
Acetic Acid: An Underestimated Metabolite in Ethanol-Induced Changes in Regulating Cardiovascular Function



Abstract
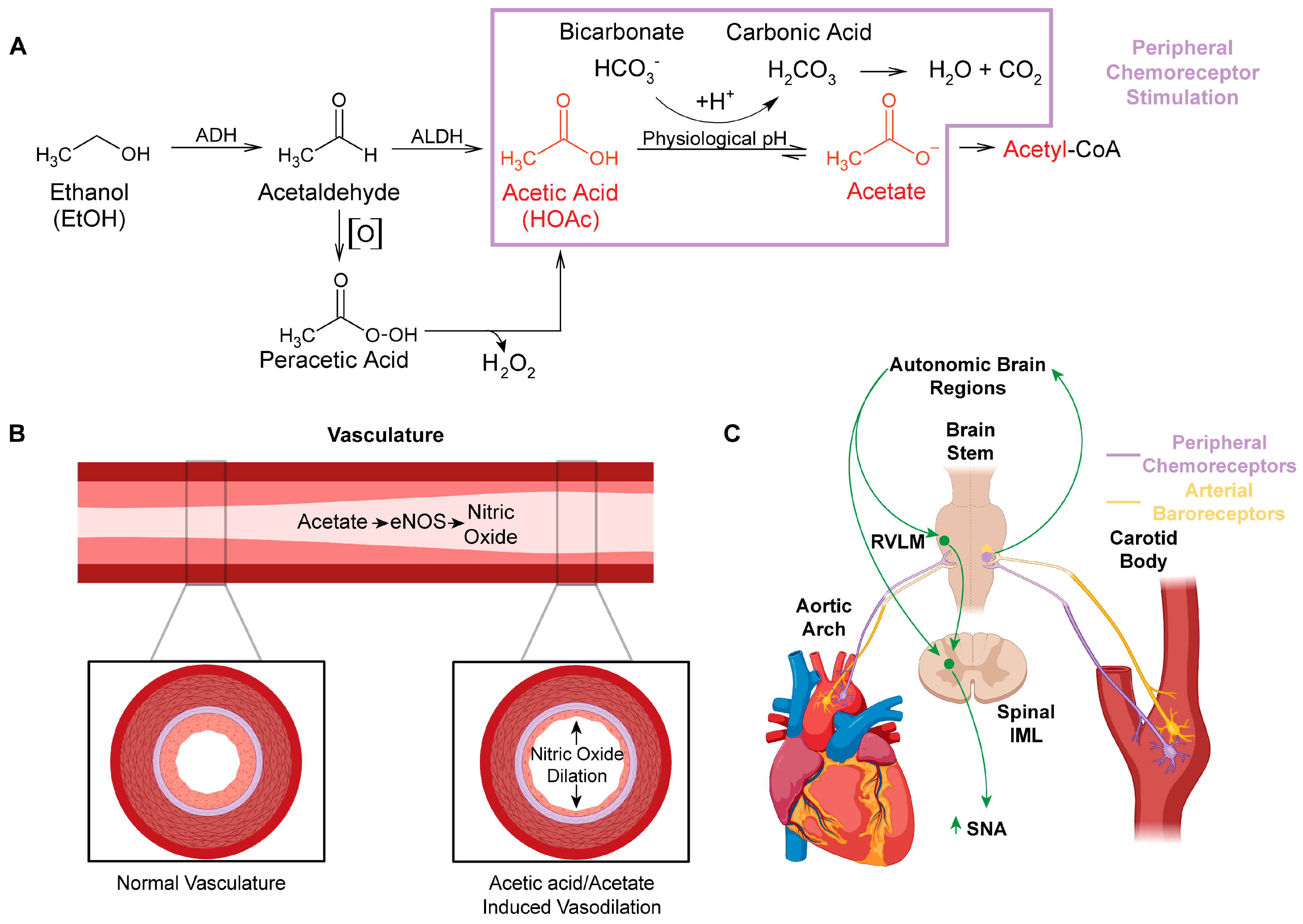
1. Introduction
2. Acetaldehyde and Peracetic Acid: All Roads Lead to Acetic Acid/Acetate
3. Active Metabolites of Ethanol
4. Peripheral Actions of Acetic Acid/Acetate: Impact on Nitric Oxide Synthase (NOS) and Nitric Oxide (NO)
5. Direct Effects of Ethanol and Acetic Acid/Acetate on Cardiac Function
6. Ethanol Metabolism to Acetic Acid Alters Acid/Base Homeostasis and Likely Engages Peripheral Chemoreceptors
7. Integration of Arterial Baroreceptors, Chemoreceptors, and NO Signaling in Response to Acetic Acid Generated by Ethanol Metabolism
8. Acetic Acid-Induced Changes in Neural Control of Cardiovascular Regulation
9. NMDAR–NO Interactions
10. Intersection of Alcohol Cardiovascular Research and AUD Research

{kind=link}
{kind=link}
| Organ/Organ System | Acetic Acid/Acetate | Ethanol |
|---|---|---|
| Brain | ↑GABA [161,204], ↑Glutamate [164,204], ↑NMDAR [2,84,139,151], ↑Dopamine [203], ↑Neuropathology [34,35,84], ↑Cerebral blood flow [205] | ↑GABA [195,206], ↓Glutamate [207,208], ↑Glutamate [206,209], ↑Dopamine [209,210,211], ↑Serotonin [211,212] ↓NMDAR [163,213], ↑Neuropathology [13,214,215], ↑Cerebral blood flow [216,217] |
| Heart | ↓Contractility [119,218], ↑O2 consumption [116], ↑Coronary flow [116], ↑Cardiac output [116] | ↓Contractility [92,219], ↑Coronary flow [220], Arrhythmia [221], ↑Cardiac output [107] |
| Gastrointestinal | ↑Inflammation: Oral cavity [222,223], Esophagus [224], Stomach [225], Small intestine [226], Liver [8], Colon [227] | ↑Inflammation: Oral cavity [228], Esophagus [228], Stomach [229], Small intestine [230,231], Liver [8,232,233], Colon [230] |
| Vasculature | ↑Vasodilation [98,99,102,106,119], ↑NOS [102,106], ↑NO [102] | ↑Vasodilation [85,93,146,234], ↑NOS [235,236], ↑NO [93,95,236] |
11. Conclusions and Future Research Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol. Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Chapp, A.D.; Nwakama, C.A.; Collins, A.R.; Mermelstein, P.G.; Thomas, M.J. Physiological acetic acid concentrations from ethanol metabolism stimulate accumbens shell medium spiny neurons via NMDAR activation in a sex-dependent manner. Neuropsychopharmacology 2023. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef]
- Fernández-Checa, J.C.; Kaplowitz, N.; Colell, A.; García-Ruiz, C. Oxidative stress and alcoholic liver disease. Alcohol. Health Res. World 1997, 21, 321–324. [Google Scholar]
- Tan, H.K.; Yates, E.; Lilly, K.; Dhanda, A.D. Oxidative stress in alcohol-related liver disease. World J. Hepatol. 2020, 12, 332–349. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Mandrekar, P. Oxidative Stress and Inflammation: Essential Partners in Alcoholic Liver Disease. Int. J. Hepatol. 2012, 2012, 853175. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef]
- Kendrick, S.F.; O’Boyle, G.; Mann, J.; Zeybel, M.; Palmer, J.; Jones, D.E.; Day, C.P. Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology 2010, 51, 1988–1997. [Google Scholar] [CrossRef]
- Comporti, M.; Signorini, C.; Leoncini, S.; Gardi, C.; Ciccoli, L.; Giardini, A.; Vecchio, D.; Arezzini, B. Ethanol-induced oxidative stress: Basic knowledge. Genes. Nutr. 2010, 5, 101–109. [Google Scholar] [CrossRef]
- Sun, A.Y.; Sun, G.Y. Ethanol and oxidative mechanisms in the brain. J. Biomed. Sci. 2001, 8, 37–43. [Google Scholar] [CrossRef]
- Crews, F.T. Alcohol-related neurodegeneration and recovery: Mechanisms from animal models. Alcohol. Res. Health 2008, 31, 377–388. [Google Scholar] [PubMed]
- Kamal, H.; Tan, G.C.; Ibrahim, S.F.; Shaikh, M.F.; Mohamed, I.N.; Mohamed, R.M.P.; Hamid, A.A.; Ugusman, A.; Kumar, J. Alcohol Use Disorder, Neurodegeneration, Alzheimer’s and Parkinson’s Disease: Interplay Between Oxidative Stress, Neuroimmune Response and Excitotoxicity. Front. Cell Neurosci. 2020, 14, 282. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Nixon, K. Mechanisms of Neurodegeneration and Regeneration in Alcoholism. Alcohol. Alcohol. 2008, 44, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Piano, M.R. Alcohol’s Effects on the Cardiovascular System. Alcohol. Res. 2017, 38, 219–241. [Google Scholar]
- Biddinger, K.J.; Emdin, C.A.; Haas, M.E.; Wang, M.; Hindy, G.; Ellinor, P.T.; Kathiresan, S.; Khera, A.V.; Aragam, K.G. Association of Habitual Alcohol Intake With Risk of Cardiovascular Disease. JAMA Netw. Open 2022, 5, e223849. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Burgess, S.; Mason, A.M.; Michaëlsson, K. Alcohol Consumption and Cardiovascular Disease. Circ. Genom. Precis. Med. 2020, 13, e002814. [Google Scholar] [CrossRef]
- Toma, A.; Paré, G.; Leong, D.P. Alcohol and Cardiovascular Disease: How Much is Too Much? Curr. Atheroscler. Rep. 2017, 19, 13. [Google Scholar] [CrossRef]
- Sutanto, H.; Cluitmans, M.J.M.; Dobrev, D.; Volders, P.G.A.; Bébarová, M.; Heijman, J. Acute effects of alcohol on cardiac electrophysiology and arrhythmogenesis: Insights from multiscale in silico analyses. J. Mol. Cell Cardiol. 2020, 146, 69–83. [Google Scholar] [CrossRef]
- Kupari, M.; Koskinen, P. Alcohol, cardiac arrhythmias and sudden death. Novartis Found. Symp. 1998, 216, 68–79; discussion 79–85. [Google Scholar] [CrossRef]
- Mustroph, J.; Baier, M.J.; Unsin, D.; Provaznik, Z.; Kozakov, K.; Lebek, S.; Tarnowski, D.; Schildt, S.; Voigt, N.; Wagner, S.; et al. Ethanol-Induced Atrial Fibrillation Results From Late INa and Can Be Prevented by Ranolazine. Circulation 2023, 148, 698–700. [Google Scholar] [CrossRef]
- Agarwal, D.P. Cardioprotective effects of light–moderate consumption of alcohol: A review of putative mechanisms. Alcohol. Alcohol. 2002, 37, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Gray, M.O.; Mochly-Rosen, D. Cardioprotection from ischemia by a brief exposure to physiological levels of ethanol: Role of epsilon protein kinase C. Proc. Natl. Acad. Sci. USA 1999, 96, 12784–12789. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.A. AHA Science Advisory. Alcohol and heart disease. Nutrition Committee of the American Heart Association. Am. J. Clin. Nutr. 1997, 65, 1567–1569. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Husain, K.; Ansari, R.A.; Ferder, L. Alcohol-induced hypertension: Mechanism and prevention. World J. Cardiol. 2014, 6, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Beilin, L.J.; Puddey, I.B. Alcohol and Hypertension. Hypertension 2006, 47, 1035–1038. [Google Scholar] [CrossRef]
