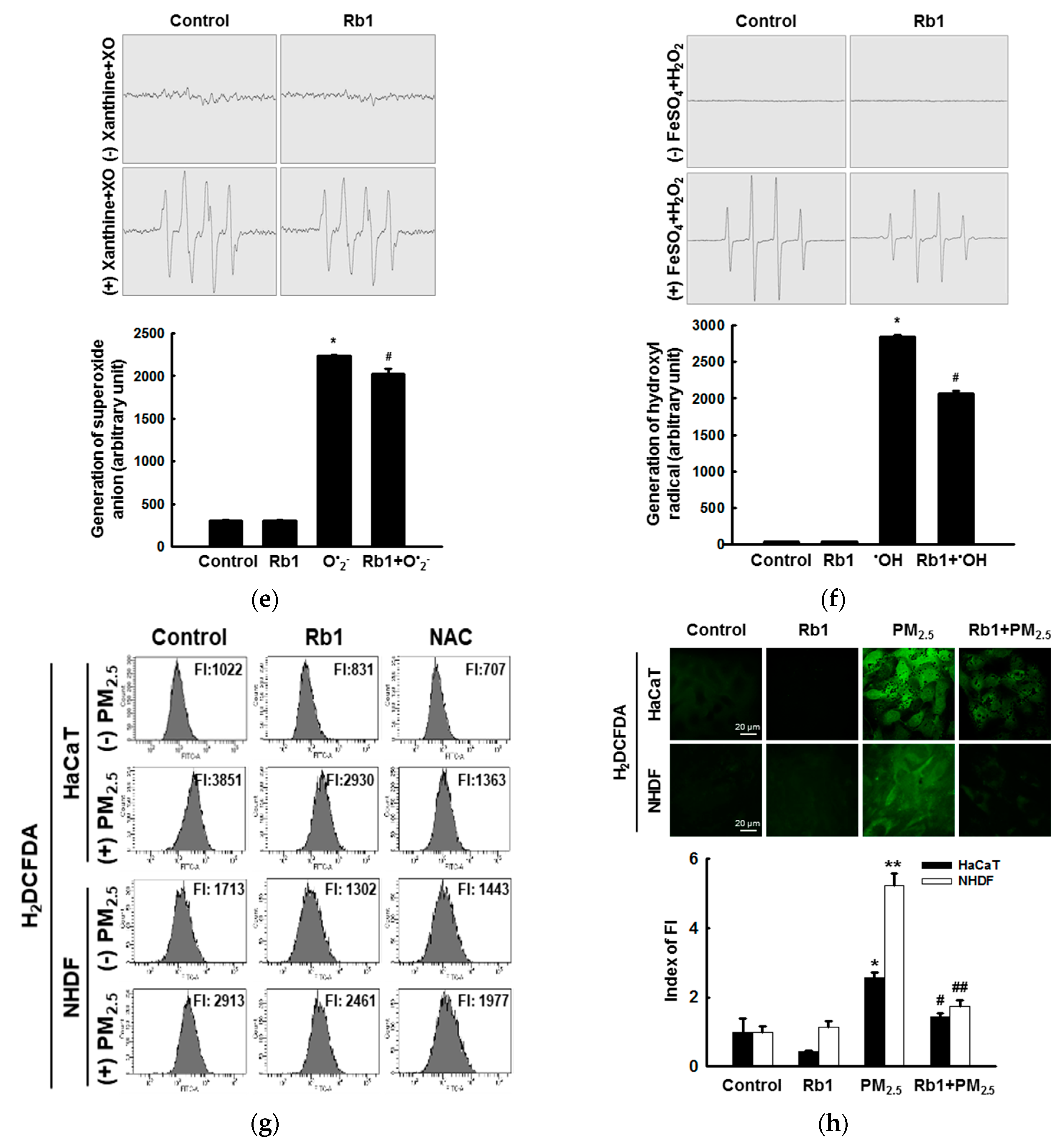
3.2. Ginsenoside Rb1 Confers Protection against PM2.5-Induced ROS
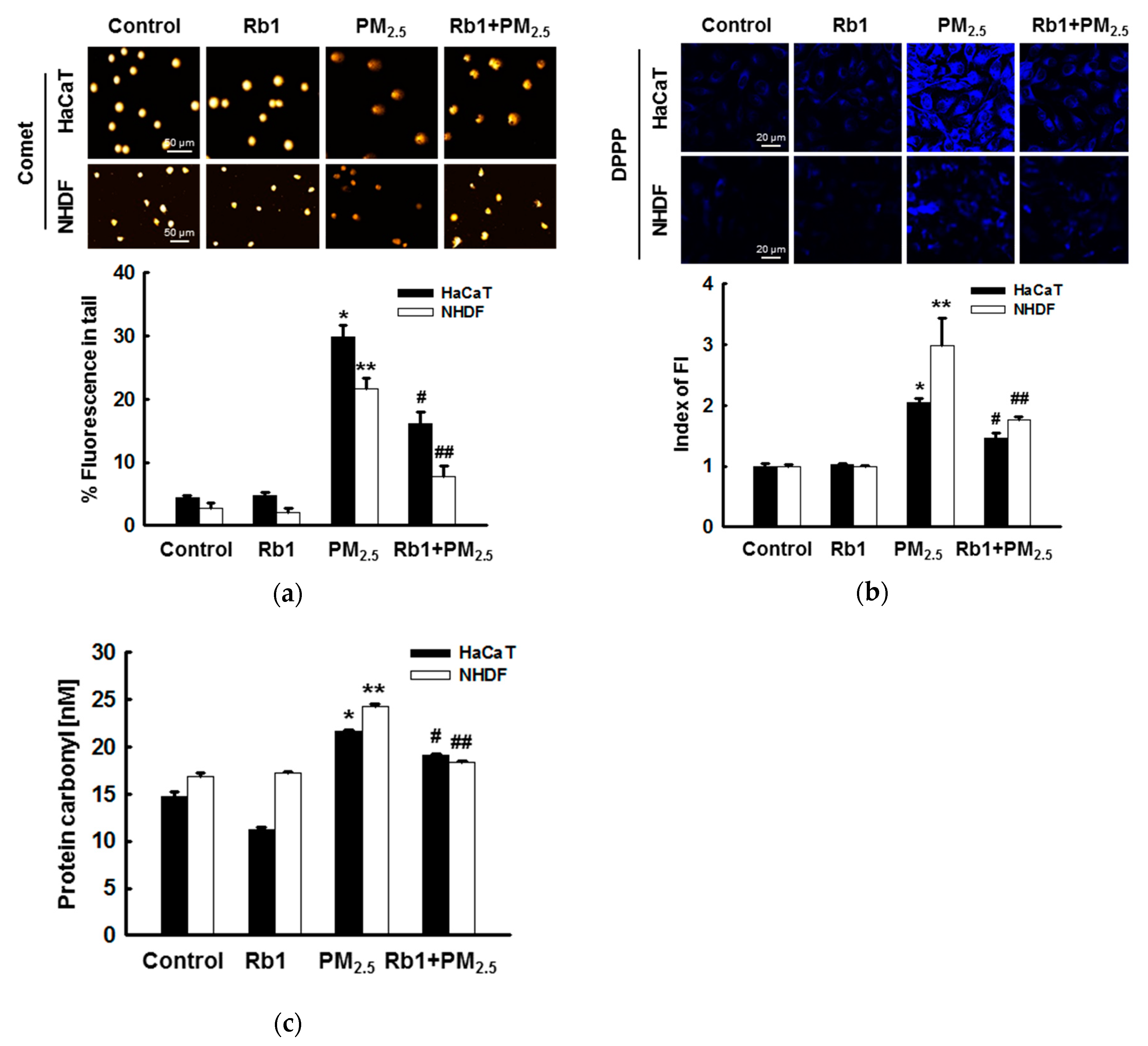
3.3. Ginsenoside Rb1 Confers Protection against PM2.5-Induced Oxidative Stress
3.4. Ginsenoside Rb1 Confers Protection against PM2.5-Induced ER Stress
3.5. Ginsenoside Rb1 Protects against PM2.5-Induced Mitochondrial Damage
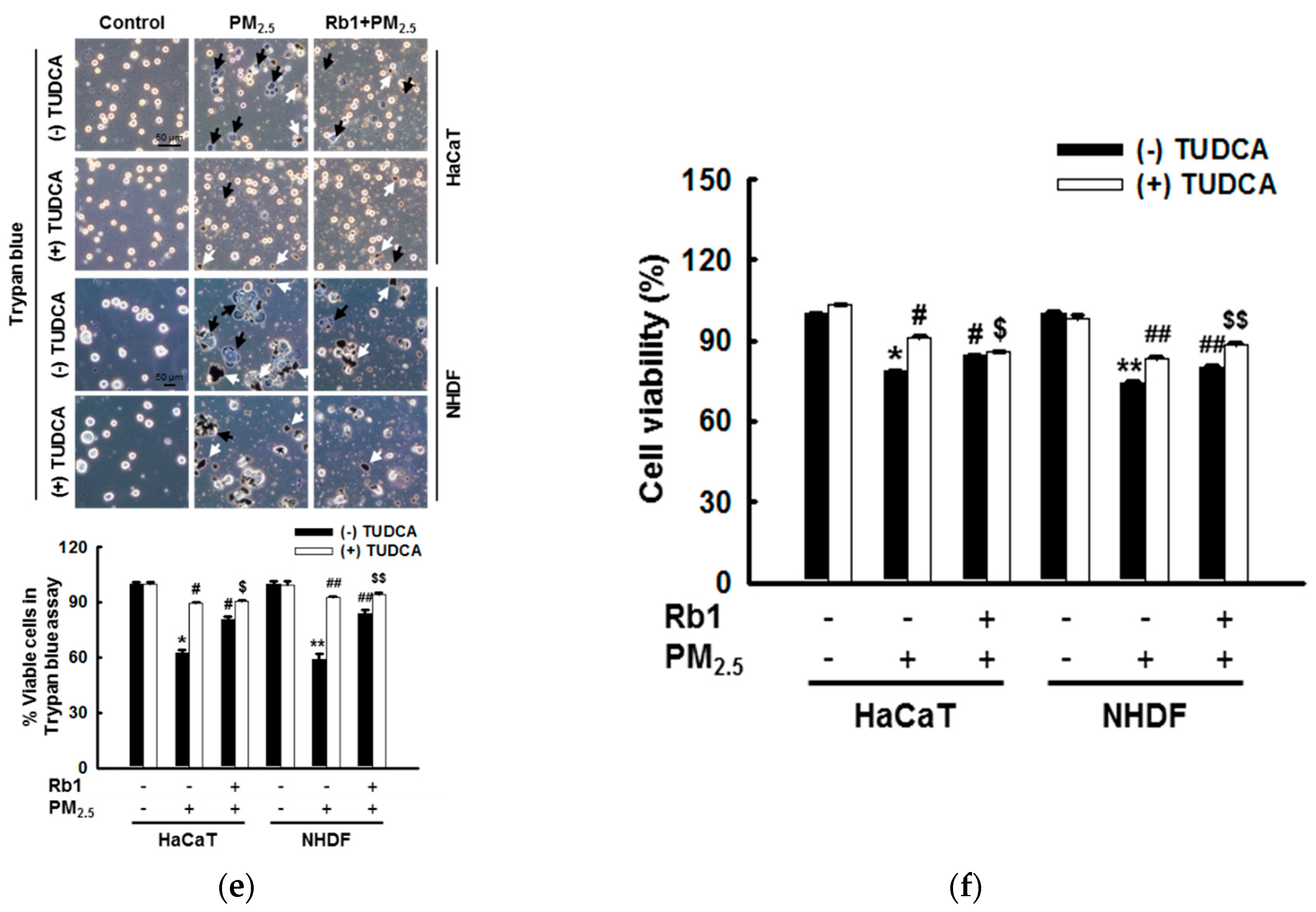
3.6. Ginsenoside Rb1 Protects against PM2.5-Induced Apoptotic Cell Death
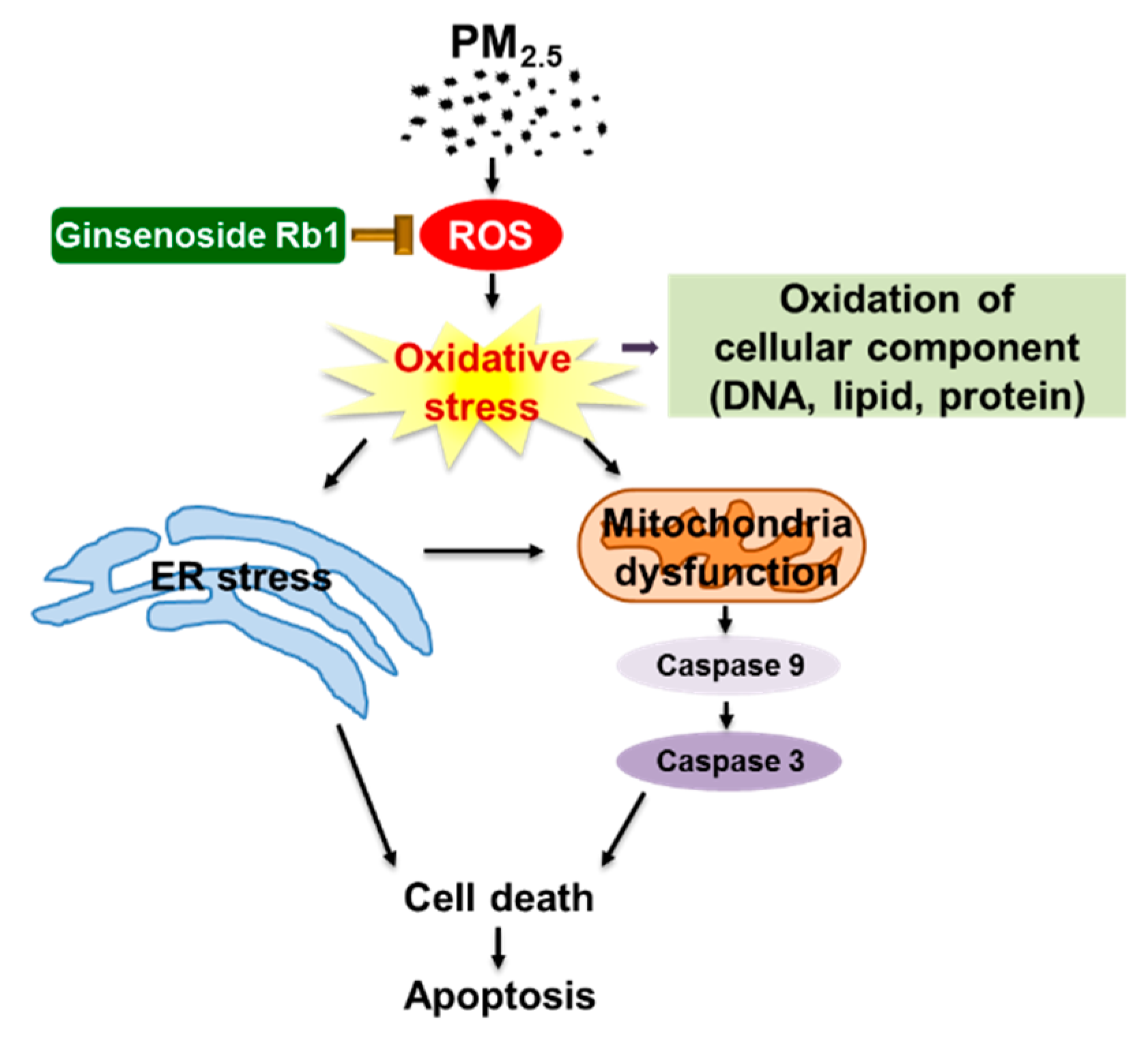
3.7. Ginsenoside Rb1 Protects against Mitochondria-Associated ER Stress-Mediated Apoptosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reid, C.E.; Brauer, M.; Johnston, F.H.; Jerrett, M.; Balmes, J.R.; Elliott, C.T. Critical review of health impacts of wildfire smoke exposure. Environ. Health Perspect. 2016, 124, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Trejo Bittar, H.E.; Yousem, S.A.; Wenzel, S.E. Pathobiology of severe asthma. Annu. Rev. Pathol. 2015, 10, 511–545. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Daiber, A. Environmental stressors and their impact on health and disease with focus on oxidative stress. Antioxid. Redox Signal. 2018, 28, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Cho, H.G.; Park, C.K.; Park, K.H.; Lim, H.B. Comparative in vitro biological toxicity of four kinds of air pollution particles. Toxicol. Res. 2017, 33, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Park, C.G.; Cho, H.K.; Shin, H.J.; Park, K.H.; Lim, H.B. Comparison