
Modeling Cardiotoxicity in Pediatric Oncology Patients Using Patient-Specific iPSC-Derived Cardiomyocytes Reveals Downregulation of Cardioprotective microRNAs

 , , , , ,
, , , , ,  ,
,  and
and
Abstract
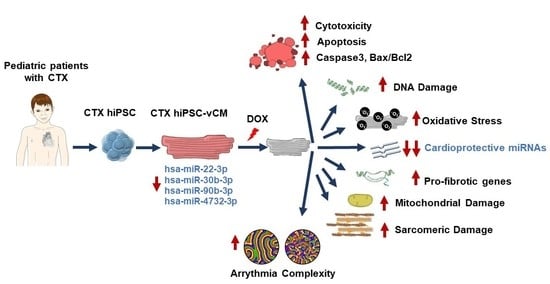
1. Introduction
2. Methods
2.1. Patients and Ethical Statements
2.2. Human iPSC Generation and Maintenance
2.3. Genetic Testing of iPSCs
2.4. Induction of Cardiac Differentiation
2.5. Immunostaining and Flow Cytometry
2.6. Immunocytochemistry
2.7. mRNA Analysis
2.8. Cell Viability
2.9. Lactate Dehydrogenase Assay
2.10. Annexin-V Staining
2.11. Caspase 3/7 Activity Assay
2.12. Western Blotting
2.13. Oxidative Stress Assessment
2.14. Mitochondrial Membrane Potential
2.15. Optical Mapping Acquisition and Image Processing
2.16. miRNA Analysis by RT-qPCR
2.17. Statistical Analysis
3. Results
3.1. Donors
3.2. Generation of hiPSCs from Pediatric Oncology Patients
3.3. Differentiation of iPSCs into Ventricular Cardiomyocytes
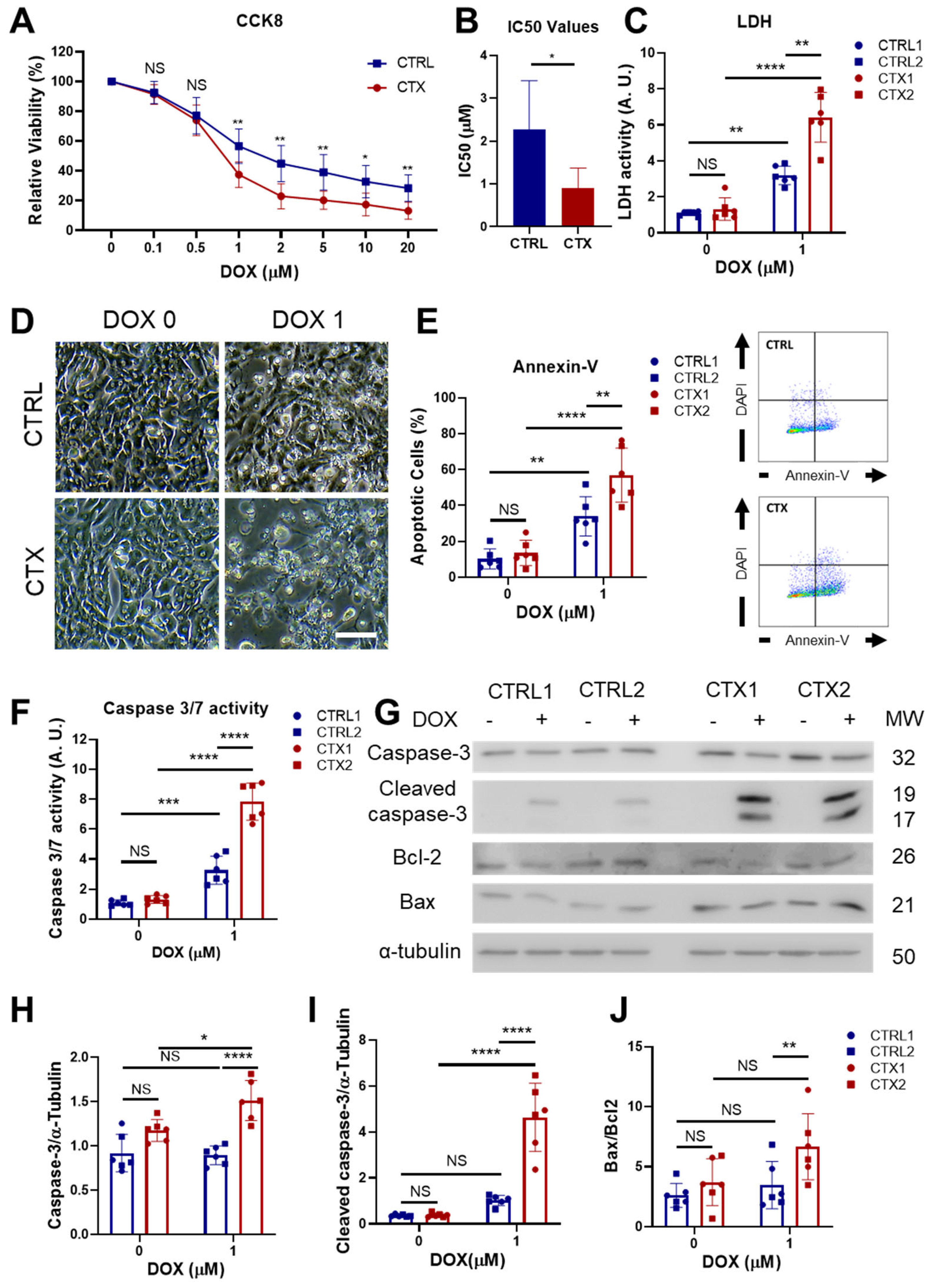
3.4. Analysis of DOX-Induced Cytotoxicity and Apoptosis in hiPSC-CMs
3.5. Effects of DOX on Oxidative Stress
3.6. DOX-Induced Genotoxic and Mitochondrial Damage in hiPSC-CMs
3.7. Sarcomere Disassembly and Gene Expression in hiPSC-CMs
3.8. TGF-β Signaling in hiPSC-CMs
3.9. Differences in Arrhythmogenicity between CTRL and CTX hiPSC-CMs Treated with DOX
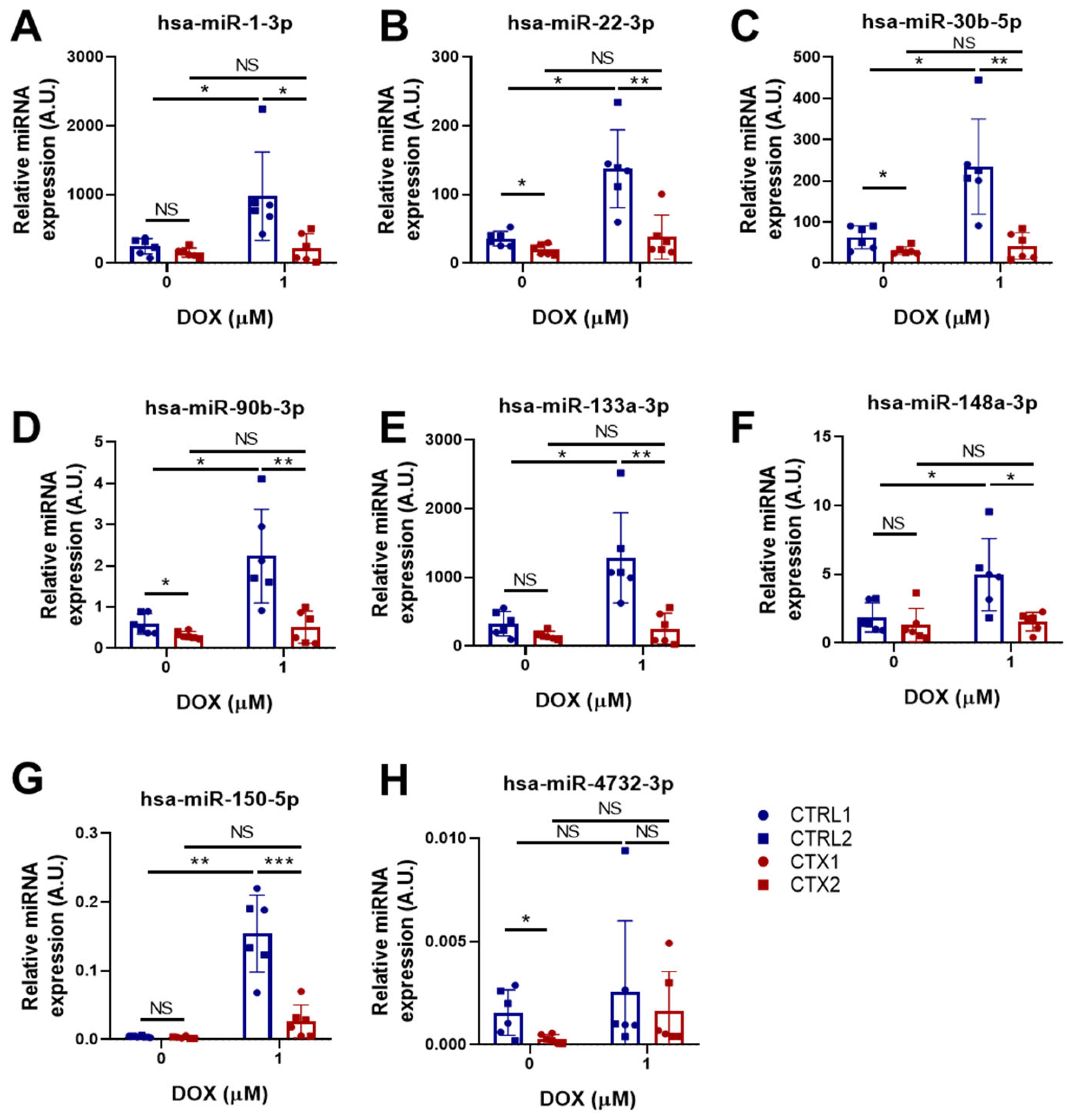
3.10. miRNA Expression Analysis in hiPSC-CMs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chow, E.J.; Leger, K.J.; Bhatt, N.S.; Mulrooney, D.A.; Ross, C.J.; Aggarwal, S.; Bansal, N.; Ehrhardt, M.J.; Armenian, S.H.; Scott, J.M.; et al. Paediatric cardio-oncology: Epidemiology, screening, prevention, and treatment. Cardiovasc. Res. 2019, 115, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Blanco, J.G.; Sharma, U.C.; Pokharel, S.; Shisler, S.; Lipshultz, S.E. Cardiovascular diseases in survivors of childhood cancer. Cancer Metastasis Rev. 2020, 39, 55–68. [Google Scholar] [CrossRef]
- Mancilla, T.R.; Iskra, B.; Aune, G.J. Doxorubicin-Induced Cardiomyopathy in Children. Compr. Physiol. 2019, 9, 905–931. [Google Scholar] [CrossRef]
- Tripaydonis, A.; Conyers, R.; Elliott, D.A. Pediatric Anthracycline Induced Cardiotoxicity: Mechanisms, Pharmacogenomics, and Pluripotent Stem-Cell Modeling. Clin. Pharmacol. Ther. 2019, 105, 614–624. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Sambatakos, P. Cardiotoxicity and Cardioprotection in childhood cancer. Acta Haematol. 2014, 132, 391–399. [Google Scholar] [CrossRef]
