KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced S-Glutathionylation of Distinct Structural and Regulatory Proteins

 , , , , ,
, , , , ,  , , ,
, , , 

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Treatment
2.3. Cell Transfection and siRNA-Mediated Gene Silencing
2.4. Preparation of Liposomes
2.5. Preparation of Cellular Extracts
2.6. Measurement of Cellular Levels of Reactive Oxidative Species
2.7. Total Oxyradical Scavenging Capacity Assay
2.8. Quantitative Determination of Glutathione and Glutathione Disulfide Levels
2.9. Enzymatic Activity Assays
2.10. Western Blotting Analysis
2.11. Immunoprecipitation
2.12. Two-Dimensional Polyacrylamide Gel Electrophoresis (2D PAGE) Analysis
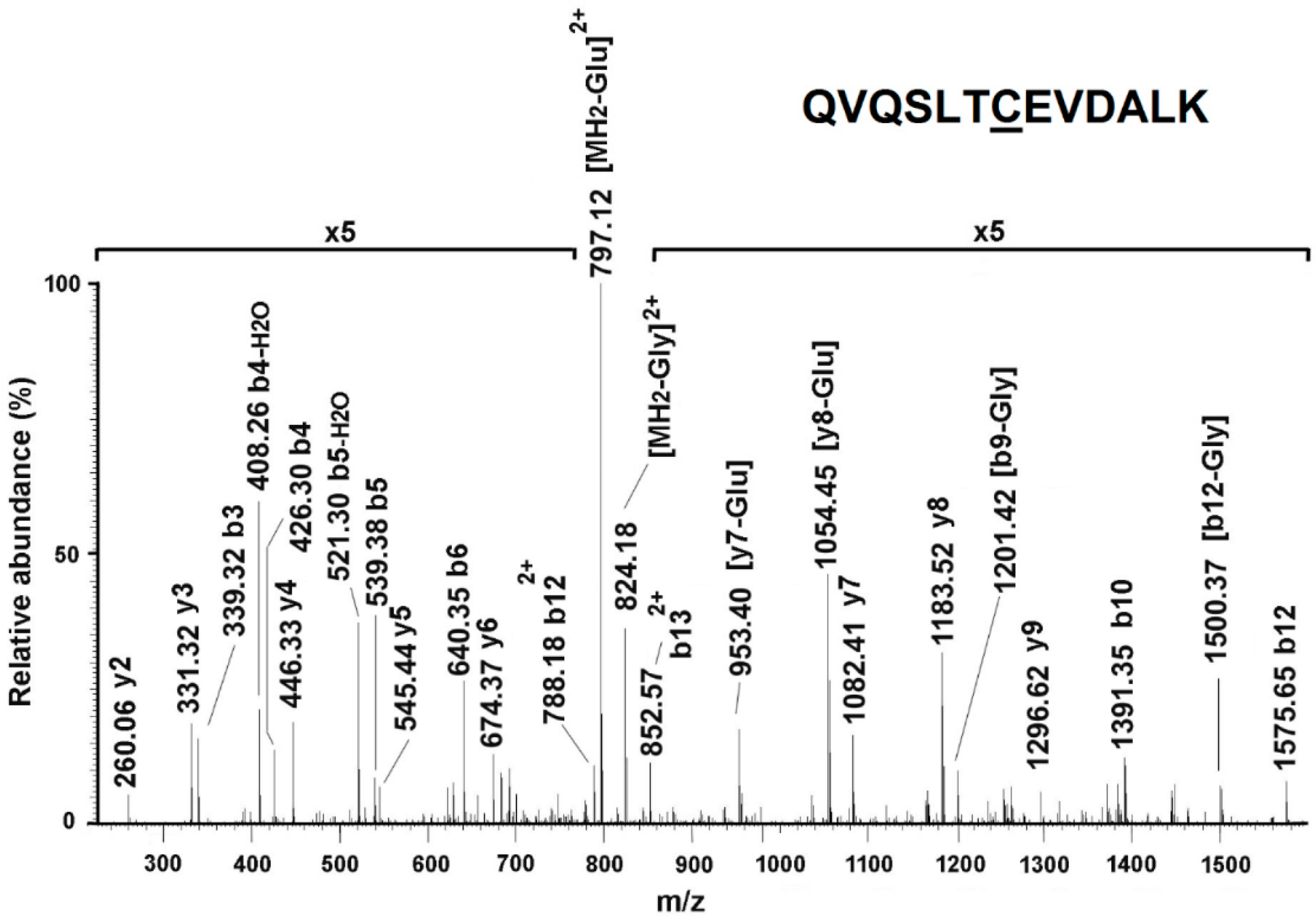
2.13. Identification of S-Glutathionylated Proteins by Mass Spectrometry Analysis
2.14. Immunohistochemical Analysis
2.15. Statistical Analysis
2.16. Ethics Statement
3. Results
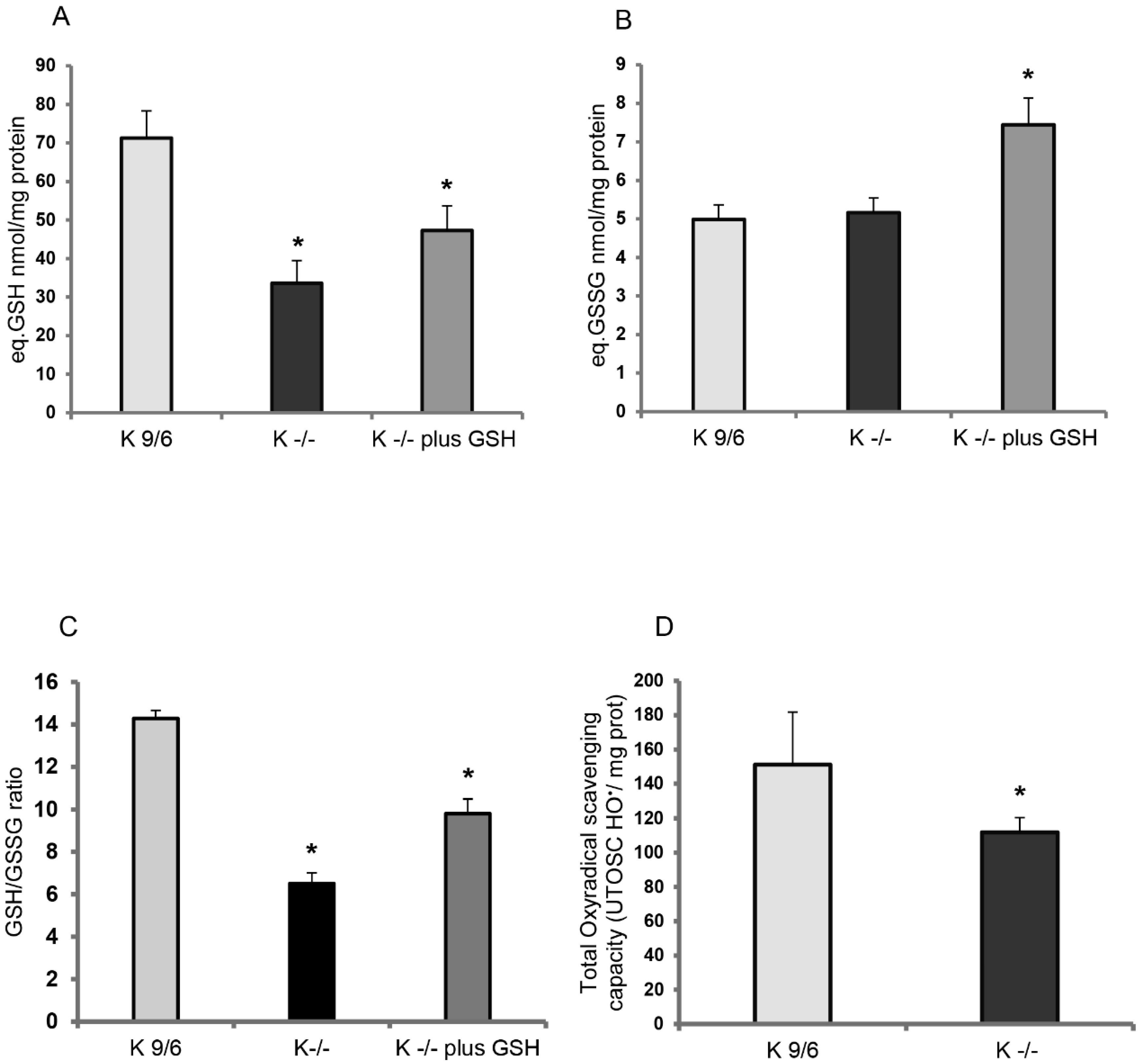
3.1. KRIT1 Loss-Of-Function Is Associated with Intracellular Glutathione Depletion
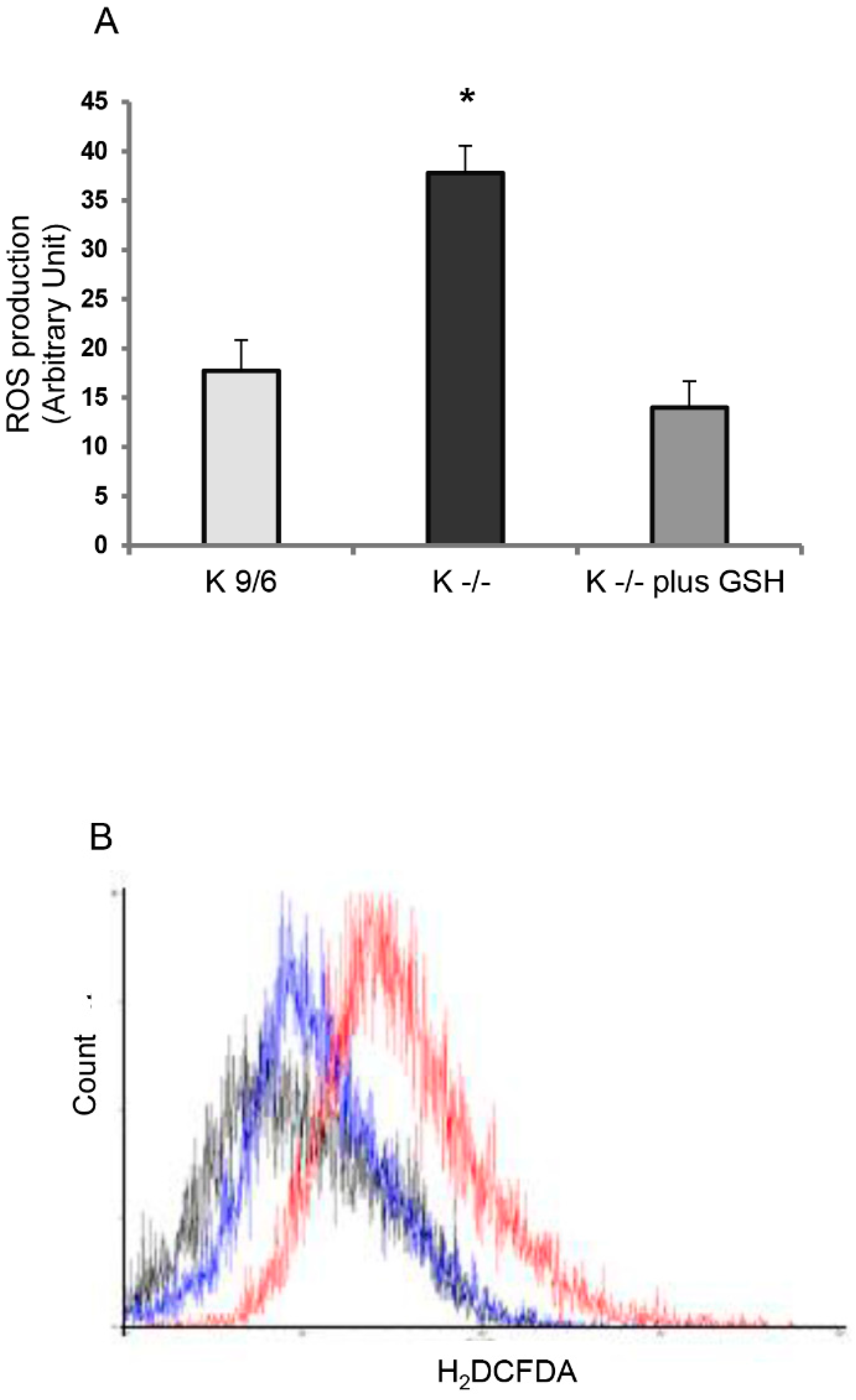
3.2. GSH Delivery Rescues the Cellular Redox Imbalance Induced by KRIT1 Loss-Of-Function
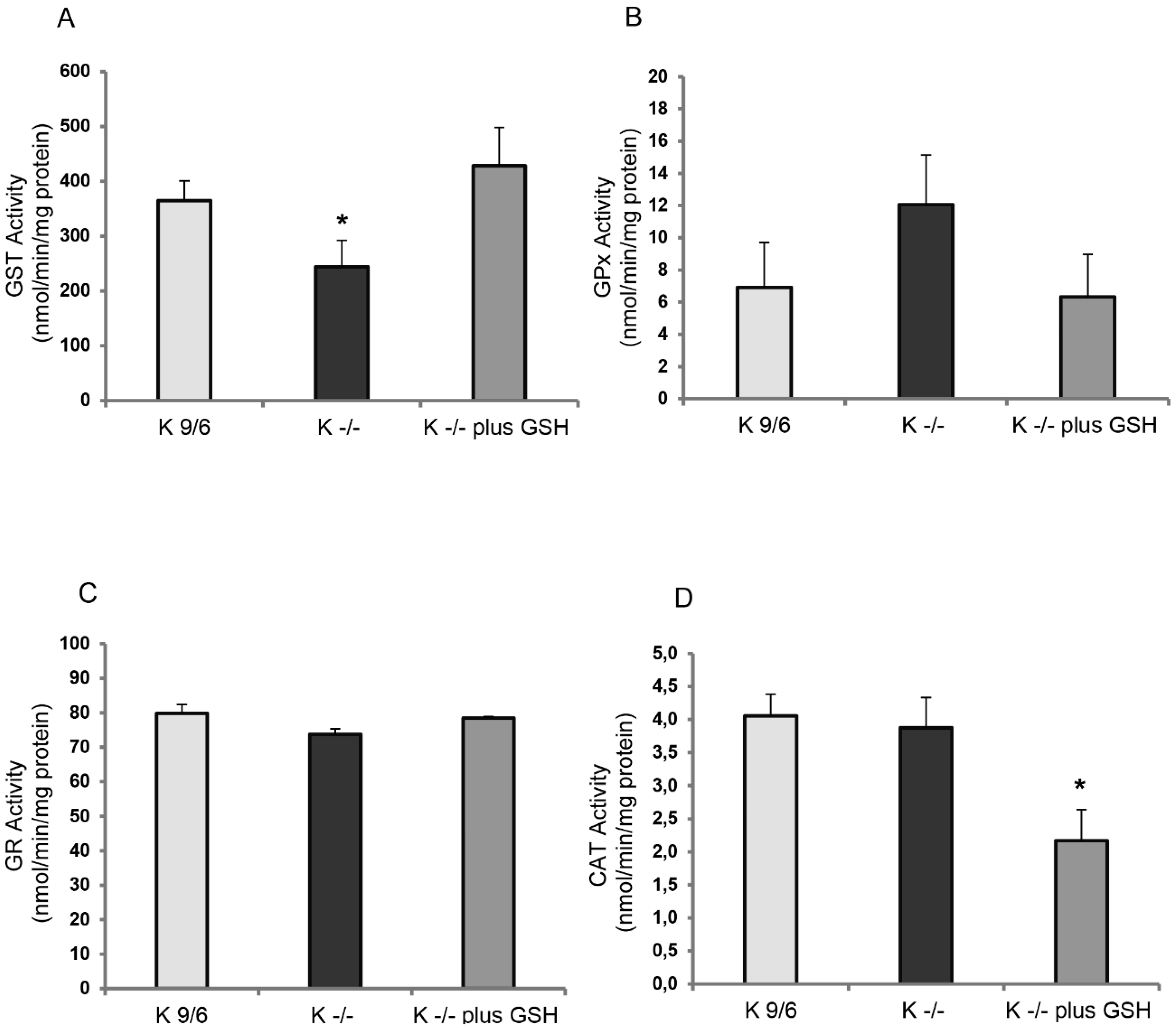
3.3. KRIT1 Loss-Of-Function Affects the Activity of GSH-Dependent Antioxidant Enzymes
3.4. KRIT1 Loss-Of-Function Induces Changes in Protein S-Glutathionylation Pattern
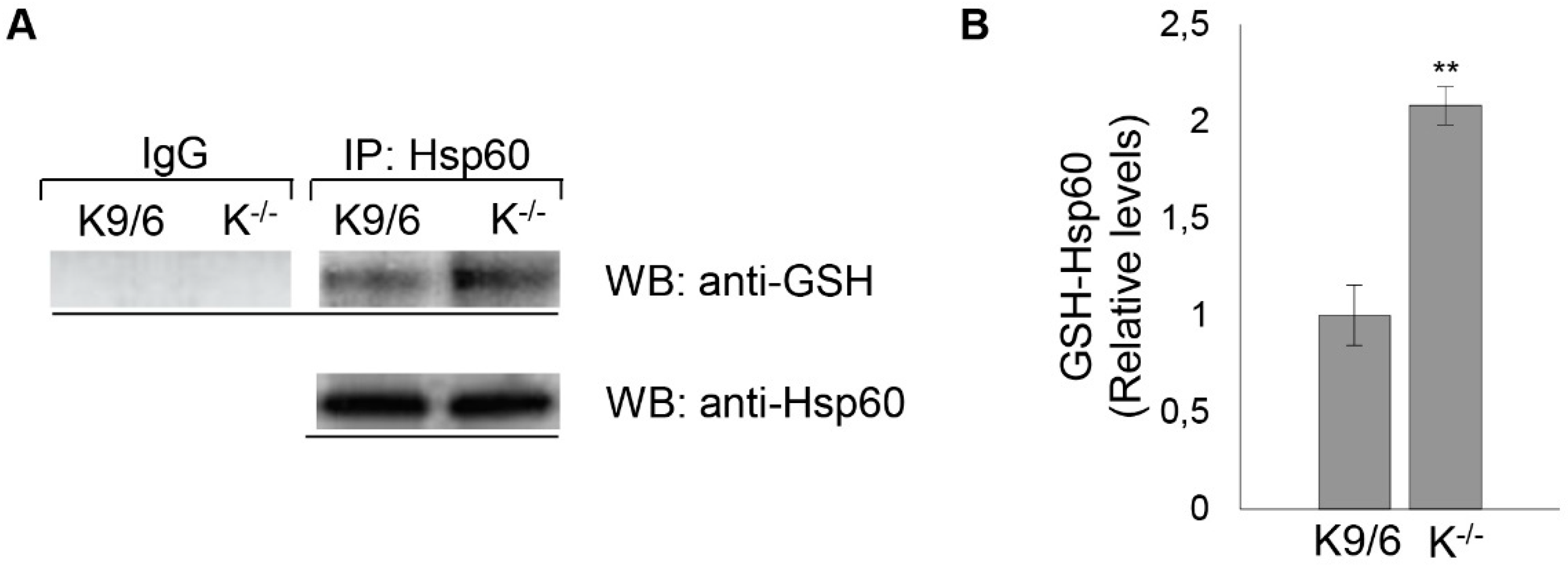
3.5. Confirmation of HSP60 S-Glutathionylation Induced by KRIT1 Loss-Of-Function
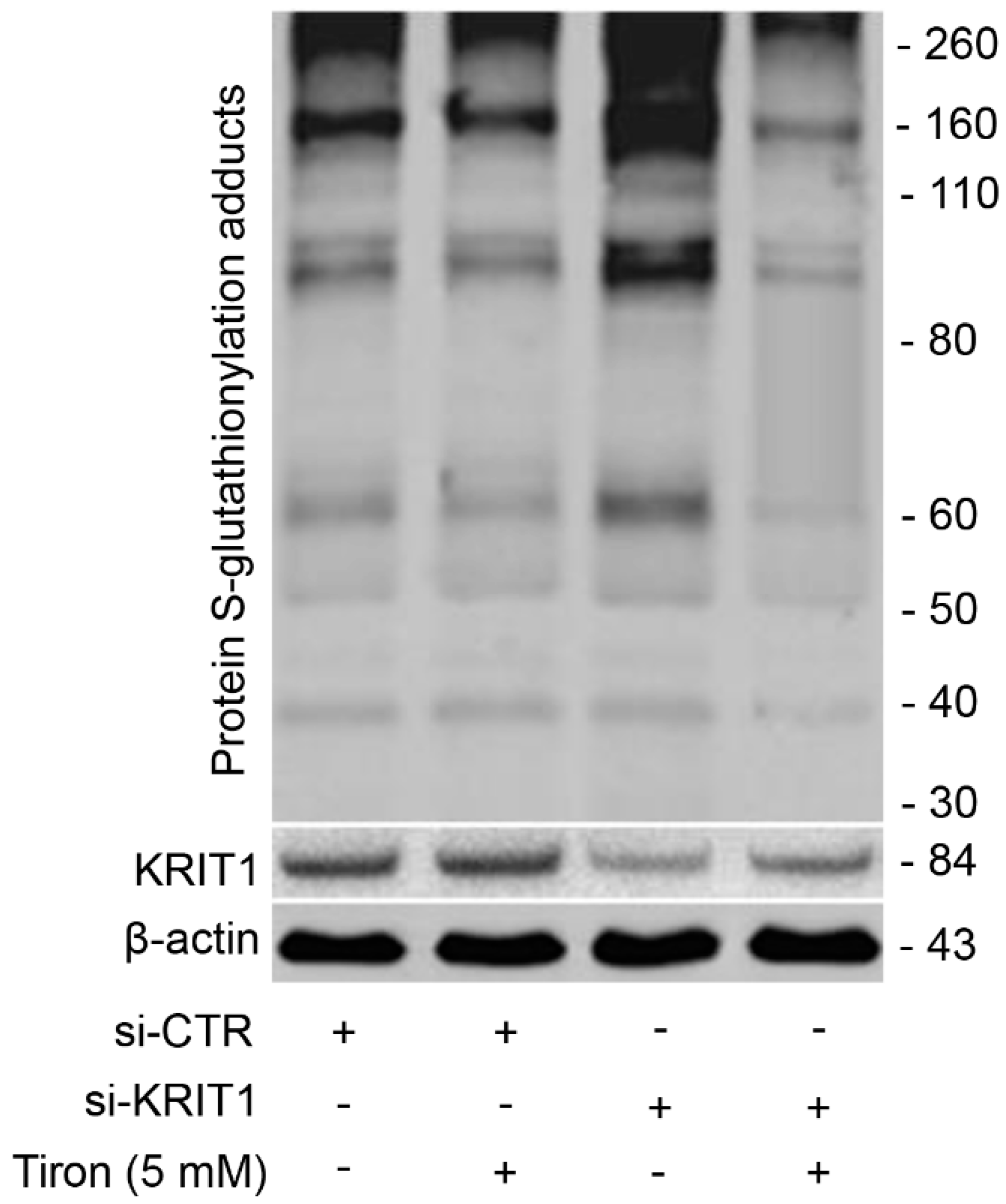
3.6. Redox-Dependent Changes in PSSG Occur in Human Brain Microvascular Endothelial Cells upon KRIT1 Knockdown
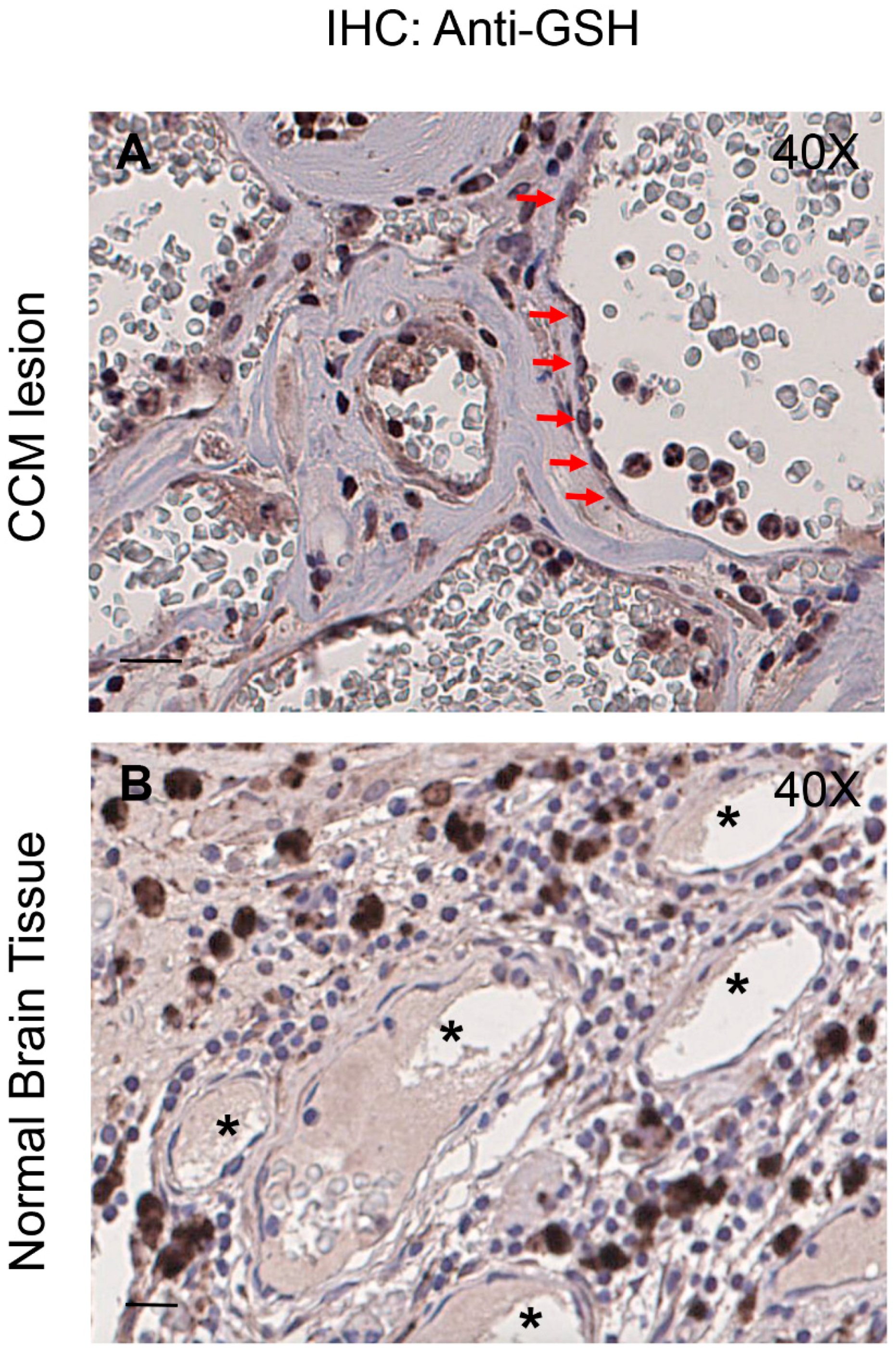
3.7. Enhanced Protein S-Glutathionylation Occurs in Endothelial Cells Lining Human CCM Lesions
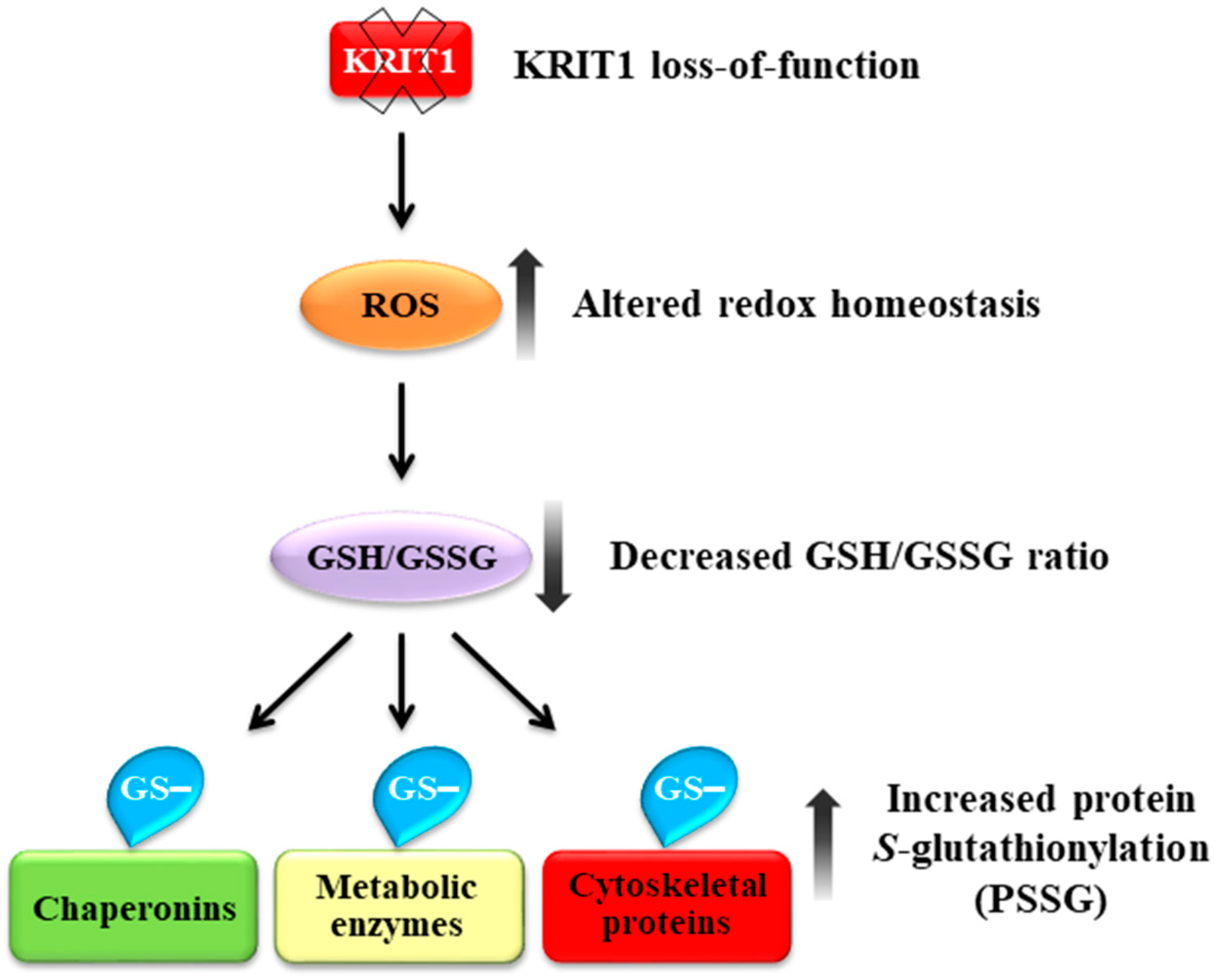
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Batra, S.; Lin, D.; Recinos, P.F.; Zhang, J.; Rigamonti, D. Cavernous malformations: Natural history, diagnosis and treatment. Nat. Rev. Neurol. 2009, 5, 659–670. [Google Scholar] [CrossRef]
- Choquet, H.; Pawlikowska, L.; Lawton, M.T.; Kim, H. Genetics of cerebral cavernous malformations: Current status and future prospects. J. Neurosurg. Sci. 2015, 59, 211–220. [Google Scholar] [PubMed]
- Retta, S.F.; Glading, A.J. Oxidative stress and inflammation in cerebral cavernous malformation disease pathogenesis: Two sides of the same coin. Int. J. Biochem. Cell Biol. 2016, 81, 254–270. [Google Scholar] [CrossRef]
- Trapani, E.; Retta, S.F. Cerebral cavernous malformation (CCM) disease: From monogenic forms to genetic susceptibility factors. J. Neurosurg. Sci. 2015, 59, 201–209. [Google Scholar] [PubMed]
- Fontanella, M.; Bacigaluppi, S. Treatment of cerebral cavernous malformations: Where do we stand. J. Neurosurg. Sci. 2015, 59, 199–200. [Google Scholar]
- Gibson, C.C.; Zhu, W.; Davis, C.T.; Bowman-Kirigin, J.A.; Chan, A.C.; Ling, J.; Walker, A.E.; Goitre, L.; Monache, S.D.; Retta, S.F.; et al. Strategy for Identifying Repurposed Drugs for the Treatment of Cerebral Cavernous Malformation. Circulation 2014, 131, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; Balzac, F.; Degani, S.; Degan, P.; Marchi, S.; Pinton, P.; Retta, S.F. KRIT1 Regulates the Homeostasis of Intracellular Reactive Oxygen Species. PLoS ONE 2010, 5, e11786. [Google Scholar] [CrossRef]