of mutagenic activities of various ultra-fine particles. Toxicol. Res. 2018, 34, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Krutmann, J.; Liu, W.; Li, L.; Pan, X.; Crawford, M.; Sore, G.; Seite, S. Pollution and skin: From epidemiological and mechanistic studies to clinical implications. J. Dermatol. Sci. 2014, 76, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Lee, J.E.; Myung, C.H.; Park, J.I.; Jo, C.S.; Hwang, J.S. Particulate matter-induced aryl hydrocarbon receptor regulates autophagy in keratinocytes. Biomol. Ther. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Shin, D.W.; Kim, W.; Doh, S.J.; Lee, S.H.; Noh, M. Asian dust storm particles induce a broad toxicological transcriptional program in human epidermal keratinocytes. Toxicol. Lett. 2011, 200, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Xie, X.Y.; Xu, S.K.; Wang, Y.N.; Jiang, M.; Wen, L.R.; Lai, W.; Guan, L. PM2.5 exposure elicits oxidative stress responses and mitochondrial apoptosis pathway activation in HaCaT keratinocytes. Chin. Med. J. (Engl). 2017, 130, 2205–2214. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kang, Z.; Jiang, S.; Zhao, J.; Yan, S.; Xu, F.; Xu, J. Effects of ambient fine particles PM2.5 on human HaCaT cells. Int. J. Environ. Res. Public Health 2017, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Ryu, Y.S.; Hyun, Y.J.; Shilnikova, K.; Zhen, A.X.; Jeong, J.W.; Choi, Y.H.; Kang, H.K.; et al. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch. Toxicol. 