- Leerink, J.M.; de Baat, E.C.; Feijen, E.A.M.; Bellersen, L.; van Dalen, E.C.; Grotenhuis, H.B.; Kapusta, L.; Kok, W.E.M.; Loonen, J.; van der Pal, H.J.H.; et al. Cardiac Disease in Childhood Cancer Survivors: Risk Prediction, Prevention, and Surveillance: JACC CardioOncology State-of-the-Art Review. JACC Cardio Oncol. 2020, 2, 363–378. [Google Scholar] [CrossRef]
- Spallarossa, P.; Meliota, G.; Brunelli, C.; Arboscello, E.; Ameri, P.; Dessalvi, C.C.; Grossi, F.; Deidda, M.; Mele, D.; Sarocchi, M.; et al. Potential cardiac risk of immune-checkpoint blockade as anticancer treatment: What we know, what we do not know, and what we can do to prevent adverse effects. Med. Res. Rev. 2018, 38, 1447–1468. [Google Scholar] [CrossRef]
- Mulrooney, D.A.; Yeazel, M.W.; Kawashima, T.; Mertens, A.C.; Mitby, P.; Stovall, M.; Donaldson, S.S.; Green, D.M.; Sklar, C.A.; Robison, L.L.; et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: Retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ 2009, 339, 34. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid. Med. Cell. Longev. 2017, 2017, 363–378. [Google Scholar] [CrossRef]
- Koleini, N.; Nickel, B.E.; Edel, A.L.; Fandrich, R.R.; Ravandi, A.; Kardami, E. Chemico-Biological Interactions Oxidized phospholipids in Doxorubicin-induced cardiotoxicity. Chem. Biol. Interact. 2019, 303, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Catanzaro, M.P.; Weiner, A.; Kaminaris, A.; Li, C.; Cai, F.; Zhao, F.; Kobayashi, S.; Kobayashi, T.; Huang, Y.; Sesaki, H.; et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. 2019, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.F.; Li, V.W.Y.; Lai, C.T.M.; Shin, V.Y.; Keung, W.; Cheuk, D.K.L.; Kwong, A.; Li, R.A.; Chan, G.C.F. Circulating high-sensitivity troponin T and microRNAs as markers of myocardial damage during childhood leukaemia treatment. Pediatr. Res. 2021, 89, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Magdy, T.; Burmeister, B.T.; Burridge, P.W. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol. Ther. 2016, 168, 113–125. [Google Scholar] [CrossRef]
- Aminkeng, F.; Ross, C.J.D.; Rassekh, S.R.; Rieder, M.J.; Bhavsar, A.P.; Sanatani, S.; Bernstein, D.; Hayden, M.R.; Amstutz, U.; Carleton, B.C. Pharmacogenomic screening for anthracycline-induced cardiotoxicity in childhood cancer. Br. J. Clin. Pharmacol. 2017, 83, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S. Genetics of Anthracycline Cardiomyopathy in Cancer Survivors JACC: CardioOncology State-of-the-Art Review. JACC Cardio Oncol. 2020, 2, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Benzoni, P.; Campostrini, G.; Landi, S.; Bertini, V.; Marchina, E.; Iascone, M.; Ahlberg, G.; Olesen, M.S.; Crescini, E.; Mora, C.; et al. Human iPSC modelling of a familial form of atrial fibrillation reveals a gain of function of If and ICaL in patient-derived cardiomyocytes. Cardiovasc. Res. 2020, 116, 1147–1160. [Google Scholar] [CrossRef]
