Identification of Small Peptides that Inhibit NADPH Oxidase (Nox2) Activation


Abstract
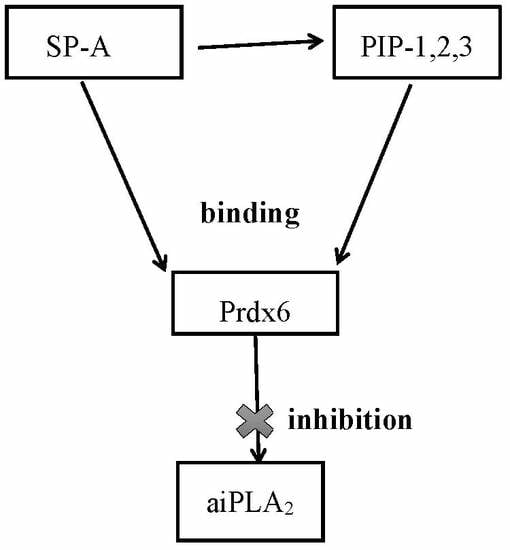
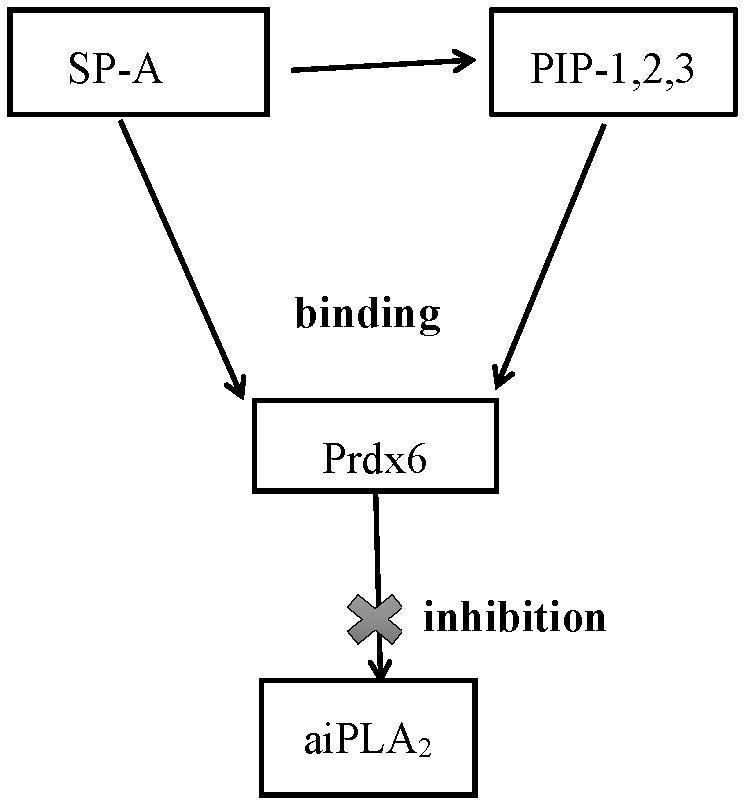
1. Introduction
2. Methods
3. Results
3.1. Minimal Effective Peptide Sequence
3.2. Species Variability
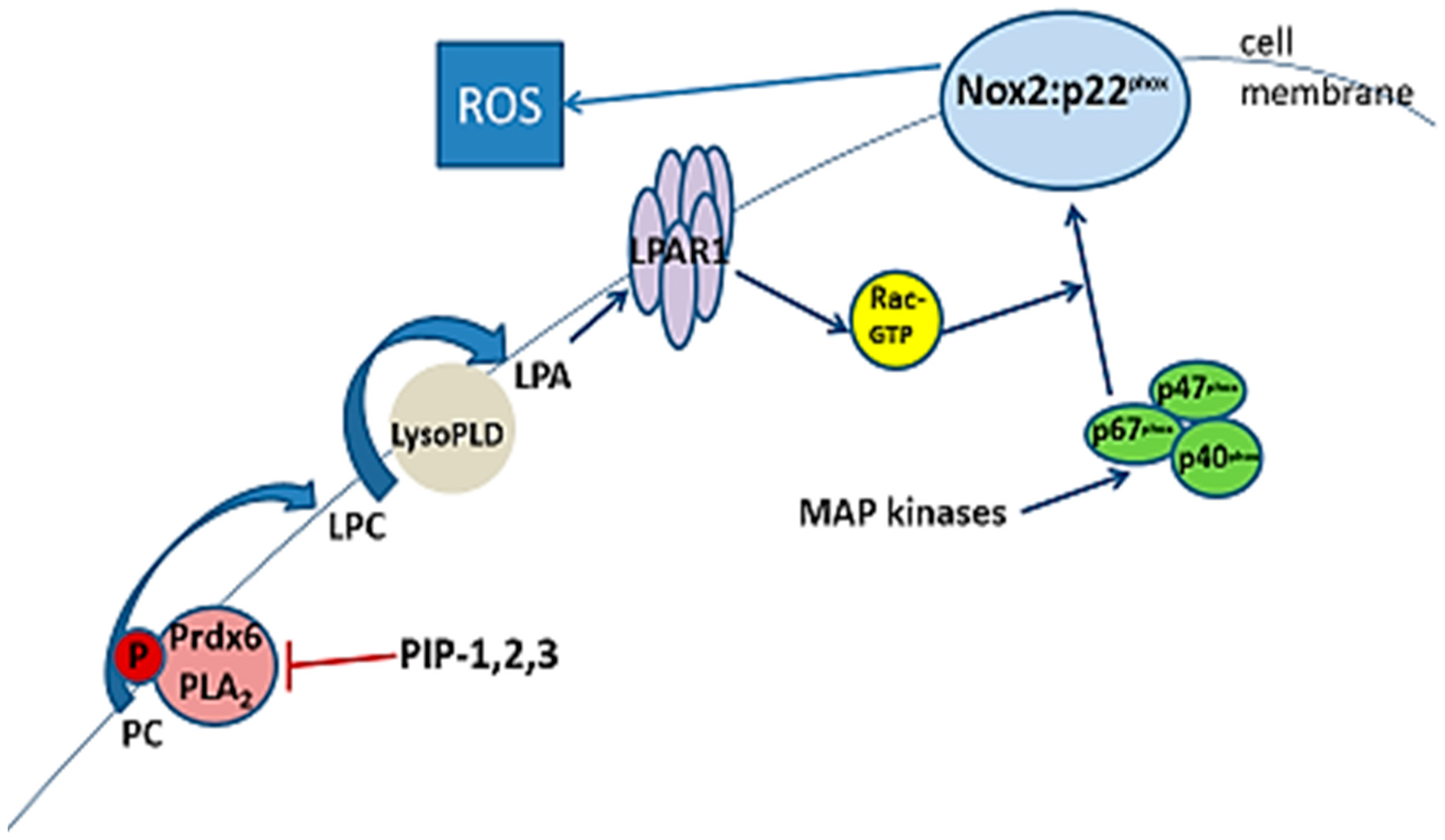
3.3. Physical Properties of Inhibitory Peptides
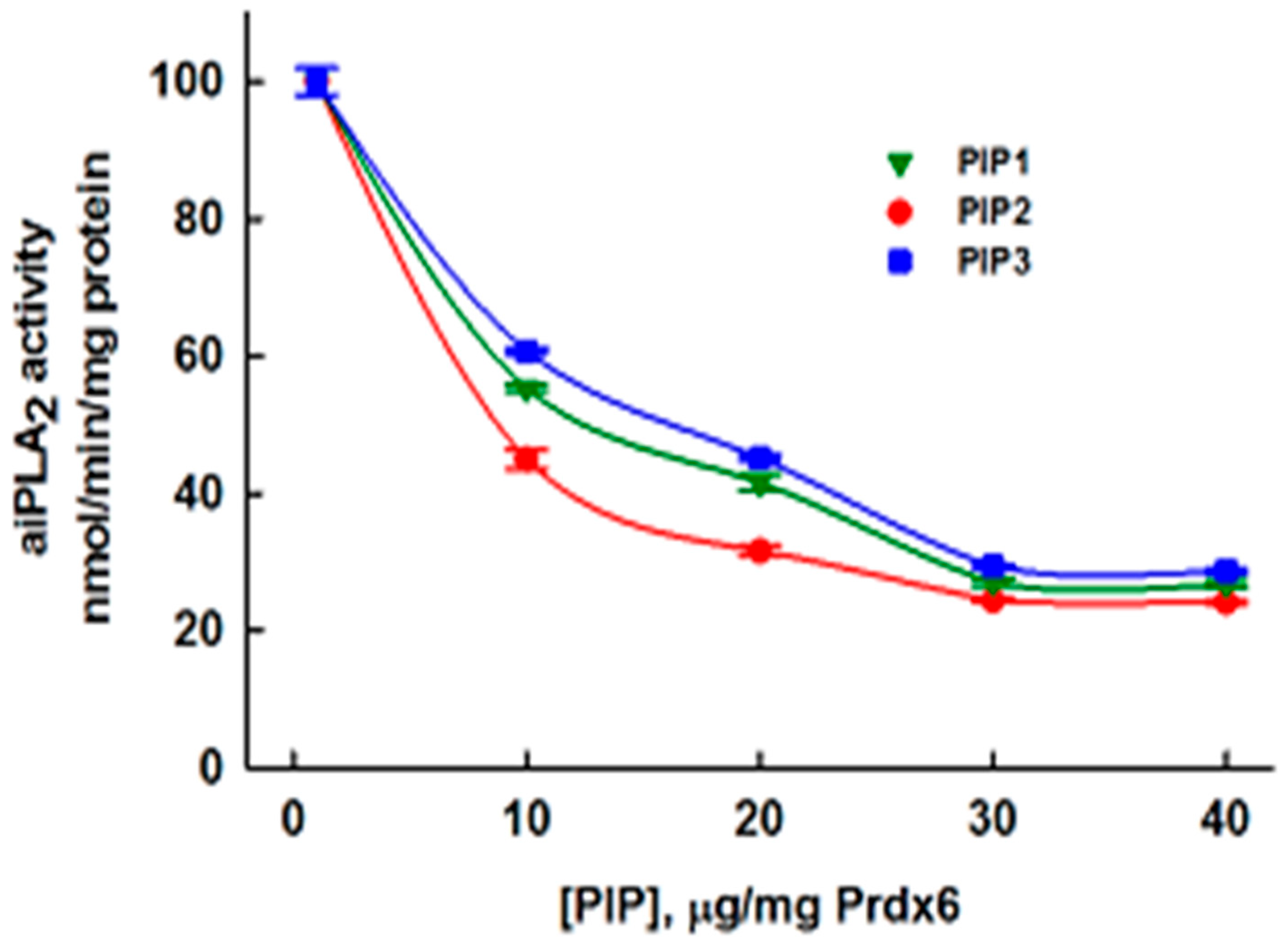
3.5. Inhibition of aiPLA2 Activity In Vitro
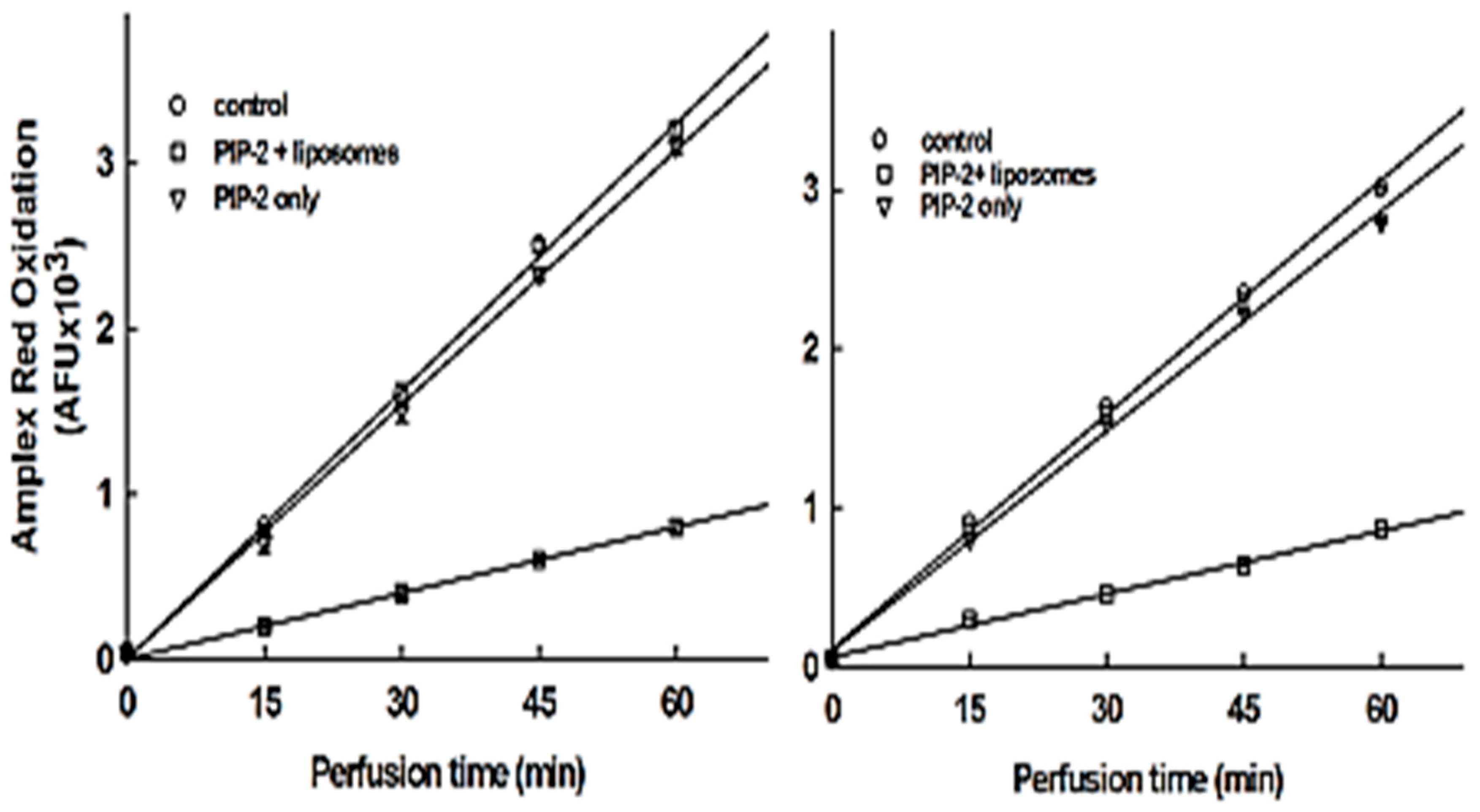
3.6. Intracellular Delivery of PIP-2
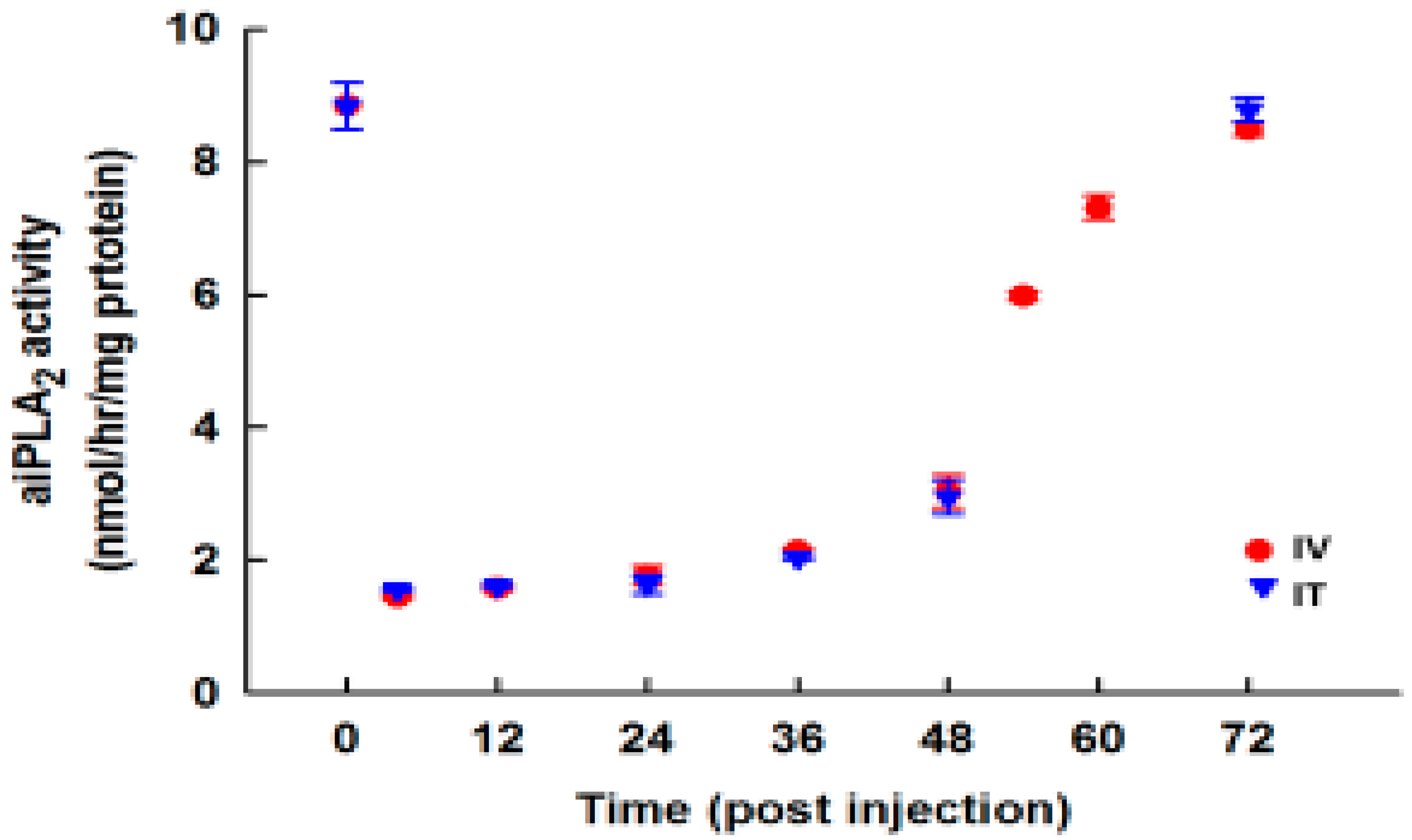
3.7. Effect of PIP-2 on aiPLA2 Activity In Vivo and its Biological Stability
3.8. Specificity of PLA2 Inhibition
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. NADPH oxidase: An update. Blood 1999, 93, 1464–1476. [Google Scholar] [PubMed]
- Diebold, B.A.; Smith, S.M.; Li, Y.; Lambeth, J.D. NOX2 As a Target for Drug Development: Indications, Possible Complications, and Progress. Antioxid. Redox Signal. 2015, 23, 375–405. [Google Scholar] [CrossRef] [PubMed]