2018, 92, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Hyun, Y.M.; Kim, S.H.; Ko, M.K.; Park, C.O.; Hyun, J.W. Particulate matter induces inflammatory cytokine production via activation of NFκB by TLR5-NOX4-ROS signaling in human skin keratinocyte and mouse skin. Redox Biol. 2019, 21, 101080. [Google Scholar] [CrossRef] [PubMed]
- Soldati, F.; Sticher, O. HPLC separation and quantitative determination of ginsenosides from Panax ginseng, Panax quinquefolium and from ginseng drug preparations. 2nd communication. Planta Med. 1980, 39, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Niu, X.Y.; He, X.J.; Shu, J. Ginsenoside Rg1 reduces toxicity of fine particulate matter on human alveolar epithelial cells: A preliminary observation. Mol. Med. Rep. 2014, 9, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Li, C.P.; Qin, G.; Shi, R.Z.; Zhang, M.S.; Lv, J.Y. Ginsenoside Rg1 reduces toxicity of PM(2.5) on human umbilical vein endothelial cells by upregulating intracellular antioxidative state. Environ. Toxicol. Pharmacol. 2013, 35, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Kim, K.; Lim, C.J. Protective properties of ginsenoside Rb1 against UV-B radiation-induced oxidative stress in human dermal keratinocytes. Pharmazie 2015, 70, 381–387. [Google Scholar]
- Huang, J.; Qiu, L.; Ding, L.; Wang, S.; Wang, J.; Zhu, Q.; Song, F.; Hu, J. Ginsenoside Rb1 and paeoniflorin inhibit transient receptor potential vanilloid-1-activated IL-8 and PGE2 production in a human keratinocyte cell line HaCaT. Int. Immunopharmacol. 