- Ontoria-Oviedo, I.; Földes, G.; Tejedor, S.; Panadero, J.; Kitani, T.; Vázquez, A.; Wu, J.C.; Harding, S.E.; Sepúlveda, P. Modeling transposition of the great arteries with patient-specific induced pluripotent stem cells. Int. J. Mol. Sci. 2021, 22, 3270. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, U.; Nemade, H.; Wagh, V.; Gaspar, J.A.; Ellis, J.K.; Srinivasan, S.P.; Spitkovski, D.; Nguemo, F.; Louisse, J.; Bremer, S.; et al. Identification of genomic biomarkers for anthracycline-induced cardiotoxicity in human iPSC-derived cardiomyocytes: An in vitro repeated exposure toxicity approach for safety assessment. Arch. Toxicol. 2016, 90, 2763–2777. [Google Scholar] [CrossRef]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef]
- Kitani, T.; Ong, S.G.; Lam, C.K.; Rhee, J.W.; Zhang, J.Z.; Oikonomopoulos, A.; Ma, N.; Tian, L.; Lee, J.; Telli, M.L.; et al. Human-Induced Pluripotent Stem Cell Model of Trastuzumab-Induced Cardiac Dysfunction in Patients With Breast Cancer. Circulation 2019, 139, 2451–2465. [Google Scholar] [CrossRef] [PubMed]
- Magdy, T.; Jouni, M.; Kuo, H.H.; Weddle, C.J.; Lyra-Leite, D.; Fonoudi, H.; Romero-Tejeda, M.; Gharib, M.; Javed, H.; Fajardo, G.; et al. Identification of Drug Transporter Genomic Variants and Inhibitors That Protect against Doxorubicin-Induced Cardiotoxicity. Circulation 2022, 145, 279–294. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kleinsorge, M.; Cyganek, L. Subtype-Directed Differentiation of Human iPSCs into Atrial and Ventricular Cardiomyocytes. STAR Protoc. 2020, 1, 100026. [Google Scholar] [CrossRef] [PubMed]
- Garreta, E.; de Oñate, L.; Fernández-Santos, M.E.; Oria, R.; Tarantino, C.; Climent, A.M.; Marco, A.; Samitier, M.; Martínez, E.; Valls-Margarit, M.; et al. Myocardial commitment from human pluripotent stem cells: Rapid production of human heart grafts. Biomaterials 2016, 98, 64–78. [Google Scholar] [CrossRef]
- Climent, A.M.; Guillem, M.S.; Fuentes, L.; Lee, P.; Bollensdorff, C.; Fernández-Santos, M.E.; Suárez-Sancho, S.; Sanz-Ruiz, R.; Sánchez, P.L.; Atienza, F.; et al. Role of atrial tissue remodeling on rotor dynamics: An in vitro study. Am. J. Physiol.-Heart Circ. Physiol. 2015, 309, H1964–H1973. [Google Scholar] [CrossRef]
- Bray, M.A.; Wikswo, J.P. Considerations in phase plane analysis for nonstationary reentrant cardiac behavior. Phys. Rev. E-Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 2002, 65, 051902. [Google Scholar] [CrossRef] [PubMed]