- Goitre, L.; De Luca, E.; Braggion, S.; Trapani, E.; Guglielmotto, M.; Biasi, F.; Forni, M.; Moglia, A.; Trabalzini, L.; Retta, S.F. KRIT1 loss of function causes a ROS-dependent upregulation of c-Jun. Free Radic. Biol. Med. 2014, 68, 134–147. [Google Scholar] [CrossRef]
- Marchi, S.; Corricelli, M.; Trapani, E.; Bravi, L.; Pittaro, A.; Monache, S.D.; Ferroni, L.; Patergnani, S.; Missiroli, S.; Goitre, L.; et al. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol. Med. 2015, 7, 1403–1417. [Google Scholar] [CrossRef]
- Marchi, S.; Retta, S.F.; Pinton, P. Cellular processes underlying cerebral cavernous malformations: Autophagy as another point of view. Autophagy 2016, 12, 424–425. [Google Scholar] [CrossRef]
- Marchi, S.; Trapani, E.; Corricelli, M.; Goitre, L.; Pinton, P.; Retta, S.F. Beyond Multiple Mechanisms and Unique Drugs: Defective Autophagy as Pivotal Player in Cerebral Cavernous Malformation Pathogenesis and Implications for Targeted Therapies. Rare Dis. 2016, 4, e1142640. [Google Scholar] [CrossRef] [PubMed]
- Choquet, H.; Trapani, E.; Goitre, L.; Trabalzini, L.; Akers, A.; Fontanella, M.; Hart, B.L.; Morrison, L.A.; Pawlikowska, L.; Kim, H.; et al. Cytochrome P450 and matrix metalloproteinase genetic modifiers of disease severity in Cerebral Cavernous Malformation type 1. Free Radic. Biol. Med. 2016, 92, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Wang, S.-B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine Oxidative Posttranslational Modifications: Emerging Regulation in the Cardiovascular System. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A.D.G. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular Mechanisms and Clinical Implications of Reversible Protein S-Glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef]
- Pimentel, D.; Haeussler, D.J.; Matsui, R.; Burgoyne, J.R.; Cohen, R.A.; Bachschmid, M.M. Regulation of Cell Physiology and Pathology by Protein S-Glutathionylation: Lessons Learned from the Cardiovascular System. Antioxid. Redox Signal. 2012, 16, 524–542. [Google Scholar] [CrossRef]
- Armeni, T.; Cianfruglia, L.; Piva, F.; Urbanelli, L.; Caniglia, M.L.; Pugnaloni, A.; Principato, G. S-D-Lactoylglutathione can be an alternative supply of mitochondrial glutathione. Free Radic. Biol. Med. 2014, 67, 451–459. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Monache, S.D.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef]
- Yang, H.; Paromov, V.; Smith, M.; Stone, W.L. Preparation, Characterization, and Use of Antioxidant-Liposomes. Methods Mol. Biol. 2008, 477, 277–292. [Google Scholar] [PubMed]