- Pick, E.; Gorzalczany, Y.; Engel, S. Role of the rac1 p21-GDP-dissociation inhibitor for rho heterodimer in the activation of the superoxide-forming NADPH oxidase of macrophages. Eur. J. Biochem. 1993, 217, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Pick, E. Cell-free NADPH oxidase activation assays: "In vitro veritas". Methods Mol. Biol. 2014, 1124, 339–403. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Medina, J.P.; Dodia, C.; Weng, L.; Mesaros, C.; Blair, I.; Feinstein, S.I.; Chatterjee, C.; Fisher, A. The phospholipase A2 activity of peroxiredoxin 6 modulates NADPH oxidase 2 activation via lysophosphatidic acid receptor signaling in the pulmonary endothelium and alveolar macrophages. FASEB J. 2016, 30, 2885–2898. [Google Scholar] [CrossRef]
- Nauseef, W.M. Biological roles for the NOX family NADPH oxidases. J. Biol. Chem. 2008, 283, 16961–16965. [Google Scholar] [CrossRef]
- Bernard, K.; Hecker, L.; Luckhardt, T.R.; Cheng, G.; Thannickal, V.J. NADPH oxidases in lung health and disease. Antioxid. Redox Signal. 2014, 20, 2838–2853. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Bromberg, Y.; Pick, E. Unsaturated fatty acids as second messengers of superoxide generation by macrophages. Cell Immunol. 1983, 79, 240–252. [Google Scholar] [CrossRef]
- Henderson, L.M.; Chappell, J.B.; Jones, O.T. Superoxide generation is inhibited by phospholipase A2 inhibitors. Role for phospholipase A2 in the activation of the NADPH oxidase. Biochem. J. 1989, 264, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Dana, R.; Malech, H.L.; Levy, R. The requirement for phospholipase A2 for activation of the assembled NADPH oxidase in human neutrophils. Biochem. J. 1994, 297, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef] [PubMed]
- Dana, R.; Leto, T.L.; Malech, H.L.; Levy, R. Essential requirement of cytosolic phospholipase A2 for activation of the phagocyte NADPH oxidase. J. Biol. Chem. 1998, 273, 441–445. [Google Scholar] [CrossRef]