- Welch, P.D. The Use of Fast Fourier Transform for the Estimation of Power Spectra: A Method Based on Time Averaging Over Short, Modified Periodograms. IEEE Trans. Audio Electroacoust. 1967, 15, 70–73. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologie developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Y.; Park, K.M.; Hu, Q.; Teoh, J.P.; Broskova, Z.; Ranganathan, P.; Jayakumar, C.; Li, J.; Su, H.; et al. MicroRNA-150 protects the mouse heart from ischaemic injury by regulating cell death. Cardiovasc. Res. 2015, 106, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, S.; Chang, S.; Ren, D.; Shali, S.; Li, C.; Yang, H.; Huang, Z.; Ge, J. M2 macrophage-derived exosomes carry microRNA-148a to alleviate myocardial ischemia/reperfusion injury via inhibiting TXNIP and the TLR4/NF-κB/NLRP3 inflammasome signaling pathway. J. Mol. Cell. Cardiol. 2020, 142, 65–79. [Google Scholar] [CrossRef]
- Nazari-Shafti, T.Z.; Exarchos, V.; Biefer, H.R.C.; Cesarovic, N.; Meyborg, H.; Falk, V.; Emmert, M.Y. MicroRNA Mediated Cardioprotection—Is There a Path to Clinical Translation? Front. Bioeng. Biotechnol. 2020, 8, 149. [Google Scholar] [CrossRef] [PubMed]
- Razavi-Azarkhiavi, K.; Jaafari, M.R.; Abnous, K.; Razavi, B.M.; Jafarian, A.H.; Hassani, F.V.; Shirani, K.; Karimi, G. The Cardiotoxic Mechanism of Doxorubicin (DOX) and Pegylated Liposomal DOX in Mice Bearing C-26 Colon Carcinoma: A Study Focused on microRNA Role for Toxicity Assessment of New Formulations. Pharm. Res. 2017, 34, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhong, T.; Ma, Y.; Wan, X.; Qin, A.; Yao, B.; Zou, H.; Song, Y.; Yin, D. Bnip3 mediates doxorubicin-induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci. 2020, 242, 117186. [Google Scholar] [CrossRef]
- Yang, S.; Thor, A.D.; Edgerton, S.; Yang, X. Caspase-3 mediated feedback activation of apical caspases in doxorubicin and TNF-alpha induced apoptosis. Apoptosis 2006, 11, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Sawai, T.; Hashidume, Y.; Ohkawa, Y.; Yokoyama, S.; Ohkawa, K. Caspase-3 activation during apoptosis caused by glutathione-doxorubicin conjugate. Br. J. Cancer 1999, 80, 711–715. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wang, X.Q.; Oveisi, F.; Rad, B. Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats. Hypertens 2000, 36, 142–146. [Google Scholar] [CrossRef]
- Xu, J.; Su, X.; Burley, S.K.; Zheng, X.F.S. Nuclear SOD1 in Growth Control, Oxidative Stress Response, Amyotrophic Lateral Sclerosis, and Cancer. Antioxidants 2022, 11, 427. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Long, D.J.; Jaiswal, A.K. NRH:quinone oxidoreductase2 (NQO2). Chem. Biol. Interact. 2000, 129, 99–112. [Google Scholar] [CrossRef]