- Armeni, T.; Battino, M.; Stronati, A.; Pugnaloni, A.; Tomassini, G.; Rosi, G.; Biagini, G.; Principato, G. Total Antioxidant Capacity and Nuclear DNA Damage in Keratinocytes after Exposure to H2O2. Biol. Chem. 2001, 382, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Trapani, E.; Monache, S.D.; Perrelli, A.; Fornelli, C.; Retta, F.; Cassoni, P.; Talesa, V.N.; Retta, S.F. Data in support of sustained upregulation of adaptive redox homeostasis mechanisms caused by KRIT1 loss-of-function. Data Brief 2018, 16, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; Distefano, P.V.; Moglia, A.; Nobiletti, N.; Baldini, E.; Trabalzini, L.; Keubel, J.; Trapani, E.; Shuvaev, V.V.; Muzykantov, V.R.; et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci. Rep. 2017, 7, 8296. [Google Scholar] [CrossRef] [PubMed]
- Regoli, F.; Winston, G.W. Quantification of Total Oxidant Scavenging Capacity of Antioxidants for Peroxynitrite, Peroxyl Radicals, and Hydroxyl Radicals. Toxicol. Appl. Pharmacol. 1999, 156, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Armeni, T.; Ercolani, L.; Urbanelli, L.; Magini, A.; Magherini, F.; Pugnaloni, A.; Piva, F.; Modesti, A.; Emiliani, C.; Principato, G.; et al. Cellular Redox Imbalance and Changes of Protein S-glutathionylation Patterns Are Associated with Senescence Induced by Oncogenic H-Ras. PLoS ONE 2012, 7, e52151. [Google Scholar] [CrossRef]
- Brigelius, R.; Muckel, C.; Akerboom, T.P.; Sies, H. Identification and quantitation of glutathione in hepatic protein mixed disulfides and its relationship to glutathione disulfide. Biochem. Pharmacol. 1983, 32, 2529–2534. [Google Scholar] [CrossRef]
- Giustarini, D.; Galvagni, F.; Tesei, A.; Farolfi, A.; Zanoni, M.; Pignatta, S.; Milzani, A.D.G.; Marone, I.M.; Dalle-Donne, I.; Nassini, R.; et al. Glutathione, glutathione disulfide, and S -glutathionylated proteins in cell cultures. Free Radic. Biol. Med. 2015, 89, 972–981. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Purification and characterization of the flavoenzyme glutathione reductase from rat liver. J. Biol. Chem. 1975, 250, 5475–5480. [Google Scholar]
- Habig, W.H.; Pabst, M.J.; Fleischner, G.; Gatmaitan, Z.; Arias, I.M.; Jakoby, W.B. The Identity of Glutathione S-Transferase B with Ligandin, a Major Binding Protein of Liver. Proc. Natl. Acad. Sci. USA 1974, 71, 3879–3882. [Google Scholar] [CrossRef] [PubMed]
- Gauci, V.J.; Padula, M.P.; Coorssen, J.R. Coomassie blue staining for high sensitivity gel-based proteomics. J. Proteom. 2013, 90, 96–106. [Google Scholar] [CrossRef]
- D’Ambrosio, C.; Arena, S.; Salzano, A.M.; Renzone, G.; Ledda, L.; Scaloni, A. A proteomic characterization of water buffalo milk fractions describing PTM of major species and the identification of minor components involved in nutrient delivery and defense against pathogens. Proteomics 2008, 8, 3657–3666. [Google Scholar]
- Salzano, A.M.; Novi, G.; Arioli, S.; Corona, S.; Mora, D.; Scaloni, A. Mono-dimensional blue native-PAGE and bi-dimensional blue native/urea-PAGE or/SDS-PAGE combined with nLC–ESI-LIT-MS/MS unveil membrane protein heteromeric and homomeric complexes in Streptococcus thermophilus. J. Proteom. 2013, 94, 240–261. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Busu, C.; Circu, M.L.; Li, W.; Aw, T.Y. Glutathione in Cerebral Microvascular Endothelial Biology and Pathobiology: Implications for Brain Homeostasis. Int. J. Cell Biol. 2012, 2012, 434971. [Google Scholar]