- Rubin, B.B.; Downey, G.P.; Koh, A.; Degousee, N.; Ghomashchi, F.; Nallan, L.; Stefanski, E.; Harkin, D.W.; Sun, C.; Smart, B.P.; et al. Cytosolic phospholipase A2-alpha is necessary for platelet-activating factor biosynthesis, efficient neutrophil-mediated bacterial killing, and the innate immune response to pulmonary infection: cPLA2-alpha does not regulate neutrophil NADPH oxidase activity. J. Biol. Chem. 2005, 280, 7519–7529. [Google Scholar] [CrossRef]
- Chatterjee, S.; Feinstein, S.I.; Dodia, C.; Sorokina, E.; Lien, Y.C.; Nguyen, S.; Debolt, K.; Speicher, D.; Fisher, A.B. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J. Biol. Chem. 2011, 286, 11696–11706. [Google Scholar] [CrossRef]
- Fisher, A.B. The Phospholipase A2 Activity of Peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef]
- Vazquez-Medina, J.P.; Tao, J.Q.; Patel, P.; Bannitz-Fernandes, R.; Dodia, C.; Sorokina, E.M.; Feinstein, S.I.; Chatterjee, S.; Fisher, A.B. Genetic inactivation of the phospholipase A2 activity of peroxiredoxin 6 in mice protects against LPS-induced acute lung injury. Am. J. Physiol. Lung 2018. under review. [Google Scholar]