- Pacher, P.; Liaudet, L.; Bai, P.; Mabley, J.G.; Kaminski, P.M.; Virág, L.; Deb, A.; Szabó, É.; Ungvári, Z.; Wolin, M.S.; et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation 2003, 107, 896–904. [Google Scholar] [CrossRef]
- Neilan, T.G.; Blake, S.L.; Ichinose, F.; Raher, M.J.; Buys, E.S.; Jassal, D.S.; Furutani, E.; Perez-Sanz, T.M.; Graveline, A.; Janssens, S.P.; et al. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation 2007, 116, 506–514. [Google Scholar] [CrossRef]
- Biswas, M.; Chan, J.Y. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 2010, 244, 16–20. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–310. [Google Scholar]
- Holmgren, G.; Sartipy, P.; Andersson, C.X.; Lindahl, A.; Synnergren, J. Expression profiling of human pluripotent stem cell-derived cardiomyocytes exposed to doxorubicin-Integration and visualization of multi-omics data. Toxicol. Sci. 2018, 163, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Tonomura, Y.; Matsushima, S.; Kashiwagi, E.; Fujisawa, K.; Takagi, S.; Nishimura, Y.; Fukushima, R.; Torii, M.; Matsubara, M. Biomarker panel of cardiac and skeletal muscle troponins, fatty acid binding protein 3 and myosin light chain 3 for the accurate diagnosis of cardiotoxicity and musculoskeletal toxicity in rats. Toxicology 2012, 302, 179–189. [Google Scholar] [CrossRef]
- Norton, N.; Bruno, K.A.; Di Florio, D.N.; Whelan, E.R.; Hill, A.R.; Morales-Lara, A.C.; Mease, A.A.; Sousou, J.M.; Malavet, J.A.; Dorn, L.E.; et al. Trpc6 Promotes Doxorubicin-Induced Cardiomyopathy in Male Mice With Pleiotropic Differences between Males and Females. Front. Cardiovasc. Med. 2022, 8, 757784. [Google Scholar] [CrossRef]
- Pierantozzi, E.; Szentesi, P.; Paolini, C.; Dienes, B.; Fodor, J.; Oláh, T.; Colombini, B.; Rassier, D.E.; Rubino, E.M.; Lange, S.; et al. Impaired Intracellular Ca2+ Dynamics, M-Band and Sarcomere Fragility in Skeletal Muscles of Obscurin KO Mice. Int. J. Mol. Sci. 2022, 23, 1319. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; Esposito, G.; Piegari, E.; Russo, R.; Ciuffreda, L.P.; Rivellino, A.; Berrino, L.; Rossi, F.; De Angelis, A.; Urbanek, K. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int. J. Cardiol. 2016, 205, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Xu, J.; Kaiser, R.A.; Melendez, J.; Hambleton, M.; Sargent, M.A.; Lorts, A.; Brunskill, E.W.; Dorn, G.W.; Conway, S.J.; et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 2007, 101, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef]