- Okouchi, M.; Okayama, N.; Aw, T.Y. Preservation of cellular glutathione status and mitochondrial membrane potential by N-acetylcysteine and insulin sensitizers prevent carbonyl stress-induced human brain endothelial cell apoptosis. Curr. Neurovasc. Res. 2009, 6, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubálek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R.; et al. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Distefano, P.V.; Kuebel, J.M.; Sarelius, I.H.; Glading, A. KRIT1 Protein Depletion Modifies Endothelial Cell Behavior via Increased Vascular Endothelial Growth Factor (VEGF) Signaling. J. Biol. Chem. 2014, 289, 33054–33065. [Google Scholar] [CrossRef]
- Akerboom, T.P.; Sies, H. Assay of glutathione, glutathione disulfide, and glutathione mixed disulfides in biological samples. Methods Enzymol. 1981, 77, 373–382. [Google Scholar]
- Balcerczyk, A.; Bartosz, G. Thiols are Main Determinants of Total Antioxidant Capacity of Cellular Homogenates. Free Radic. Res. 2003, 37, 537–541. [Google Scholar] [CrossRef]
- Arthur, J.R. The glutathione peroxidases. Cell. Mol. Life Sci. 2001, 57, 1825–1835. [Google Scholar] [CrossRef]
- DePonte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta (BBA) Bioenergy 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. J. Biol. Inorg. Chem. 2000, 267, 4928–4944. [Google Scholar] [CrossRef]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-Glutathionylation: From Molecular Mechanisms to Health Outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Chock, P.B. Posttranslational Modification of Cysteine in Redox Signaling and Oxidative Stress: Focus on S-Glutathionylation. Antioxid. Redox Signal. 2012, 16, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-Glutathionylation: From redox regulation of protein functions to human diseases. J. Cell. Mol. Med. 2004, 8, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Eriksson, S.; Wang, L. Oxidative Stress InducedS-glutathionylation and Proteolytic Degradation of Mitochondrial Thymidine Kinase 2. J. Biol. Chem. 2012, 287, 24304–24312. [Google Scholar] [CrossRef] [PubMed]
- Cabiscol, E.; Bellí, G.; Tamarit, J.; Echave, P.; Herrero, E.; Ros, J. Mitochondrial Hsp60, Resistance to Oxidative Stress, and the Labile Iron Pool Are Closely Connected in Saccharomyces cerevisiae. J. Biol. Chem. 2002, 277, 44531–44538. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Weisbrod, R.M.; Shao, D.; Watanabe, Y.; Yin, X.; Bachschmid, M.M.; Seta, F.; Janssen-Heininger, Y.M.; Matsui, R.; Zang, M.; et al. The redox mechanism for vascular barrier dysfunction associated with metabolic disorders: Glutathionylation of Rac1 in endothelial cells. Redox Biol. 2016, 9, 306–319. [Google Scholar] [CrossRef]
- Galougahi, K.K.; Liu, C.; Gentile, C.; Kok, C.; Nunez, A.; Garcia, A.; Fry, N.A.S.; Davies, M.J.; Hawkins, C.L.; Rasmussen, H.H.; et al. Glutathionylation Mediates Angiotensin II–Induced eNOS Uncoupling, Amplifying NADPH Oxidase-Dependent Endothelial Dysfunction. J. Am. Heart Assoc. 2014, 3, 000731. [Google Scholar] [CrossRef]
- Pastore, A.; Piemonte, F. Protein Glutathionylation in Cardiovascular Diseases. IJMS 2013, 14, 20845–20876. [Google Scholar] [CrossRef]