- Tsuruta, M.; Adachi, H.; Hirai, Y.; Fujiura, Y.; Imaizumi, T. Association Between Alcohol Intake and Development of Hypertension in Japanese Normotensive Men: 12-Year Follow-Up Study. Am. J. Hypertens. 2000, 13, 482–487. [Google Scholar] [CrossRef]
- Roerecke, M.; Kaczorowski, J.; Tobe, S.W.; Gmel, G.; Hasan, O.S.M.; Rehm, J. The effect of a reduction in alcohol consumption on blood pressure: A systematic review and meta-analysis. Lancet Public. Health 2017, 2, e108–e120. [Google Scholar] [CrossRef]
- Federico, S.D.; Filippini, T.; Whelton, P.K.; Cecchini, M.; Iamandii, I.; Boriani, G.; Vinceti, M. Alcohol Intake and Blood Pressure Levels: A Dose-Response Meta-Analysis of Nonexperimental Cohort Studies. Hypertension 2023, 80, 1961–1969. [Google Scholar] [CrossRef]
- Association, A.H. Even Just 1 Alcoholic Drink a Day May Increase Blood Pressure. Available online: https://www.heart.org/en/news/2023/07/31/even-just-1-alcoholic-drink-a-day-may-increase-blood-pressure (accessed on 19 December 2023).
- Zakhari, S. Alcohol metabolism and epigenetics changes. Alcohol. Res. 2013, 35, 6–16. [Google Scholar]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Mews, P.; Egervari, G.; Nativio, R.; Sidoli, S.; Donahue, G.; Lombroso, S.I.; Alexander, D.C.; Riesche, S.L.; Heller, E.A.; Nestler, E.J.; et al. Alcohol metabolism contributes to brain histone acetylation. Nature 2019, 574, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Dokalis, N.; Mezö, C.; Castoldi, A.; Mossad, O.; Staszewski, O.; Frosch, M.; Villa, M.; Fuchs, V.; Mayer, A.; et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021, 33, 2260–2276.e7. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.V.; Sadler, R.K.; Llovera, G.; Singh, V.; Roth, S.; Heindl, S.; Sebastian Monasor, L.; Verhoeven, A.; Peters, F.; Parhizkar, S.; et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 2021, 10, e59826. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef]
- Tuma, D.J.; Hoffman, T.; Sorrell, M.F. The chemistry of acetaldehyde-protein adducts. Alcohol. Alcohol. Suppl. 1991, 1, 271–276. [Google Scholar]
- Rintala, J.; Jaatinen, P.; Parkkila, S.; Sarviharju, M.; Kiianmaa, K.; Hervonen, A.; Niemelä, O. Evidence of acetaldehyde–protein adduct formation in rat brain after lifelong consumption of ethanol. Alcohol. Alcohol. 2000, 35, 458–463. [Google Scholar] [CrossRef]
- Ito, A.; Jamal, M.; Ameno, K.; Tanaka, N.; Takakura, A.; Kawamoto, T.; Kitagawa, K.; Nakayama, K.; Matsumoto, A.; Miki, T.; et al. Acetaldehyde administration induces salsolinol formation in vivo in the dorsal striatum of Aldh2-knockout and C57BL/6N mice. Neurosci. Lett. 2018, 685, 50–54. [Google Scholar] [CrossRef]
- Peana, A.T.; Rosas, M.; Porru, S.; Acquas, E. From Ethanol to Salsolinol: Role of Ethanol Metabolites in the Effects of Ethanol. J. Exp. Neurosci. 2016, 10, 137–146. [Google Scholar] [CrossRef]
- Bassareo, V.; Frau, R.; Maccioni, R.; Caboni, P.; Manis, C.; Peana, A.T.; Migheli, R.; Porru, S.; Acquas, E. Ethanol-Dependent Synthesis of Salsolinol in the Posterior Ventral Tegmental Area as Key Mechanism of Ethanol’s Action on Mesolimbic Dopamine. Front. Neurosci. 2021, 15, 675061. [Google Scholar] [CrossRef] [PubMed]
- Correa, M.; Acquas, E.; Salamone, J.D. The renaissance of acetaldehyde as a psychoactive compound: Decades in the making. Front. Behav. Neurosci. 2014, 8, 249. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, K.; Ohkuma, S.; Taguchi, J.-I.; Hashimoto, T. Alcohol, acetaldehyde and salsolinol-induced alterations in functions of cerebral GABA/benzodiazepine receptor complex. Physiol. Behav. 1987, 40, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Deehan Jr, G.A.; Engleman, E.A.; Ding, Z.M.; McBride, W.J.; Rodd, Z.A. Microinjections of acetaldehyde or salsolinol into the posterior ventral tegmental area increase dopamine release in the nucleus accumbens shell. Alcohol. Clin. Exp. Res. 2013, 37, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Jamal, M.; Ameno, K.; Ameno, S.; Okada, N.; Ijiri, I. In vivo study of salsolinol produced by a high concentration of acetaldehyde in the striatum and nucleus accumbens of free-moving rats. Alcohol. Clin. Exp. Res. 2003, 27, 79S–84S. [Google Scholar] [CrossRef]
- Zuddas, A.; Corsini, G.U.; Schinelli, S.; Barker, J.L.; Kopin, I.J.; di Porzio, U. Acetaldehyde directly enhances MPP+ neurotoxicity and delays its elimination from the striatum. Brain Res. 1989, 501, 11–22. [Google Scholar] [CrossRef]
- Cui, J.; Liu, Y.; Chang, X.; Gou, W.; Zhou, X.; Liu, Z.; Li, Z.; Wu, Y.; Zuo, D. Acetaldehyde Induces Neurotoxicity In Vitro via Oxidative Stress- and Ca(2+) Imbalance-Mediated Endoplasmic Reticulum Stress. Oxid. Med. Cell Longev. 2019, 2019, 2593742. [Google Scholar] [CrossRef]
- Yan, T.; Zhao, Y. Acetaldehyde induces phosphorylation of dynamin-related protein 1 and mitochondrial dysfunction via elevating intracellular ROS and Ca2+ levels. Redox Biol. 2020, 28, 101381. [Google Scholar] [CrossRef]
- Peana, A.T.; Sánchez-Catalán, M.J.; Hipólito, L.; Rosas, M.; Porru, S.; Bennardini, F.; Romualdi, P.; Caputi, F.F.; Candeletti, S.; Polache, A.; et al. Mystic Acetaldehyde: The Never-Ending Story on Alcoholism. Front. Behav. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef]
- Matsumura, Y.; Stiles, K.M.; Reid, J.; Frenk, E.Z.; Cronin, S.; Pagovich, O.E.; Crystal, R.G. Gene Therapy Correction of Aldehyde Dehydrogenase 2 Deficiency. Mol. Ther. Methods Clin. Dev. 2019, 15, 72–82. [Google Scholar] [CrossRef]
- Goldman, D. Aldehyde Dehydrogenase Deficiency as Cause of Facial Flushing Reaction to Alcohol in Japanese. Alcohol. Health Res. World 1995, 19, 48–49. [Google Scholar] [PubMed]
- Chen, C.H.; Kraemer, B.R.; Mochly-Rosen, D. ALDH2 variance in disease and populations. Dis. Model. Mech. 2022, 15, dmm049601. [Google Scholar] [CrossRef]
- Ohsawa, I.; Kamino, K.; Nagasaka, K.; Ando, F.; Niino, N.; Shimokata, H.; Ohta, S. Genetic deficiency of a mitochondrial aldehyde dehydrogenase increases serum lipid peroxides in community-dwelling females. J. Hum. Genet. 2003, 48, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.; Abdijadid, S. Disulfiram. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Rosman, A.S.; Waraich, A.; Baraona, E.; Lieber, C.S. Disulfiram treatment increases plasma and red blood cell acetaldehyde in abstinent alcoholics. Alcohol. Clin. Exp. Res. 2000, 24, 958–964. [Google Scholar] [CrossRef]
- Kleczkowska, P.; Sulejczak, D.; Zaremba, M. Advantages and disadvantages of disulfiram coadministered with popular addictive substances. Eur. J. Pharmacol. 2021, 904, 174143. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, C.J. Measurement of acetaldehyde: What levels occur naturally and in response to alcohol? Novartis Found. Symp. 2007, 285, 247–255; discussion 256–260. [Google Scholar] [CrossRef]