2010, 10, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Kwok, H.H.; Yue, P.Y.; Mak, N.K.; Wong, R.N. Ginsenoside Rb1 induces type I collagen expression through peroxisome proliferator-activated receptor-delta. Biochem. Pharmacol. 2012, 84, 532–539. [Google Scholar] [CrossRef]
- Hou, J.; Kim, S. Possible role of ginsenoside Rb1 in skin wound healing via regulating senescent skin dermal fibroblast. Biochem. Biophys. Res. Commun. 2018, 499, 381–388. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Ma, Z.; Liang, Q.; Tang, X.; Tan, H.; Xiao, C.; Gao, Y. Ginsenoside Rb1 inhibits doxorubicin-triggered H9C2 cell apoptosis via aryl hydrocarbon receptor. Biomol. Ther. 2017, 25, 202–212. [Google Scholar] [CrossRef]
- Kohno, M.; Mizuta, Y.; Kusai, M.; Masumizu, T.; Makino, K. Measurements of superoxide anion radical and superoxide anion scavenging activity by electron spin resonance spectroscopy coupled with DMPO spin trapping. Bull. Chem. Soc. Jpn. 1994, 67, 1085–1090. [Google Scholar] [CrossRef]
- Li, L.; Abe, Y.; Kanagawa, K.; Usui, N.; Imai, K.; Mashino, T.; Mochizuki, M.; Miyata, N. Distinguishing the 5,5-dimethyl-1-pyrroline N-oxide (DMPO)-OH radical quenching effect from the hydroxyl radical scavenging effect in the ESR spin-trapping method. Anal. Chim. Acta 2004, 512, 121–124. [Google Scholar] [CrossRef]