- Lee, I.; Dodia, C.; Chatterjee, S.; Zagorski, J.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Jain, M.; Fisher, A.B. A novel nontoxic inhibitor of the activation of NADPH oxidase reduces reactive oxygen species production in mouse lung. J. Pharmacol. Exp. Ther. 2013, 345, 284–296. [Google Scholar] [CrossRef]
- Lee, I.; Dodia, C.; Chatterjee, S.; Feinstein, S.I.; Fisher, A.B. Protection against LPS-induced acute lung injury by a mechanism based inhibitor of NADPH-oxidase (Type 2). Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Dodia, C.; Chander, A.; Beers, M.F.; Bates, S.R. Inhibition of Trimeresurus flavoviridis phospholipase A2 by lung surfactant protein A (SP-A). Biochim. Biophys. Acta 1994, 1211, 256–262. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Manevich, Y.; Baldwin, J.L.; Dodia, C.; Yu, K.; Feinstein, S.I.; Fisher, A.B. Interaction of surfactant protein A with peroxiredoxin 6 regulates phospholipase A2 activity. J. Biol. Chem. 2006, 281, 7515–7525. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Dodia, C.; Chander, A. Inhibition of lung calcium-independent phospholipase A2 by surfactant protein A. Am. J. Physiol. Lung Cell Mol. Physiol. 1994, 267, L335–L341. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, E.M.; Feinstein, S.I.; Zhou, S.; Fisher, A.B. Intracellular targeting of peroxiredoxin 6 to lysosomal organelles requires MAPK activity and binding to 14-3-3epsilon. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, C1430–C1441. [Google Scholar] [CrossRef] [PubMed]
- Krishnaiah, S.; Dodia, C.; Sorokina, E.; Feinstein, S.I.; Fisher, A. Binding sites for interaction of peroxiredoxin 6 with surfactatn protein A. BBA Proteins Proteom. 2015, 1864, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Protein Docking and Design program. Available online: https://zlab.bu.edu/zlab/index.shtml (accessed on 26 October 2018).