- Lindros, K.O. Human blood acetaldehyde levels: With improved methods, a clearer picture emerges. Alcohol. Clin. Exp. Res. 1983, 7, 70–75. [Google Scholar] [CrossRef]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 78th ed.; CRC Press: Boca Raton, FL, USA, 1997; p. 3. [Google Scholar]
- Bowen, E.J.; Tietz, E.L. The Oxidation of Acetaldehyde by Oxygen. Nature 1929, 124, 914. [Google Scholar] [CrossRef]
- Bawn, C.E.H.; Williamson, J.B. The oxidation of acetaldehyde in solution. Part I.—The chemistry of the intermediate stages. Trans. Faraday Soc. 1951, 47, 721–734. [Google Scholar] [CrossRef]
- Sánchez, M.; Sabio, L.; Gálvez, N.; Capdevila, M.; Dominguez-Vera, J.M. Iron chemistry at the service of life. IUBMB Life 2017, 69, 382–388. [Google Scholar] [CrossRef]
- Koivunen, J.; Heinonen-Tanski, H. Peracetic acid (PAA) disinfection of primary, secondary and tertiary treated municipal wastewaters. Water Res. 2005, 39, 4445–4453. [Google Scholar] [CrossRef] [PubMed]
- Viola, K.S.; Rodrigues, E.M.; Tanomaru-Filho, M.; Carlos, I.Z.; Ramos, S.G.; Guerreiro-Tanomaru, J.M.; Faria, G. Cytotoxicity of peracetic acid: Evaluation of effects on metabolism, structure and cell death. Int. Endod. J. 2018, 51, e264–e277. [Google Scholar] [CrossRef]
- Turrà, N.; Neuenschwander, U.; Hermans, I. Molecule-induced peroxide homolysis. Chemphyschem 2013, 14, 1666–1669. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, R.R.; Irwin, K.C.; Gould, C.W. Homolytic decompositions of hydroperoxides. IV. Metal-catalyzed decompositions. J. Org. Chem. 1968, 33, 1430–1435. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Machlin, L.J.; Bendich, A. Free radical tissue damage: Protective role of antioxidant nutrients. Faseb J. 1987, 1, 441–445. [Google Scholar] [CrossRef]
- Wu, M.-L.; Tsai, K.-L.; Wang, S.-M.; Wu, J.-C.; Wang, B.-S.; Lee, Y.-T. Mechanism of Hydrogen Peroxide and Hydroxyl Free Radical–Induced Intracellular Acidification in Cultured Rat Cardiac Myoblasts. Circ. Res. 1996, 78, 564–572. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Carlos, T.D.; Bezerra, L.B.; Vieira, M.M.; Sarmento, R.A.; Pereira, D.H.; Cavallini, G.S. Fenton-type process using peracetic acid: Efficiency, reaction elucidations and ecotoxicity. J. Hazard. Mater. 2021, 403, 123949. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Goodwin, M.L.; Harris, J.E.; Hernández, A.; Gladden, L.B. Blood lactate measurements and analysis during exercise: A guide for clinicians. J. Diabetes Sci. Technol. 2007, 1, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Mathew, T.K.; Zubair, M.; Tadi, P. Blood Glucose Monitoring. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Foucher, C.D.; Tubben, R.E. Lactic Acidosis. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Hosios, A.M.; Vander Heiden, M.G. Acetate metabolism in cancer cells. Cancer Metab. 2014, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.H.; Dowling, J.A.; Vreman, H.J.; Feldman, C.; Weiner, M.W. Acetate levels in human plasma. Proc. Clin. Dial. Transpl. Forum 1976, 6, 73–79. [Google Scholar]
- Davies, P.G.; Venkatesh, B.; Morgan, T.J.; Presneill, J.J.; Kruger, P.S.; Thomas, B.J.; Roberts, M.S.; Mundy, J. Plasma acetate, gluconate and interleukin-6 profiles during and after cardiopulmonary bypass: A comparison of Plasma-Lyte 148 with a bicarbonate-balanced solution. Crit. Care 2011, 15, R21. [Google Scholar] [CrossRef] [PubMed]
- Pomare, E.W.; Branch, W.J.; Cummings, J.H. Carbohydrate fermentation in the human colon and its relation to acetate concentrations in venous blood. J. Clin. Investig. 1985, 75, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Tollinger, C.D.; Vreman, H.J.; Weiner, M.W. Measurement of acetate in human blood by gas chromatography: Effects of sample preparation, feeding, and various diseases. Clin. Chem. 1979, 25, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Nuutinen, H.; Lindros, K.; Hekali, P.; Salaspuro, M. Elevated blood acetate as indicator of fast ethanol elimination in chronic alcoholics. Alcohol 1985, 2, 623–626. [Google Scholar] [CrossRef]
- Chapp, A.D.; Schum, S.; Behnke, J.E.; Hahka, T.; Huber, M.J.; Jiang, E.; Larson, R.A.; Shan, Z.; Chen, Q.H. Measurement of cations, anions, and acetate in serum, urine, cerebrospinal fluid, and tissue by ion chromatography. Physiol. Rep. 2018, 6, e13666. [Google Scholar] [CrossRef]
- Chapp, A.D.; Behnke, J.E.; Driscoll, K.M.; Fan, Y.; Hoban, E.; Shan, Z.; Zhang, L.; Chen, Q.H. Acetate Mediates Alcohol Excitotoxicity in Dopaminergic-like PC12 Cells. ACS Chem. Neurosci. 2019, 10, 235–245. [Google Scholar] [CrossRef]
- van de Borne, P.; Mark, A.L.; Montano, N.; Mion, D.; Somers, V.K. Effects of alcohol on sympathetic activity, hemodynamics, and chemoreflex sensitivity. Hypertension 1997, 29, 1278–1283. [Google Scholar] [CrossRef]
- Morvai, V.; Nádházi, Z.; Molnár, G.Y.; Ungváry, G.Y.; Folly, G. Acute effects of low doses of alcohol on the cardiovascular system in young men. Acta Med. Hung. 1988, 45, 339–348. [Google Scholar] [PubMed]
- Sagawa, Y.; Kondo, H.; Matsubuchi, N.; Takemura, T.; Kanayama, H.; Kaneko, Y.; Kanbayashi, T.; Hishikawa, Y.; Shimizu, T. Alcohol has a dose-related effect on parasympathetic nerve activity during sleep. Alcohol. Clin. Exp. Res. 2011, 35, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Julian, T.H.; Syeed, R.; Glascow, N.; Zis, P. Alcohol-induced autonomic dysfunction: A systematic review. Clin. Auton. Res. 2020, 30, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Greenlund, I.M.; Cunningham, H.A.; Tikkanen, A.L.; Bigalke, J.A.; Smoot, C.A.; Durocher, J.J.; Carter, J.R. Morning sympathetic activity after evening binge alcohol consumption. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H305–H315. [Google Scholar] [CrossRef] [PubMed]
- Randin, D.; Vollenweider, P.; Tappy, L.; Jéquier, E.; Nicod, P.; Scherrer, U. Suppression of Alcohol-Induced Hypertension by Dexamethasone. N. Engl. J. Med. 1995, 332, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.R.; Stream, S.F.; Durocher, J.J.; Larson, R.A. Influence of acute alcohol ingestion on sympathetic neural responses to orthostatic stress in humans. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E771–E778. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y. Physio-pathological effects of alcohol on the cardiovascular system: Its role in hypertension and cardiovascular disease. Hypertens. Res. 2010, 33, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Tawakol, A.; Omland, T.; Creager, M.A. Direct effect of ethanol on human vascular function. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H2468–H2473. [Google Scholar] [CrossRef][Green Version]