- Cardin, S.; Libby, E.; Pelletier, P.; Le Bouter, S.; Shiroshita-Takeshita, A.; Le Meur, N.; Léger, J.; Demolombe, S.; Ponton, A.; Glass, L.; et al. Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ. Res. 2007, 100, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 2012, 92, 635–688. [Google Scholar] [CrossRef]
- Bouzeghrane, F.; Reinhardt, D.P.; Reudelhuber, T.L.; Thibault, G. Enhanced expression of fibrillin-1, a constituent of the myocardial extracellular matrix in fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H982–H991. [Google Scholar] [CrossRef]
- Safa, A.; Bahroudi, Z.; Shoorei, H.; Majidpoor, J.; Abak, A.; Taheri, M.; Ghafouri-Fard, S. miR-1: A comprehensive review of its role in normal development and diverse disorders. Biomed. Pharmacother. 2020, 132, 110903. [Google Scholar] [CrossRef]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Izarra, A.; Moscoso, I.; Levent, E.; Cañón, S.; Cerrada, I.; Díez-Juan, A.; Blanca, V.; Núñez-Gil, I.J.; Valiente, I.; Ruíz-Sauri, A.; et al. MiR-133a enhances the protective capacity of cardiac progenitors cells after myocardial infarction. Stem Cell Rep. 2014, 3, 1029–1042. [Google Scholar] [CrossRef]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.-H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol. Cell. Biol. 2009, 29, 2193–2204. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Chaanine, A.H.; Kang, S.; Mukete, B.N.; Jeong, D.; Zhang, S.; Hajjar, R.J.; Lebeche, D. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J. Am. Heart Assoc. 2013, 2, e000078. [Google Scholar] [CrossRef]
- Zhao, X.S.; Ren, Y.; Wu, Y.; Ren, H.K.; Chen, H. MiR-30b-5p and miR-22-3p restrain the fibrogenesis of post-myocardial infarction in mice via targeting PTAFR. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3993–4004. [Google Scholar] [CrossRef]
- Wang, K.; An, T.; Zhou, L.Y.; Liu, C.Y.; Zhang, X.J.; Feng, C.; Li, P.F. E2F1-regulated miR-30b suppresses Cyclophilin D and protects heart from ischemia/reperfusion injury and necrotic cell death. Cell Death Differ. 2015, 22, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Raso, A.; Dirkx, E.; Philippen, L.E.; Fernandez-Celis, A.; De Majo, F.; Sampaio-Pinto, V.; Sansonetti, M.; Juni, R.; el Azzouzi, H.; Calore, M.; et al. Therapeutic Delivery of miR-148a Suppresses Ventricular Dilation in Heart Failure. Mol. Ther. 2019, 27, 584–599. [Google Scholar] [CrossRef]