- Fisher, O.S.; Boggon, T.J. Signaling pathways and the cerebral cavernous malformations proteins: Lessons from structural biology. Cell. Mol. Life Sci. 2013, 71, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Glading, A.; Han, J.; Stockton, R.A.; Ginsberg, M.H. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 2007, 179, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G.; Orsenigo, F.; Rudini, N.; Maddaluno, L.; Boulday, G.; Chapon, F.; Dejana, E. CCM1 regulates vascular-lumen organization by inducing endothelial polarity. J. Cell Sci. 2010, 123, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Faurobert, E.; Rome, C.; Lisowska, J.; Manet-Dupé, S.; Boulday, G.; Malbouyres, M.; Balland, M.; Bouin, A.-P.; Kéramidas, M.; Bouvard, D.; et al. CCM1-ICAP-1 complex controls beta1 integrin-dependent endothelial contractility and fibronectin remodeling. J. Cell Biol. 2013, 202, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Draheim, K.M.; Zhang, R.; Calderwood, D.A.; Boggon, T.J. Mechanism for KRIT1 Release of ICAP1-Mediated Suppression of Integrin Activation. Mol. Cell 2013, 49, 719–729. [Google Scholar] [CrossRef]
- Jilkova, Z.M.; Lisowska, J.; Manet, S.; Verdier, C.; Deplano, V.; Geindreau, C.; Faurobert, E.; Albigès-Rizo, C.; Duperray, A. CCM proteins control endothelial beta1 integrin dependent response to shear stress. Biol. Open 2014, 3, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Borikova, A.L.; Dibble, C.F.; Sciaky, N.; Welch, C.M.; Abell, A.N.; Bencharit, S.; Johnson, G.L. Rho Kinase Inhibition Rescues the Endothelial Cell Cerebral Cavernous Malformation Phenotype. J. Biol. Chem. 2010, 285, 11760–11764. [Google Scholar] [CrossRef] [PubMed]
- Stockton, R.A.; Shenkar, R.; Awad, I.A.; Ginsberg, M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010, 207, 881–896. [Google Scholar] [CrossRef]
- Whitehead, K.J.; Chan, A.C.; Navankasattusas, S.; Koh, W.; London, N.R.; Ling, J.; Mayo, A.H.; Drakos, S.G.; Jones, C.A.; Zhu, W.; et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 2009, 15, 177–184. [Google Scholar] [CrossRef]
- Zheng, X.; Xu, C.; Di Lorenzo, A.; Kleaveland, B.; Zou, Z.; Seiler, C.; Chen, M.; Cheng, L.; Xiao, J.; He, J.; et al. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J. Clin. Investig. 2010, 120, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Stockton, R.A.; Gingras, A.; Ablooglu, A.J.; Han, J.; Bobkov, A.A.; Ginsberg, M.H. A mechanism of Rap1-induced stabilization of endothelial cell–cell junctions. Mol. Biol. Cell 2011, 22, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Schulz, G.B.; Wieland, E.; Wüstehube-Lausch, J.; Boulday, G.; Moll, I.; Tournier-Lasserve, E.; Fischer, A. Cerebral Cavernous Malformation-1 Protein Controls DLL4-Notch3 Signaling Between the Endothelium and Pericytes. Stroke 2015, 46, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Wüstehube, J.; Bartol, A.; Liebler, S.S.; Brütsch, R.; Zhu, Y.; Felbor, U.; Sure, U.; Augustin, H.G.; Fischer, A. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 12640–12645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Rigamonti, D.; Badr, A. Ccm1 Assures Microvascular Integrity during Angiogenesis. Transl. Stroke Res. 2010, 1, 146–153. [Google Scholar]