- Rezvani, A.H.; Grady, D.R.; Peek, A.E.; Pucilowski, O. Inhibition of nitric oxide synthesis attenuates alcohol consumption in two strains of alcohol-preferring rats. Pharmacol. Biochem. Behav. 1995, 50, 265–270. [Google Scholar] [CrossRef]
- Baraona, E.; Zeballos, G.A.; Shoichet, L.; Mak, K.M.; Lieber, C.S. Ethanol Consumption Increases Nitric Oxide Production in Rats, and Its Peroxynitrite-Mediated Toxicity Is Attenuated by Polyenylphosphatidylcholine. Alcohol. Clin. Exp. Res. 2002, 26, 883–889. [Google Scholar] [CrossRef]
- Hakim, R.M.; Pontzer, M.A.; Tilton, D.; Lazarus, J.M.; Gottlieb, M.N. Effects of acetate and bicarbonate dialysate in stable chronic dialysis patients. Kidney Int. 1985, 28, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Leunissen, K.M.L.; Hoorntje, S.J.; Fiers, H.A.; Dekkers, W.T.; Mulder, A.W. Acetate versus Bicarbonate Hemodialysis in Critically Ill Patients. Nephron 2008, 42, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Keshaviah, P.R. The role of acetate in the etiology of symptomatic hypotension. Artif. Organs 1982, 6, 378–387. [Google Scholar] [CrossRef]
- Vinay, P.; Cardoso, M.; Tejedor, A.; Prud’homme, M.; Levelillee, M.; Vinet, B.; Courteau, M.; Gougoux, A.; Rengel, M.; Lapierre, L.; et al. Acetate metabolism during hemodialysis: Metabolic considerations. Am. J. Nephrol. 1987, 7, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Keshaviah, P.; Shapiro, F.L. A critical examination of dialysis-induced hypotension. Am. J. Kidney Dis. 1982, 2, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Todeschini, M.; Casiraghi, F.; Roccatello, D.; Martina, G.; Minetti, L.; Imberti, B.; Gaspari, F.; Atti, M.; Remuzzi, G. Effect of acetate, bicarbonate dialysis, and acetate-free biofiltration on nitric oxide synthesis: Implications for dialysis hypotension. Am. J. Kidney Dis. 1998, 32, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Amore, A.; Cirina, P.; Mitola, S.; Peruzzi, L.; Bonaudo, R.; Gianoglio, B.; Coppo, R. Acetate intolerance is mediated by enhanced synthesis of nitric oxide by endothelial cells. J. Am. Soc. Nephrol. 1997, 8, 1431–1436. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S193–S201. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Biosynthesis of nitric oxide from L-arginine. A pathway for the regulation of cell function and communication. Biochem. Pharmacol. 1989, 38, 1709–1715. [Google Scholar] [CrossRef]
- Sakakibara, S.; Murakami, R.; Takahashi, M.; Fushimi, T.; Murohara, T.; Kishi, M.; Kajimoto, Y.; Kitakaze, M.; Kaga, T. Vinegar intake enhances flow-mediated vasodilatation via upregulation of endothelial nitric oxide synthase activity. Biosci. Biotechnol. Biochem. 2010, 74, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Riff, D.P.; Jain, A.C.; Doyle, J.T. Acute hemodynamic effects of ethanol on normal human volunteers. Am. Heart J. 1969, 78, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Puddey, I.B.; Vandongen, R.; Beilin, L.J.; Rouse, I.L. Alcohol stimulation of renin release in man: Its relation to the hemodynamic, electrolyte, and sympatho-adrenal responses to drinking. J. Clin. Endocrinol. Metab. 1985, 61, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Yelamanchili, V.S.; Brown, K.N.; Goel, A. Holiday Heart Syndrome. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Kupari, M. Acute cardiovascular effects of ethanol A controlled non-invasive study. Br. Heart J. 1983, 49, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Matsukawa, T.; Ishihara, S.; Tanaka, A.; Tanabe, K.; Danbara, A.; Matsuo, M.; Sugiyama, Y.; Mano, T. Effect of oral ethanol intake on muscle sympathetic nerve activity and cardiovascular functions in humans. J. Auton. Nerv. Syst. 1995, 54, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, L.M.; Connell, P.J.; Harris, P.J.; Morgan, T.O. Ethanol effects on cardiomyocyte contractility. Clin. Sci. 2000, 98, 401–407. [Google Scholar] [CrossRef][Green Version]
- Ahmed, S.S.; Levinson, G.E.; Regan, T.J. Depression of myocardial contractility with low doses of ethanol in normal man. Circulation 1973, 48, 378–385. [Google Scholar] [CrossRef]
- Fernández-Solà, J. The Effects of Ethanol on the Heart: Alcoholic Cardiomyopathy. Nutrients 2020, 12, 572. [Google Scholar] [CrossRef]
- Figueredo, V.M.; Chang, K.C.; Baker, A.J.; Camacho, S.A. Chronic alcohol-induced changes in cardiac contractility are not due to changes in the cytosolic Ca2+ transient. Am. J. Physiol. 1998, 275, H122–H130. [Google Scholar] [CrossRef]
- Kiviluoma, K.T.; Karhunen, M.; Lapinlampi, T.; Peuhkurinen, K.J.; Hassinen, I.E. Acetate-induced changes in cardiac energy metabolism and hemodynamics in the rat. Basic. Res. Cardiol. 1988, 83, 431–444. [Google Scholar] [CrossRef]
- Suokas, A.; Kupari, M.; Heikkilä, J.; Lindros, K.; Ylikahri, R. Acute Cardiovascular and Metabolic Effects of Acetate in Men. Alcohol. Clin. Exp. Res. 1988, 12, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, Y.; Zhang, H.; Zhang, X.; Yin, X.; Yuan, F.; Wang, S.; Tian, Y. Acetate suppresses myocardial contraction via the short-chain fatty acid receptor GPR43. Front. Physiol. 2022, 13, 1111156. [Google Scholar] [CrossRef] [PubMed]
- Poll, B.G.; Xu, J.; Jun, S.; Sanchez, J.; Zaidman, N.A.; He, X.; Lester, L.; Berkowitz, D.E.; Paolocci, N.; Gao, W.D.; et al. Acetate, a Short-Chain Fatty Acid, Acutely Lowers Heart Rate and Cardiac Contractility Along with Blood Pressure. J. Pharmacol. Exp. Ther. 2021, 377, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Massion, P.B.; Feron, O.; Dessy, C.; Balligand, J.-L. Nitric Oxide and Cardiac Function. Circ. Res. 2003, 93, 388–398. [Google Scholar] [CrossRef]
- Finkel, M.S.; Oddis, C.V.; Jacob, T.D.; Watkins, S.C.; Hattler, B.G.; Simmons, R.L. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 1992, 257, 387–389. [Google Scholar] [CrossRef]
- Shaaban, A.; Gangwani, M.K.; Pendela, V.S.; Vindhyal, M.R. Alcoholic Cardiomyopathy. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Andersson, C.; Schou, M.; Gustafsson, F.; Torp-Pedersen, C. Alcohol Intake in Patients With Cardiomyopathy and Heart Failure: Consensus and Controversy. Circ. Heart Fail. 2022, 15, e009459. [Google Scholar] [CrossRef]
- Riddick, J.A.; Bunger, W.B.; Sakano, T.K. Organic Solvents: Physical Properties and Methods of Purification, 4th ed.; Wiley: Hoboken, NJ, USA, 1986. [Google Scholar]