- Aonuma, T.; Moukette, B.; Kawaguchi, S.; Barupala, N.P.; Sepúlveda, M.N.; Frick, K.; Tang, Y.; Guglin, M.; Raman, S.V.; Cai, C.; et al. MiR-150 Attenuates Maladaptive Cardiac Remodeling Mediated by Long Noncoding RNA MIAT and Directly Represses Profibrotic Hoxa4. Circ. Heart Fail. 2022, 15, E008686. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Xing, W.; Gong, F.; Wang, W.; Yan, Y.; Zhang, Y.; Xie, C.; Fu, S. MiR-150-5p retards the progression of myocardial fibrosis by targeting EGR1. Cell Cycle 2019, 18, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.G.; Zhang, T.N.; Wen, R.; Liu, C.F. Overexpression of miR-150-5p Alleviates Apoptosis in Sepsis-Induced Myocardial Depression. Biomed Res. Int. 2020, 2020, 3023186. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, W.; Wang, L.; Li, Y. Circ_0001052 promotes cardiac hypertrophy via elevating Hipk3. Aging 2023, 15, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA. 2006, 103, 18255–18260. [Google Scholar] [CrossRef]
- Roca-Alonso, L.; Castellano, L.; Mills, A.; Dabrowska, A.F.; Sikkel, M.B.; Pellegrino, L.; Jacob, J.; Frampton, A.E.; Krell, J.; Coombes, R.C.; et al. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in β-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015, 6, e1754. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, R.; Reinal, I.; Peiró-Molina, E.; Buigues, M.; Tejedor, S.; Hernándiz, A.; Selva, M.; Hervás, D.; Cañada, A.J.; Dorronsoro, A.; et al. MicroRNA-4732-3p Is Dysregulated in Breast Cancer Patients with Cardiotoxicity, and Its Therapeutic Delivery Protects the Heart from Doxorubicin-Induced Oxidative Stress in Rats. Antioxidants 2022, 11, 1955. [Google Scholar] [CrossRef]
- Doss, J.F.; Corcoran, D.L.; Jima, D.D.; Telen, M.J.; Dave, S.S.; Chi, J.T. A comprehensive joint analysis of the long and short RNA transcriptomes of human erythrocytes. BMC Genom. 2015, 16, 952. [Google Scholar] [CrossRef]
- Abu-Halima, M.; Weidinger, J.; Poryo, M.; Henn, D.; Keller, A.; Meese, E.; Abdul-Khaliq, H. Micro-RNA signatures in monozygotic twins discordant for congenital heart defects. PLoS ONE 2019, 14, e0226164. [Google Scholar] [CrossRef]
- Su, Y.; Sun, Y.; Tang, Y.; Li, H.; Wang, X.; Pan, X.; Liu, W.; Zhang, X.; Zhang, F.; Xu, Y.; et al. Circulating miR-19b-3p as a Novel Prognostic Biomarker for Acute Heart Failure. J. Am. Heart Assoc. 2021, 10, 22304. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, R.; Gómez-Ferrer, M.; Reinal, I.; Buigues, M.; Villanueva-Bádenas, E.; Ontoria-Oviedo, I.; Hernándiz, A.; González-King, H.; Peiró-Molina, E.; Dorronsoro, A.; et al. miR-4732-3p in Extracellular Vesicles From Mesenchymal Stromal Cells Is Cardioprotective During Myocardial Ischemia. Front. Cell Dev. Biol. 2021, 9, 734143. [Google Scholar] [CrossRef]