- Head, J.F.; Mealy, T.R.; McCormack, F.X.; Seaton, B.A. Crystal structure of trimeric carbohydrate recognition and neck domains of surfactant protein A. J. Biol. Chem. 2003, 278, 43254–43260. [Google Scholar] [CrossRef]
- Li, H.; Benipal, B.; Zhou, S.; Dodia, C.; Chatterjee, S.; Tao, J.Q.; Sorokina, E.M.; Raabe, T.; Feinstein, S.I.; Fisher, A.B. Critical role of peroxiredoxin 6 in the repair of peroxidized cell membranes following oxidative stress. Free Radic. Biol. Med. 2015, 87, 356–365. [Google Scholar] [CrossRef]
- Fisher, A.B.; Dodia, C.; Feinstein, S.I.; Ho, Y.S. Altered lung phospholipid metabolism in mice with targeted deletion of lysosomal-type phospholipase A2. J. Lipid Res. 2005, 46, 1248–1256. [Google Scholar] [CrossRef]
- Tools for Antimicrobial Peptides. Available online: aps.unmc.edu/AP/tools.php (accessed on 26 October 2018).
- Predicted Antigenic Peptides. Available online: http://imed.med.ucm.es/Tools/antigenic.pl (accessed on 26 October 2018).
- Manevich, Y.; Reddy, K.S.; Shuvaeva, T.; Feinstein, S.I.; Fisher, A.B. Structure and phospholipase function of peroxiredoxin 6: Identification of the catalytic triad and its role in phospholipid substrate binding. J. Lipid Res. 2007, 48, 2306–2318. [Google Scholar] [CrossRef]
- Jain, M.K.; Tao, W.J.; Rogers, J.; Arenson, C.; Eibl, H.; Yu, B.Z. Active-site-directed specific competitive inhibitors of phospholipase A2: Novel transition-state analogues. Biochemistry 1991, 30, 10256–10268. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Dodia, C.; Chander, A. Beta-adrenergic mediators increase pulmonary retention of instilled phospholipids. J. Appll. Physiol. 1985, 59, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Wang, A.; Burke, D.J.; Boudreau, H.E.; Lekstrom, K.J.; Korzeniowska, A.; Sugamata, R.; Kim, Y.S.; Yi, L.; Ersoy, I.; et al. Peroxiredoxin 6 (Prdx6) supports NADPH oxidase1 (Nox1)-based superoxide generation and cell migration. Free Radic. Biol. Med. 2016, 96, 99–115. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Length | Sequence Delete from N-term | Activity, nmol/min/mg Prot. | Sequence Delete from C-term | Activity, nmol/min/mg Prot. |
|---|---|---|---|---|
| control | No peptide | 94.8 | Scrambled peptide: LELDEEITEYQKQLHI | 93.5 |
| 16 aa | DEELQTELYEIKHQIL | 32.0 | DEELQTELYEIKHQIL | 32.0 |
| 14 aa | ELQTELYEIKHQIL | 33.2 | DEELQTELYEIKHQ | 102 |
| 12 aa | QTELYEIKHQIL | 31.5 | No entry | |
| 10 aa | ELYEIKHQIL | 28.6 | No entry | |
| 9 aa | LYEIKHQIL (PIP-1) | 32.3 | ELYEIKHQI | 89.6 |
| 8 aa | YEIKHQIL | 94.4 | DEELQTEL | 93.6 |
| Peptide, Number of Amino Acids | Sequence | Activity, nmol/min/mg Prot. | Comment |
|---|---|---|---|
| No peptide | Control | 92.0 | No added peptide |
| 16 aa | DEELQATLHDFRHQIL | 45.0 | 16 aa human peptide |
| 10 aa | TLHDFRHQIL | 31.5 | Delete from N-term |