- Serjeant, E.P.; Dempsey, B. Ionisation constants of organic acids in aqueous solution. IUPAC Chem. Data Ser. 1979, 23, 160–190. [Google Scholar]
- Bjarnsholt, T.; Alhede, M.; Jensen, P.; Nielsen, A.K.; Johansen, H.K.; Homøe, P.; Høiby, N.; Givskov, M.; Kirketerp-Møller, K. Antibiofilm Properties of Acetic Acid. Adv. Wound Care 2015, 4, 363–372. [Google Scholar] [CrossRef]
- Koob, G.F.; Arends, M.A.; Le Moal, M. Chapter 6—Alcohol. In Drugs, Addiction, and the Brain; Koob, G.F., Arends, M.A., Le Moal, M., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 173–219. [Google Scholar] [CrossRef]
- Gemma, S.; Vichi, S.; Testai, E. Individual susceptibility and alcohol effects:biochemical and genetic aspects. Ann. Ist. Super. Sanita 2006, 42, 8–16. [Google Scholar]
- Raphael, K.L.; Murphy, R.A.; Shlipak, M.G.; Satterfield, S.; Huston, H.K.; Sebastian, A.; Sellmeyer, D.E.; Patel, K.V.; Newman, A.B.; Sarnak, M.J.; et al. Bicarbonate Concentration, Acid-Base Status, and Mortality in the Health, Aging, and Body Composition Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 308–316. [Google Scholar] [CrossRef]
- Burger, M.; Schaller, D.J. Metabolic Acidosis. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Hopkins, E.; Sanvictores, T.; Sharma, S. Physiology, Acid Base Balance. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Raphael, K.L. Metabolic Acidosis in CKD: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 74, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.; Kerndt, C.C.; Moore, R.A. Physiology, Baroreceptors. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Suarez-Roca, H.; Mamoun, N.; Sigurdson, M.I.; Maixner, W. Baroreceptor Modulation of the Cardiovascular System, Pain, Consciousness, and Cognition. Compr. Physiol. 2021, 11, 1373–1423. [Google Scholar] [CrossRef]
- Caverson, M.M.; Ciriello, J.; Calaresu, F.R. Chemoreceptor and baroreceptor inputs to ventrolateral medullary neurons. Am. J. Physiol. 1984, 247, R872–R879. [Google Scholar] [CrossRef] [PubMed]
- Cooper, V.L.; Pearson, S.B.; Bowker, C.M.; Elliott, M.W.; Hainsworth, R. Interaction of chemoreceptor and baroreceptor reflexes by hypoxia and hypercapnia—A mechanism for promoting hypertension in obstructive sleep apnoea. J. Physiol. 2005, 568, 677–687. [Google Scholar] [CrossRef]
- Guyenet, P.G.; Bayliss, D.A. Neural Control of Breathing and CO2 Homeostasis. Neuron 2015, 87, 946–961. [Google Scholar] [CrossRef] [PubMed]
- Toledo, C.; Andrade, D.C.; Lucero, C.; Schultz, H.D.; Marcus, N.; Retamal, M.; Madrid, C.; Del Rio, R. Contribution of peripheral and central chemoreceptors to sympatho-excitation in heart failure. J. Physiol. 2017, 595, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Chapp, A.D.; Gui, L.; Huber, M.J.; Liu, J.; Larson, R.A.; Zhu, J.; Carter, J.R.; Chen, Q.H. Sympathoexcitation and pressor responses induced by ethanol in the central nucleus of amygdala involves activation of NMDA receptors in rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H701–H709. [Google Scholar] [CrossRef]
- Brizzolara, A.L.; Morris, D.G.; Burnstock, G. Ethanol affects sympathetic cotransmission and endothelium-dependent relaxation in the rat. Eur. J. Pharmacol. 1994, 254, 175–181. [Google Scholar] [CrossRef]
- El-Mas, M.M.; Abdel-Rahman, A.A. Enhanced catabolism to acetaldehyde in rostral ventrolateral medullary neurons accounts for the pressor effect of ethanol in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H837–H844. [Google Scholar] [CrossRef]
- Chan, T.C.; Wall, R.A.; Sutter, M.C. Chronic ethanol consumption, stress, and hypertension. Hypertension 1985, 7, 519–524. [Google Scholar] [CrossRef]
- Grassi, G.M.; Somers, V.K.; Renk, W.S.; Abboud, F.M.; Mark, A.L. Effects of alcohol intake on blood pressure and sympathetic nerve activity in normotensive humans: A preliminary report. J. Hypertens. Suppl. 1989, 7, S20–S21. [Google Scholar] [CrossRef] [PubMed]
- Hering, D.; Kucharska, W.; Kara, T.; Somers, V.K.; Narkiewicz, K. Potentiated sympathetic and hemodynamic responses to alcohol in hypertensive vs. normotensive individuals. J. Hypertens. 2011, 29, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Resstel, L.B.; Scopinho, A.A.; Lopes da Silva, A.; Antunes-Rodrigues, J.; Corrêa, F.M. Increased circulating vasopressin may account for ethanol-induced hypertension in rats. Am. J. Hypertens. 2008, 21, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Tirapelli, C.R.; Leone, A.F.; Coelho, E.B.; Resstel, L.B.; Corrêa, F.M.; Lanchote, V.L.; Uyemura, S.A.; Padovan, C.M.; de Oliveira, A.M. Effect of ethanol consumption on blood pressure and rat mesenteric arterial bed, aorta and carotid responsiveness. J. Pharm. Pharmacol. 2007, 59, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Ireland, M.A.; Vandongen, R.; Davidson, L.; Beilin, L.J.; Rouse, I.L. Acute effects of moderate alcohol consumption on blood pressure and plasma catecholamines. Clin. Sci. 1984, 66, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Crandall, D.L.; Ferraro, G.D.; Lozito, R.J.; Cervoni, P.; Clark, L.T. Cardiovascular effects of intermittent drinking: Assessment of a novel animal model of human alcoholism. J. Hypertens. 1989, 7, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Pronko, P.S.; Velichko, M.G.; Moroz, A.R.; Rubanovich, N.N. Low-molecular-weight metabolites relevant to ethanol metabolism: Correlation with alcohol withdrawal severity and utility for identification of alcoholics. Alcohol. Alcohol. 1997, 32, 761–768. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiang, L.; Gulanski, B.I.; De Feyter, H.M.; Weinzimer, S.A.; Pittman, B.; Guidone, E.; Koretski, J.; Harman, S.; Petrakis, I.L.; Krystal, J.H.; et al. Increased brain uptake and oxidation of acetate in heavy drinkers. J. Clin. Investig. 2013, 123, 1605–1614. [Google Scholar] [CrossRef]
- Chapp, A.D.; Collins, A.R.; Driscoll, K.M.; Behnke, J.E.; Shan, Z.; Zhang, L.; Chen, Q.-H. Ethanol Metabolite, Acetate, Increases Excitability of the Central Nucleus of Amygdala Neurons through Activation of NMDA Receptors. ACS Chem. Neurosci. 2023, 14, 1278–1290. [Google Scholar] [CrossRef]
- Chapp, A.D.; Wang, R.; Cheng, Z.J.; Shan, Z.; Chen, Q.H. Long-Term High Salt Intake Involves Reduced SK Currents and Increased Excitability of PVN Neurons with Projections to the Rostral Ventrolateral Medulla in Rats. Neural Plast. 2017, 2017, 7282834. [Google Scholar] [CrossRef]