- Goukassian, D.; Arakelyan, A.; Brojakowska, A.; Bisserier, M.; Hakobyan, S.; Hadri, L.; Rai, A.K.; Evans, A.; Sebastian, A.; Truongcao, M.; et al. Space flight associated changes in astronauts’ plasma-derived small extracellular vesicle microRNA: Biomarker identification. Clin. Transl. Med. 2022, 12, e845. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTX | Sex | Type of Tumor | Cumulative AC Dose | Age at Diagnosis | Age at iPSC Generation | Tissue | |
|---|---|---|---|---|---|---|---|
| CTRL 1 | No | M | None | - | - | 9 | Foreskin |
| CTRL 2 | No | M | None | - | - | 2.5 | Scrotum |
| CTX 1 | Yes | M | Embryonal rhabdomyosarcoma | >150 mg/m2 | 3.9 | 4 | PBMCs |
| CTX 2 | Yes | M | AML | >250 mg/m2 | 14 | 14 | PBMCs |
| Control | Cardiotoxicity | ||||
|---|---|---|---|---|---|
| Cell Lines | CRTL 1 | CTRL 2 | CTX1 | CTX2 | Total |
| No DOX | 9 (3) | 8 (4) | 12 (4) | 14 (5) | 42 |
| DOX | 5 (3) | 7 (3) | 11 (3) | 12 (5) | 36 |
| Total | 14 | 15 | 23 | 26 | 78 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinal, I.; Ontoria-Oviedo, I.; Selva, M.; Casini, M.; Peiró-Molina, E.; Fambuena-Santos, C.; Climent, A.M.; Balaguer, J.; Cañete, A.; Mora, J.; et al. Modeling Cardiotoxicity in Pediatric Oncology Patients Using Patient-Specific iPSC-Derived Cardiomyocytes Reveals Downregulation of Cardioprotective microRNAs. Antioxidants 2023, 12, 1378. https://doi.org/10.3390/antiox12071378
Reinal I, Ontoria-Oviedo I, Selva M, Casini M, Peiró-Molina E, Fambuena-Santos C, Climent AM, Balaguer J, Cañete A, Mora J, et al. Modeling Cardiotoxicity in Pediatric Oncology Patients Using Patient-Specific iPSC-Derived Cardiomyocytes Reveals Downregulation of Cardioprotective microRNAs. Antioxidants. 2023; 12(7):1378. https://doi.org/10.3390/antiox12071378
Chicago/Turabian StyleReinal, Ignacio, Imelda Ontoria-Oviedo, Marta Selva, Marilù Casini, Esteban Peiró-Molina, Carlos Fambuena-Santos, Andreu M. Climent, Julia Balaguer, Adela Cañete, Jaume Mora, and et al. 2023. "Modeling Cardiotoxicity in Pediatric Oncology Patients Using Patient-Specific iPSC-Derived Cardiomyocytes Reveals Downregulation of Cardioprotective microRNAs" Antioxidants 12, no. 7: 1378. https://doi.org/10.3390/antiox12071378
APA StyleReinal, I., Ontoria-Oviedo, I., Selva, M., Casini, M., Peiró-Molina, E., Fambuena-Santos, C., Climent, A. M., Balaguer, J., Cañete, A., Mora, J., Raya, Á., & Sepúlveda, P. (2023). Modeling Cardiotoxicity in Pediatric Oncology Patients Using Patient-Specific iPSC-Derived Cardiomyocytes Reveals Downregulation of Cardioprotective microRNAs. Antioxidants, 12(7), 1378. https://doi.org/10.3390/antiox12071378