| 9 aa | LHDFRHQIL (PIP-2) | 29.9 | Delete from N-term |
| 9 aa | TLHDFRHQI | 89.6 | Delete from C-term |
| Species | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Human * | L | H | D | F | R | H | Q | I | L |
| Primates † | L | H | D | F | R | H | Q | I | L |
| Elephant (African) # | L | H | D | F | R | H | Q | I | L |
| Horse | L | H | D | I | R | H | Q | I | L |
| Wolf | L | H | D | L | R | H | Q | I | L |
| Rabbit | L | H | E | L | R | H | H | A | L |
| Chicken | L | L | N | L | R | Q | R | I | L |
| Rat | L | Y | E | I | K | H | Q | I | L |
| Mouse | L | Y | E | I | K | H | Q | I | L |
| Cotton rat | L | H | E | I | K | H | K | I | L |
| Cow | L | H | E | I | R | H | Q | V | L |
| Yak | L | H | E | I | R | H | Q | V | L |
| Sheep | L | H | E | I | R | H | Q | V | L |
| Pig | L | H | E | I | R | H | Q | I | L |
| Guinea pig | F | H | L | N | K | H | K | I | L |
| Properties | Rat/Mouse (PIP-1) | Human (PIP-2) | Hybrid (PIP-3) |
|---|---|---|---|
| Number of residues | 9 | 9 | 9 |
| Sequence | LYEIKHQIL | LHDFRHQIL | LYDIRHQIL |
| Molecular weight, g/mol | 1156 | 1178 | 1170 |
| Hydrophobic residues On same surface Grand average hydropathy | L,I,I,L I,I 0.1333 | L,F,I,L F,I −0.3333 | L,I,I,L I,I 0.0666 |
| Charged amino acids: neg pos Iso-electric point, pH | E K,H 7.7 | D R,H,H 8.0 | D R,H 7.8 |
| Protein-binding potential, kcal/mol | 0.33 | 2.3 | 1.58 |
| Extinction coeff. at 280 nm, M−1cm−1 | 1490 | 0 | 1490 |
| Antigenic propensity, average | 1.077 | 1.057 | 1.072 |
| Antigenic determinants | none | none | none |
| Conditions | aiPLA2 activity nmol/min/mg Prot. |
|---|---|
| No PIP | 8.72 ± 0.16 |
| PIP-2 in saline | 8.50 ± 0.26 |
| PIP-2 in liposomes | 1.55 ± 0.11 |
| Conditions | PLA2 activity, nmol/min/mg Prot. | |||
|---|---|---|---|---|
| pH 4 | pH 7 + Ca2+ | |||
| WT | D140A-Prdx6 | WT | D140A-Prdx6 | |
| No inhibitor | 8.7 ± 0.16 | 0.2 ± 0.03 | 8.5 ± 0.26 | 8.3 ± 0.26 |
| +PIP-2 | 1.6 ± 0.10 | 0.2 ± 0.06 | 8.5 + 0.30 | 8.5 ± 0.10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisher, A.B.; Dodia, C.; Feinstein, S.I. Identification of Small Peptides that Inhibit NADPH Oxidase (Nox2) Activation. Antioxidants 2018, 7, 181. https://doi.org/10.3390/antiox7120181
Fisher AB, Dodia C, Feinstein SI. Identification of Small Peptides that Inhibit NADPH Oxidase (Nox2) Activation. Antioxidants. 2018; 7(12):181. https://doi.org/10.3390/antiox7120181
Chicago/Turabian StyleFisher, Aron B., Chandra Dodia, and Sheldon I. Feinstein. 2018. "Identification of Small Peptides that Inhibit NADPH Oxidase (Nox2) Activation" Antioxidants 7, no. 12: 181. https://doi.org/10.3390/antiox7120181
APA StyleFisher, A. B., Dodia, C., & Feinstein, S. I. (2018). Identification of Small Peptides that Inhibit NADPH Oxidase (Nox2) Activation. Antioxidants, 7(12), 181. https://doi.org/10.3390/antiox7120181