- Larson, R.A.; Chapp, A.D.; Gui, L.; Huber, M.J.; Cheng, Z.J.; Shan, Z.; Chen, Q.-H. High Salt Intake Augments Excitability of PVN Neurons in Rats: Role of the Endoplasmic Reticulum Ca2+ Store. Front. Neurosci. 2017, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Gui, L.; Huber, M.J.; Chapp, A.D.; Zhu, J.; LaGrange, L.P.; Shan, Z.; Chen, Q.H. Sympathoexcitation in ANG II-salt hypertension involves reduced SK channel function in the hypothalamic paraventricular nucleus. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1547–H1555. [Google Scholar] [CrossRef]
- Chen, Q.H.; Haywood, J.R.; Toney, G.M. Sympathoexcitation by PVN-injected bicuculline requires activation of excitatory amino acid receptors. Hypertension 2003, 42, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.H.; Toney, G.M. Excitability of paraventricular nucleus neurones that project to the rostral ventrolateral medulla is regulated by small-conductance Ca2+-activated K+ channels. J. Physiol. 2009, 587, 4235–4247. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.H.; Toney, G.M. In vivo discharge properties of hypothalamic paraventricular nucleus neurons with axonal projections to the rostral ventrolateral medulla. J. Neurophysiol. 2010, 103, 4–15. [Google Scholar] [CrossRef]
- Adams, J.M.; McCarthy, J.J.; Stocker, S.D. Excess dietary salt alters angiotensinergic regulation of neurons in the rostral ventrolateral medulla. Hypertension 2008, 52, 932–937. [Google Scholar] [CrossRef]
- Kumagai, H.; Oshima, N.; Matsuura, T.; Iigaya, K.; Imai, M.; Onimaru, H.; Sakata, K.; Osaka, M.; Onami, T.; Takimoto, C.; et al. Importance of rostral ventrolateral medulla neurons in determining efferent sympathetic nerve activity and blood pressure. Hypertens. Res. 2012, 35, 132–141. [Google Scholar] [CrossRef]
- El-Mas, M.M.; Fan, M.; Abdel-Rahman, A.A. Role of rostral ventrolateral medullary ERK/JNK/p38 MAPK signaling in the pressor effects of ethanol and its oxidative product acetaldehyde. Alcohol. Clin. Exp. Res. 2013, 37, 1827–1837. [Google Scholar] [CrossRef][Green Version]
- Jin, S.; Cao, Q.; Yang, F.; Zhu, H.; Xu, S.; Chen, Q.; Wang, Z.; Lin, Y.; Cinar, R.; Pawlosky, R.J.; et al. Brain ethanol metabolism by astrocytic ALDH2 drives the behavioural effects of ethanol intoxication. Nat. Metab. 2021, 3, 337–351. [Google Scholar] [CrossRef]
- Jiang, E.; Chapp, A.D.; Fan, Y.; Larson, R.A.; Hahka, T.; Huber, M.J.; Yan, J.; Chen, Q.H.; Shan, Z. Expression of Proinflammatory Cytokines Is Upregulated in the Hypothalamic Paraventricular Nucleus of Dahl Salt-Sensitive Hypertensive Rats. Front. Physiol. 2018, 9, 104. [Google Scholar] [CrossRef]
- Lovinger, D.M.; White, G.; Weight, F.F. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 1989, 243, 1721–1724. [Google Scholar] [CrossRef]
- Chapp, A.D.; Mermelstein, P.G.; Thomas, M.J. The Ethanol Metabolite Acetic Acid Activates Mouse Nucleus Accumbens Shell Medium Spiny Neurons. J. Neurophysiol. 2021, 125, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 1988, 336, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Nitric oxide and neuronal death. Nitric Oxide 2010, 23, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Ledo, A.; Barbosa, R.M.; Gerhardt, G.A.; Cadenas, E.; Laranjinha, J. Concentration dynamics of nitric oxide in rat hippocampal subregions evoked by stimulation of the NMDA glutamate receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 17483–17488. [Google Scholar] [CrossRef]
- Thiel, V.E.; Audus, K.L. Nitric oxide and blood-brain barrier integrity. Antioxid. Redox Signal 2001, 3, 273–278. [Google Scholar] [CrossRef]
- Regehr, W.G.; Carey, M.R.; Best, A.R. Activity-dependent regulation of synapses by retrograde messengers. Neuron 2009, 63, 154–170. [Google Scholar] [CrossRef]
- Cserép, C.; Szőnyi, A.; Veres, J.M.; Németh, B.; Szabadits, E.; de Vente, J.; Hájos, N.; Freund, T.F.; Nyiri, G. Nitric Oxide Signaling Modulates Synaptic Transmission during Early Postnatal Development. Cereb. Cortex 2011, 21, 2065–2074. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef]
- Araki, S.; Osuka, K.; Takata, T.; Tsuchiya, Y.; Watanabe, Y. Coordination between Calcium/Calmodulin-Dependent Protein Kinase II and Neuronal Nitric Oxide Synthase in Neurons. Int. J. Mol. Sci. 2020, 21, 7997. [Google Scholar] [CrossRef]
- Joca, S.R.L.; Sartim, A.G.; Roncalho, A.L.; Diniz, C.F.A.; Wegener, G. Nitric oxide signalling and antidepressant action revisited. Cell Tissue Res. 2019, 377, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.L.; Tjen, A.L.S.C.; Fu, L.W.; Longhurst, J.C. Nitric oxide in rostral ventrolateral medulla regulates cardiac-sympathetic reflexes: Role of synthase isoforms. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1478–H1486. [Google Scholar] [CrossRef] [PubMed]
- Martins-Pinge, M.C.; Baraldi-Passy, I.; Lopes, O.U. Excitatory Effects of Nitric Oxide Within the Rostral Ventrolateral Medulla of Freely Moving Rats. Hypertension 1997, 30, 704–707. [Google Scholar] [CrossRef]
- Morimoto, S.; Sasaki, S.; Miki, S.; Kawa, T.; Nakamura, K.; Itoh, H.; Nakata, T.; Takeda, K.; Nakagawa, M.; Fushiki, S. Nitric oxide is an excitatory modulator in the rostral ventrolateral medulla in rats. Am. J. Hypertens. 2000, 13, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.M.; Patel, K.P. Post-translational regulation of neuronal nitric oxide synthase: Implications for sympathoexcitatory states. Expert. Opin. Ther. Targets 2017, 21, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Mayhan, W.G.; Patel, K.P. Nitric oxide within the paraventricular nucleus mediates changes in renal sympathetic nerve activity. Am. J. Physiol. 1997, 273, R864–R872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, Y.F.; Patel, K.P. Blunted nitric oxide-mediated inhibition of renal nerve discharge within PVN of rats with heart failure. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H995–H1004. [Google Scholar] [CrossRef]
- Northcott, C.A.; Billecke, S.; Craig, T.; Hinojosa-Laborde, C.; Patel, K.P.; Chen, A.F.; D’Alecy, L.G.; Haywood, J.R. Nitric oxide synthase, ADMA, SDMA, and nitric oxide activity in the paraventricular nucleus throughout the etiology of renal wrap hypertension. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2276–H2284. [Google Scholar] [CrossRef][Green Version]
- Crews, F.T.; Morrow, A.L.; Criswell, H.; Breese, G. Effects of ethanol on ion channels. Int. Rev. Neurobiol. 1996, 39, 283–367. [Google Scholar] [CrossRef]
- Ferguson, A.V.; Latchford, K.J.; Samson, W.K. The paraventricular nucleus of the hypothalamus—A potential target for integrative treatment of autonomic dysfunction. Expert. Opin. Ther. Targets 2008, 12, 717–727. [Google Scholar] [CrossRef]