- Bravi, L.; Malinverno, M.; Pisati, F.; Rudini, N.; Cuttano, R.; Pallini, R.; Martini, M.; LaRocca, L.M.; Locatelli, M.; Levi, V.; et al. Endothelial Cells Lining Sporadic Cerebral Cavernous Malformation Cavernomas Undergo Endothelial-to-Mesenchymal Transition. Stroke 2016, 47, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Maddaluno, L.; Rudini, N.; Cuttano, R.; Bravi, L.; Giampietro, C.; Corada, M.; Ferrarini, L.; Orsenigo, F.; Papa, E.; Boulday, G.; et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013, 498, 492–496. [Google Scholar] [CrossRef]
- Cuttano, R.; Rudini, N.; Bravi, L.; Corada, M.; Giampietro, C.; Papa, E.; Morini, M.F.; Maddaluno, L.; Baeyens, N.; Adams, R.H.; et al. KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol. Med. 2015, 8, 6–24. [Google Scholar] [CrossRef]
- Fisher, O.S.; Deng, H.; Liu, D.; Zhang, Y.; Wei, R.; Deng, Y.; Zhang, F.; Louvi, A.; Turk, B.E.; Boggon, T.J.; et al. Structure and vascular function of MEKK3–cerebral cavernous malformations 2 complex. Nat. Commun. 2015, 6, 7937. [Google Scholar] [CrossRef]
- Zhou, Z.; Rawnsley, D.R.; Goddard, L.M.; Pan, W.; Cao, X.-J.; Jakus, Z.; Zheng, H.; Yang, J.; Arthur, J.S.C.; Whitehead, K.J.; et al. The Cerebral Cavernous Malformation Pathway Controls Cardiac Development via Regulation of Endocardial MEKK3 Signaling and KLF Expression. Dev. Cell 2015, 32, 168–180. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, A.T.; Wong, W.-Y.; Bamezai, S.; Goddard, L.M.; Shenkar, R.; Zhou, S.; Yang, J.; Wright, A.C.; Foley, M.; et al. Cerebral cavernous malformations arise from endothelial gain of MEKK3–KLF2/4 signalling. Nature 2016, 532, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, P.; Goitre, L.; Ferro, E.; Cutano, V.; Martino, C.; Trabalzini, L.; Retta, S.F. Identification of the Kelch Family Protein Nd1-L as a Novel Molecular Interactor of KRIT1. PLoS ONE 2012, 7, e44705. [Google Scholar] [CrossRef] [PubMed]
- Kevil, C.G. Regulation of endothelial glutathione by ICAM-1: Implications for inflammation. FASEB J. 2004, 18, 1321–1323. [Google Scholar] [CrossRef]
- Langston, W.; Chidlow, J.H., Jr.; Booth, B.A.; Barlow, S.C.; Lefer, D.J.; Patel, R.P.; Kevil, C.G. Regulation of endothelial glutathione by ICAM-1 governs VEGF-A-mediated eNOS activity and angiogenesis. Free Radic. Biol. Med. 2007, 42, 720–729. [Google Scholar] [CrossRef]
- Chen, C.-A.; Wang, T.-Y.; Varadharaj, S.; Hemann, C.; Chen, Y.-R.; A Reyes, L.; Talukder, M.A.H.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Moglia, A.; Goitre, L.; Gianoglio, S.; Baldini, E.; Trapani, E.; Genre, A.; Scattina, A.; Dondo, G.; Trabalzini, L.; Beekwilder, J.; et al. Evaluation of the bioactive properties of avenanthramide analogs produced in recombinant yeast. BioFactors 2015, 41, 15–27. [Google Scholar] [CrossRef]
- Moglianetti, M.; De Luca, E.; Pedone, D.; Marotta, R.; Catelani, T.; Sartori, B.; Amenitsch, H.; Retta, S.F.; Pompa, P.P. Platinum nanozymes recover cellular ROS homeostasis in oxidative stress-mediated disease model. Nanoscale 2016, 8, 3739–3752. [Google Scholar] [CrossRef] [PubMed]