- Morita, M.; Naito, Y.; Yoshikawa, T.; Niki, E. Plasma lipid oxidation induced by peroxynitrite, hypochlorite, lipoxygenase and peroxyl radicals and its inhibition by antioxidants as assessed by diphenyl-1-pyrenylphosphine. Redox Biol. 2016, 8, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, D.; Varin, A.; Nicolas, V.; Courilleau, D.; Mateo, P.; Caubere, C.; Rouet, P.; Gomez, A.M.; Vandecasteele, G.; et al. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis. 2016, 7, e2198. [Google Scholar] [CrossRef] [PubMed]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012, 3, e430. [Google Scholar] [CrossRef] [PubMed]
- Milakovic, T.; Johnson, G.V. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem. 2005, 280, 30773–30782. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Invest. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Jakobsen, C.H.; Størvold, G.L.; Bremseth, H.; Follestad, T.; Sand, K.; Mack, M.; Olsen, K.S.; Lundemo, A.G.; Iversen, J.G.; Krokan, H.E.; et al. DHA induces ER stress and growth arrest in human colon cancer cells: Associations with cholesterol and calcium homeostasis. J. Lipid Res. 2008, 49, 2089–2100. [Google Scholar] [CrossRef]
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10631–10634. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Yadav, N.; Bhat, T.A.; O’Malley, J.; Kumar, S.; Chandra, D. A potential role of X-linked inhibitor of apoptosis protein in mitochondrial membrane permeabilization and its implication in cancer therapy. Drug Discov. Today 2016, 21, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Jing, G.; Wang, J.J.; Zhang, S.X. ER stress and apoptosis: A new mechanism for retinal cell death. Exp. Diabetes Res. 2012, 2012, 589589. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Plaisance, V.; Brajkovic, S.; Tenenbaum, M.; Favre, D.; Ezanno, H.; Bonnefond, A.; Bonner, C.; Gmyr, V.; Kerr-Conte, J.; Gauthier, B.R.; et al. Endoplasmic reticulum stress links oxidative stress to impaired pancreatic beta-cell function caused by human oxidized LDL. PLoS ONE 2016, 11, e0163046. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Thompson, M.D.; Cohen, R.A.; Tong, X. Endoplasmic reticulum stress and related pathological processes. J. Pharmacol. Biomed. Anal. 2013, 1, 1000107. [Google Scholar] [PubMed]
- Park, J.; Lee, J.; Choi, C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS ONE 2011, 6, e23211. [Google Scholar] [CrossRef] [PubMed]
- Contreras, L.; Drago, I.; Zampese, E.; Pozzan, T. Mitochondria: The calcium connection. Biochim. Biophys. Acta 2010, 1797, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Thomenius, M.J.; Distelhorst, C.W. Bcl-2 on the endoplasmic reticulum: Protecting the mitochondria from a distance. J. Cell Sci. 2003, 116, 4493–4499. [Google Scholar] [CrossRef]
- Yang, T.; Kozopas, K.M.; Craig, R.W. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J. Cell Biol. 1995, 128, 1173–1184. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Guerrero, A.D.; Huang, L.; Shabier, Z.; Pan, M.; Tan, T.H.; Wang, J. Caspase-9-induced mitochondrial disruption through cleavage of anti-apoptotic BCL-2 family members. J. Biol. Chem. 2007, 282, 33888–33895. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piao, M.J.; Kang, K.A.; Zhen, A.X.; Fernando, P.D.S.M.; Ahn, M.J.; Koh, Y.S.; Kang, H.K.; Yi, J.M.; Choi, Y.H.; Hyun, J.W. Particulate Matter 2.5 Mediates Cutaneous Cellular Injury by Inducing Mitochondria-Associated Endoplasmic Reticulum Stress: Protective Effects of Ginsenoside Rb1. Antioxidants 2019, 8, 383. https://doi.org/10.3390/antiox8090383
Piao MJ, Kang KA, Zhen AX, Fernando PDSM, Ahn MJ, Koh YS, Kang HK, Yi JM, Choi YH, Hyun JW. Particulate Matter 2.5 Mediates Cutaneous Cellular Injury by Inducing Mitochondria-Associated Endoplasmic Reticulum Stress: Protective Effects of Ginsenoside Rb1. Antioxidants. 2019; 8(9):383. https://doi.org/10.3390/antiox8090383
Chicago/Turabian StylePiao, Mei Jing, Kyoung Ah Kang, Ao Xuan Zhen, Pincha Devage Sameera Madushan Fernando, Mee Jung Ahn, Young Sang Koh, Hee Kyoung Kang, Joo Mi Yi, Yung Hyun Choi, and Jin Won Hyun. 2019. "Particulate Matter 2.5 Mediates Cutaneous Cellular Injury by Inducing Mitochondria-Associated Endoplasmic Reticulum Stress: Protective Effects of Ginsenoside Rb1" Antioxidants 8, no. 9: 383. https://doi.org/10.3390/antiox8090383
APA StylePiao, M. J., Kang, K. A., Zhen, A. X., Fernando, P. D. S. M., Ahn, M. J., Koh, Y. S., Kang, H. K., Yi, J. M., Choi, Y. H., & Hyun, J. W. (2019). Particulate Matter 2.5 Mediates Cutaneous Cellular Injury by Inducing Mitochondria-Associated Endoplasmic Reticulum Stress: Protective Effects of Ginsenoside Rb1. Antioxidants, 8(9), 383. https://doi.org/10.3390/antiox8090383