- Gui, L.; LaGrange, L.P.; Larson, R.A.; Gu, M.; Zhu, J.; Chen, Q.H. Role of small conductance calcium-activated potassium channels expressed in PVN in regulating sympathetic nerve activity and arterial blood pressure in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R301–R310. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Han, S.K.; Chong, W.; Li, L.H.; Lee, I.S.; Murase, K.; Ryu, P.D. Noradrenaline excites and inhibits GABAergic transmission in parvocellular neurons of rat hypothalamic paraventricular nucleus. J. Neurophysiol. 2002, 87, 2287–2296. [Google Scholar] [CrossRef]
- Huber, M.J.; Fan, Y.; Jiang, E.; Zhu, F.; Larson, R.A.; Yan, J.; Li, N.; Chen, Q.H.; Shan, Z. Increased activity of the orexin system in the paraventricular nucleus contributes to salt-sensitive hypertension. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1075–H1086. [Google Scholar] [CrossRef] [PubMed]
- Li, D.P.; Yang, Q.; Pan, H.M.; Pan, H.L. Pre- and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. J. Physiol. 2008, 586, 1637–1647. [Google Scholar] [CrossRef]
- Li, D.P.; Zhou, J.J.; Pan, H.L. Endogenous casein kinase-1 modulates NMDA receptor activity of hypothalamic presympathetic neurons and sympathetic outflow in hypertension. J. Physiol. 2015, 593, 4439–4452. [Google Scholar] [CrossRef] [PubMed]
- Li, D.P.; Zhou, J.J.; Zhang, J.; Pan, H.L. CaMKII Regulates Synaptic NMDA Receptor Activity of Hypothalamic Presympathetic Neurons and Sympathetic Outflow in Hypertension. J. Neurosci. 2017, 37, 10690–10699. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.F.; Zhang, H.; Zheng, P.; Chen, S.; Gu, Z.; Zhou, J.J.; Phaup, J.G.; Chang, H.M.; Yeh, E.T.H.; Pan, H.L.; et al. Impaired Kv7 channel activity in the central amygdala contributes to elevated sympathetic outflow in hypertension. Cardiovasc. Res. 2022, 118, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; La Fleur, S.; Van Heijningen, C.; Buijs, R.M. Suprachiasmatic GABAergic inputs to the paraventricular nucleus control plasma glucose concentrations in the rat via sympathetic innervation of the liver. J. Neurosci. 2004, 24, 7604–7613. [Google Scholar] [CrossRef]
- Park, J.B.; Jo, J.Y.; Zheng, H.; Patel, K.P.; Stern, J.E. Regulation of tonic GABA inhibitory function, presympathetic neuronal activity and sympathetic outflow from the paraventricular nucleus by astroglial GABA transporters. J. Physiol. 2009, 587, 4645–4660. [Google Scholar] [CrossRef]
- Li, Y.F.; Mayhan, W.G.; Patel, K.P. NMDA-mediated increase in renal sympathetic nerve discharge within the PVN: Role of nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2328–H2336. [Google Scholar] [CrossRef]
- Zhang, K.; Patel, K.P. Effect of nitric oxide within the paraventricular nucleus on renal sympathetic nerve discharge: Role of GABA. Am. J. Physiol. 1998, 275, R728–R734. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Varodayan, F.P.; Oleata, C.S.; Luu, G.; Kirson, D.; Heilig, M.; Ciccocioppo, R.; Roberto, M. Glutamatergic transmission in the central nucleus of the amygdala is selectively altered in Marchigian Sardinian alcohol-preferring rats: Alcohol and CRF effects. Neuropharmacology 2016, 102, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Roberto, M.; Madamba, S.G.; Stouffer, D.G.; Parsons, L.H.; Siggins, G.R. Increased GABA Release in the Central Amygdala of Ethanol-Dependent Rats. J. Neurosci. 2004, 24, 10159–10166. [Google Scholar] [CrossRef] [PubMed]
- Khom, S.; Wolfe, S.A.; Patel, R.R.; Kirson, D.; Hedges, D.M.; Varodayan, F.P.; Bajo, M.; Roberto, M. Alcohol Dependence and Withdrawal Impair Serotonergic Regulation of GABA Transmission in the Rat Central Nucleus of the Amygdala. J. Neurosci. 2020, 40, 6842–6853. [Google Scholar] [CrossRef] [PubMed]
- Downs, A.M.; McElligott, Z.A. Noradrenergic circuits and signaling in substance use disorders. Neuropharmacology 2022, 208, 108997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.H.; Zhang, J.Y.; Holmes, A.; Pan, B.X. Amygdala Circuit Substrates for Stress Adaptation and Adversity. Biol. Psychiatry 2021, 89, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.T. Alcohol-induced hypertension: Mechanisms, complications, and clinical implications. J. Natl. Med. Assoc. 1985, 77, 385–389. [Google Scholar]
- Witkiewitz, K.; Litten, R.Z.; Leggio, L. Advances in the science and treatment of alcohol use disorder. Sci. Adv. 2019, 5, eaax4043. [Google Scholar] [CrossRef]
- Patel, A.K.; Balasanova, A.A. Unhealthy Alcohol Use. J. Am. Med. Assoc. 2021, 326, 196. [Google Scholar] [CrossRef]
- Harmata, G.I.S.; Chan, A.C.; Merfeld, M.J.; Taugher-Hebl, R.J.; Harijan, A.K.; Hardie, J.B.; Fan, R.; Long, J.D.; Wang, G.Z.; Dlouhy, B.J.; et al. Intoxicating effects of alcohol depend on acid-sensing ion channels. Neuropsychopharmacology 2022, 48, 806–815. [Google Scholar] [CrossRef]
- Drapeau, P.; Nachshen, D.A. Effects of lowering extracellular and cytosolic pH on calcium fluxes, cytosolic calcium levels, and transmitter release in presynaptic nerve terminals isolated from rat brain. J. Gen. Physiol. 1988, 91, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef]
- Tanabe, J.; Yamamoto, D.J.; Sutton, B.; Brown, M.S.; Hoffman, P.L.; Burnham, E.L.; Glueck, D.H.; Tabakoff, B. Effects of Alcohol and Acetate on Cerebral Blood Flow: A Pilot Study. Alcohol. Clin. Exp. Res. 2019, 43, 2070–2078. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Wang, L.; Chen, L.; Krnjević, K.; Fu, R.; Feng, X.; He, W.; Kang, S.; Shah, A.; Bekker, A.; et al. Ethanol potentiates both GABAergic and glutamatergic signaling in the lateral habenula. Neuropharmacology 2017, 113, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Möykkynen, T.; Korpi, E.R. Acute Effects of Ethanol on Glutamate Receptors. Basic. Clin. Pharmacol. Toxicol. 2012, 111, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Roberto, M.; Schweitzer, P.; Madamba, S.G.; Stouffer, D.G.; Parsons, L.H.; Siggins, G.R. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: An in vitro and in vivo analysis. J. Neurosci. 2004, 24, 1594–1603. [Google Scholar] [CrossRef]
- Xiao, C.; Shao, X.M.; Olive, M.F.; Griffin, W.C.; Li, K.-Y.; Krnjević, K.; Zhou, C.; Ye, J.-H. Ethanol Facilitates Glutamatergic Transmission to Dopamine Neurons in the Ventral Tegmental Area. Neuropsychopharmacology 2009, 34, 307–318. [Google Scholar] [CrossRef]
- Melendez, R.I.; Rodd-Henricks, Z.A.; McBride, W.J.; Murphy, J.M. Alcohol Stimulates the Release of Dopamine in the Ventral Pallidum but not in the Globus Pallidus: A Dual-Probe Microdialysis Study. Neuropsychopharmacology 2003, 28, 939–946. [Google Scholar] [CrossRef]
- Yoshimoto, K.; McBride, W.J.; Lumeng, L.; Li, T.K. Alcohol stimulates the release of dopamine and serotonin in the nucleus accumbens. Alcohol 1992, 9, 17–22. [Google Scholar] [CrossRef]