- De Luca, E.; Pedone, D.; Moglianetti, M.; Pulcini, D.; Perrelli, A.; Retta, S.F.; Pompa, P.P. Multifunctional Platinum@BSA–Rapamycin Nanocarriers for the Combinatorial Therapy of Cerebral Cavernous Malformation. ACS Omega 2018, 3, 15389–15398. [Google Scholar] [CrossRef]
- Um, J.-Y.; Kim, H.-M.; Han, S.-H.; Cho, K.-H.; Moon, B.-S.; Hong, S.-H. Glutathione s-transferase gene polymorphism and ischemic cerebrovascular disease. Int. J. Neurosci. 2006, 116, 55–65. [Google Scholar] [CrossRef]
- Go, Y.-M.; Jones, D.P.; Go, Y. Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 2010, 12, 116–125. [Google Scholar]
- Petrini, S.; Passarelli, C.; Pastore, A.; Tozzi, G.; Coccetti, M.; Colucci, M.; Bianchi, M.; Carrozzo, R.; Bertini, E.; Piemonte, F.; et al. Protein glutathionylation in cellular compartments: A constitutive redox signal. Redox Rep. 2012, 17, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F. S-Glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci. 2012, 46, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Dalle-Donne, I. Redox proteomics: From protein modifications to cellular dysfunction and disease. Mass Spectrom. Rev. 2014, 33, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yi, M.; Liu, Y.; Wang, Q.; Hu, Y.; Deng, H. Glutaredoxin-1 Silencing Induces Cell Senescence via p53/p21/p16 Signaling Axis. J. Proteome Res. 2018, 17, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.N. Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS ONE 2010, 5, e9422. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Li, B.X.; Xiao, X. Toward Developing Chemical Modulators of Hsp60 as Potential Therapeutics. Front. Mol. Biosci. 2018, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.-W.; Zhang, J.; Ancrum, T.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxid. Redox Signal. 2017, 26, 247–261. [Google Scholar] [CrossRef]
- Eaton, P.; Wright, N.; Hearse, D.J.; Shattock, M.J. Glyceraldehyde Phosphate Dehydrogenase Oxidation during Cardiac Ischemia and Reperfusion. J. Mol. Cell. Cardiol. 2002, 34, 1549–1560. [Google Scholar] [CrossRef]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E.; et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar] [CrossRef]
- Reddy, S.; Jones, A.D.; Cross, C.E.; Wong, P.S.-Y.; Van Der Vliet, A. Inactivation of creatine kinase by S-glutathionylation of the active-site cysteine residue. Biochem. J. 2000, 347, 821–827. [Google Scholar] [CrossRef]
- Xu, Q.; Huff, L.P.; Fujii, M.; Griendling, K.K. Redox regulation of the actin cytoskeleton and its role in the vascular system. Free Radic. Biol. Med. 2017, 109, 84–107. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Bhatnagar, A. Protein S-glutathiolation: Redox-sensitive regulation of protein function. J. Mol. Cell. Cardiol. 2012, 52, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and Consequences of Cysteine S-Glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3165–3172. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot | SwissProt Accession | Protein Description | Gene Name | Mascot Score | Unique Peptides | Sequence Coverage (%) | Mass (Da) | pI |
|---|---|---|---|---|---|---|---|---|