- Lovinger, D.M. Serotonin’s role in alcohol’s effects on the brain. Alcohol. Health Res. World 1997, 21, 114–120. [Google Scholar]
- Woodward, J.J. Ethanol and NMDA receptor signaling. Crit. Rev. Neurobiol. 2000, 14, 69–89. [Google Scholar] [CrossRef]
- Lovinger, D.M. Excitotoxicity and alcohol-related brain damage. Alcohol. Clin. Exp. Res. 1993, 17, 19–27. [Google Scholar] [CrossRef]
- Crews, F.T.; Collins, M.A.; Dlugos, C.; Littleton, J.; Wilkins, L.; Neafsey, E.J.; Pentney, R.; Snell, L.D.; Tabakoff, B.; Zou, J. Alcohol-induced neurodegeneration: When, where and why? Alcohol. Clin. Exp. Res. 2004, 28, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Kuikka, J.; Tiihonen, J.; Hakola, P.; Paanila, J.; Airaksinen, J.; Eronen, M.; Hallikainen, T. Acute ethanol-induced changes in cerebral blood flow. Radioact. Isot. Clin. Med. Res. 1995, 151, 345–350. [Google Scholar]
- Lyons, D.; Miller, M.D.; Hedgecock-Rowe, A.A.; Crane, A.M.; Porrino, L.J. Time-dependent effects of acute ethanol administration on regional cerebral blood flow in the rat. Alcohol 1998, 16, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Kirkendol, R.L.; Pearson, J.E.; Bower, J.D.; Holbert, R.D. Myocardial depressant effects of sodium acetate. Cardiovasc. Res. 1978, 12, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Leiris, J.d.; Lorgeril, M.d.; Boucher, F. Ethanol and cardiac function. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H1027–H1028. [Google Scholar] [CrossRef]
- Gazzieri, D.; Trevisani, M.; Tarantini, F.; Bechi, P.; Masotti, G.; Gensini, G.F.; Castellani, S.; Marchionni, N.; Geppetti, P.; Harrison, S. Ethanol dilates coronary arteries and increases coronary flow via transient receptor potential vanilloid 1 and calcitonin gene-related peptide. Cardiovasc. Res. 2006, 70, 589–599. [Google Scholar] [CrossRef]
- Brunner, S.; Winter, R.; Werzer, C.; von Stülpnagel, L.; Clasen, I.; Hameder, A.; Stöver, A.; Graw, M.; Bauer, A.; Sinner, M.F. Impact of acute ethanol intake on cardiac autonomic regulation. Sci. Rep. 2021, 11, 13255. [Google Scholar] [CrossRef]
- Pan, Y.; Lv, H.; Zhang, F.; Chen, S.; Cheng, Y.; Ma, S.; Hu, H.; Liu, X.; Cai, X.; Fan, F.; et al. Green tea extracts alleviate acetic acid-induced oral inflammation and reconstruct oral microbial balance in mice. J. Food Sci. 2023, 88, 5291–5308. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Lin, W.; Wu, Y.; Li, Q.; Zhou, X.; Li, H.; Xiao, Q.; Wang, Y.; Shao, B.; Yuan, Q. CBD Promotes Oral Ulcer Healing via Inhibiting CMPK2-Mediated Inflammasome. J. Dent. Res. 2022, 101, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Fuse, Y.; Kawamoto, K.; Fujino, H.; Kodama, T. Healing process of experimental esophageal ulcers induced by acetic acid in rats. Scand. J. Gastroenterol. Suppl. 1989, 162, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Yamasaki, S.; Takeuchi, K.; Okabe, S. Delayed healing of acetic acid-induced gastric ulcers in rats by indomethacin. Gastroenterology 1989, 96, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Zhang, X.J.; Gu, X.A.; Clark, D.A. Acute intestinal injury induced by acetic acid and casein: Prevention by intraluminal misoprostol. Gastroenterology 1991, 101, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Fabia, R.; Willén, R.; Ar’Rajab, A.; Andersson, R.; Ahrén, B.; Bengmark, S. Acetic Acid-Induced Colitis in the Rat: A Reproducible Experimental Model for Acute Ulcerative Colitis. Eur. Surg. Res. 2008, 24, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.J.; Peng, T.K.; Yin, S.J. Expression and activities of class IV alcohol dehydrogenase and class III aldehyde dehydrogenase in human mouth. Alcohol 1996, 13, 257–262. [Google Scholar] [CrossRef]
- Andrade, M.C.; Menezes, J.S.; Cassali, G.D.; Martins-Filho, O.A.; Cara, D.C.; Faria, A.M. Alcohol-induced gastritis prevents oral tolerance induction in mice. Clin. Exp. Immunol. 2006, 146, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Jonkers, D.M. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499. [Google Scholar] [CrossRef]
- Bishehsari, F.; Magno, E.; Swanson, G.; Desai, V.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Alcohol and Gut-Derived Inflammation. Alcohol. Res. 2017, 38, 163–171. [Google Scholar]
- Lieber, C.S. Metabolism of Alcohol. Clin. Liver Dis. 2005, 9, 1–35. [Google Scholar] [CrossRef]
- Lieber, C.S. Mechanism of ethanol induced hepatic injury. Pharmacol. Ther. 1990, 46, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Malpas, S.C.; Robinson, B.J.; Maling, T.J. Mechanism of ethanol-induced vasodilation. J. Appl. Physiol. 1990, 68, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.-S.; Deitrich, R.A. Ethanol metabolism and effects: Nitric oxide and its interaction. Curr. Clin. Pharmacol. 2007, 2, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Venkov, C.D.; Myers, P.R.; Tanner, M.A.; Su, M.; Vaughan, D.E. Ethanol increases endothelial nitric oxide production through modulation of nitric oxide synthase expression. Thromb. Haemost. 1999, 81, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Dempsey, S.K.; Daneva, Z.; Azam, M.; Li, N.; Li, P.L.; Ritter, J.K. Role of Nitric Oxide in the Cardiovascular and Renal Systems. Int. J. Mol. Sci. 2018, 19, 2605. [Google Scholar] [CrossRef]
- Hermann, M.; Flammer, A.; Lüscher, T.F. Nitric oxide in hypertension. J. Clin. Hypertens. 2006, 8, 17–29. [Google Scholar] [CrossRef]
- Node, K.; Kitakaze, M.; Yoshikawa, H.; Kosaka, H.; Hori, M. Reduced plasma concentrations of nitrogen oxide in individuals with essential hypertension. Hypertension 1997, 30, 405–408. [Google Scholar] [CrossRef]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chapp, A.D.; Shan, Z.; Chen, Q.-H. Acetic Acid: An Underestimated Metabolite in Ethanol-Induced Changes in Regulating Cardiovascular Function. Antioxidants 2024, 13, 139. https://doi.org/10.3390/antiox13020139
Chapp AD, Shan Z, Chen Q-H. Acetic Acid: An Underestimated Metabolite in Ethanol-Induced Changes in Regulating Cardiovascular Function. Antioxidants. 2024; 13(2):139. https://doi.org/10.3390/antiox13020139
Chicago/Turabian StyleChapp, Andrew D., Zhiying Shan, and Qing-Hui Chen. 2024. "Acetic Acid: An Underestimated Metabolite in Ethanol-Induced Changes in Regulating Cardiovascular Function" Antioxidants 13, no. 2: 139. https://doi.org/10.3390/antiox13020139
APA StyleChapp, A. D., Shan, Z., & Chen, Q.-H. (2024). Acetic Acid: An Underestimated Metabolite in Ethanol-Induced Changes in Regulating Cardiovascular Function. Antioxidants, 13(2), 139. https://doi.org/10.3390/antiox13020139