| 1 | P16858 | Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | 165 | 3 | 7.8 | 36,072 | 8.44 |
| 2 | P16858 | Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | 95 | 2 | 7.5 | 36,072 | 8.44 |
| 3 | P16858 | Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | 100 | 2 | 7.8 | 36,072 | 8.44 |
| 4 | P17182 | Alpha-enolase | Eno1 | 553 | 12 | 29.7 | 47,453 | 6.37 |
| 5 | P17182 | Alpha-enolase | Eno1 | 818 | 15 | 42.2 | 47,453 | 6.37 |
| 6 | P17182 | Alpha-enolase | Eno1 | 710 | 14 | 39.9 | 47,453 | 6.37 |
| 7 | P63038 | 60 kDa heat shock protein | Hspd1 | 856 | 14 | 34.2 | 61,088 | 5.91 |
| 8 | P20152 | Vimentin | Vim | 993 | 22 | 50.0 | 53,712 | 5.06 |
| 9 | P20152 | Vimentin | Vim | 968 | 21 | 50.4 | 53,712 | 5.06 |
| 10 | P14211 | Calreticulin 1 | Calr | 163 | 4 | 8.4 | 48136 | 4.33 |
| 11 | Q04447 | Creatine kinase B-type | Ckb | 190 | 4 | 16.5 | 42,971 | 5.4 |
| 12 | Q04447 | Creatine kinase B-type | Ckb | 179 | 4 | 16.8 | 42,971 | 5.4 |
| 13 | P68372 | Tubulin beta-4B chain | Tubb4b | 582 | 11 | 29.4 | 50,255 | 4.79 |
| 14 | P60710 | Actin, cytoplasmic 1 | Actb | 185 | 4 | 17.3 | 42,052 | 5.29 |
| 15 | P60710 | Actin, cytoplasmic 1 | Actb | 341 | 7 | 22.7 | 42,052 | 5.29 |
| 22 | P20152 | Vimentin | Vim | 945 | 22 | 43.8 | 53,712 | 5.06 |
| 23 | P20152 | Vimentin | Vim | 961 | 20 | 47.9 | 53,712 | 5.06 |
| 24 | P60710 | Actin, cytoplasmic 1 | Actb | 276 | 5 | 16.3 | 42,052 | 5.29 |
| 25 | P60710 | Actin, cytoplasmic 1 | Actb | 276 | 5 | 16.3 | 42,052 | 5.29 |
| 26 | Q58E70 | Tpm 3 protein | Tpm3 | 502 | 10 | 37.9 | 29,231 | 4.75 |
| Q6IRU2 | Tropomyosin alpha-4 chain | Tpm4 | 308 | 6 | 27.0 | 28,564 | 4.65 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cianfruglia, L.; Perrelli, A.; Fornelli, C.; Magini, A.; Gorbi, S.; Salzano, A.M.; Antognelli, C.; Retta, F.; Benedetti, V.; Cassoni, P.; et al. KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced S-Glutathionylation of Distinct Structural and Regulatory Proteins. Antioxidants 2019, 8, 27. https://doi.org/10.3390/antiox8010027
Cianfruglia L, Perrelli A, Fornelli C, Magini A, Gorbi S, Salzano AM, Antognelli C, Retta F, Benedetti V, Cassoni P, et al. KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced S-Glutathionylation of Distinct Structural and Regulatory Proteins. Antioxidants. 2019; 8(1):27. https://doi.org/10.3390/antiox8010027
Chicago/Turabian StyleCianfruglia, Laura, Andrea Perrelli, Claudia Fornelli, Alessandro Magini, Stefania Gorbi, Anna Maria Salzano, Cinzia Antognelli, Francesca Retta, Valerio Benedetti, Paola Cassoni, and et al. 2019. "KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced S-Glutathionylation of Distinct Structural and Regulatory Proteins" Antioxidants 8, no. 1: 27. https://doi.org/10.3390/antiox8010027
APA StyleCianfruglia, L., Perrelli, A., Fornelli, C., Magini, A., Gorbi, S., Salzano, A. M., Antognelli, C., Retta, F., Benedetti, V., Cassoni, P., Emiliani, C., Principato, G., Scaloni, A., Armeni, T., & Retta, S. F. (2019). KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced S-Glutathionylation of Distinct Structural and Regulatory Proteins. Antioxidants, 8(1), 27. https://doi.org/10.3390/antiox8010027