

Targeting Triple-Negative Breast Cancer by the Phytopolyphenol Carnosol: ROS-Dependent Mechanisms


Abstract
1. Introduction
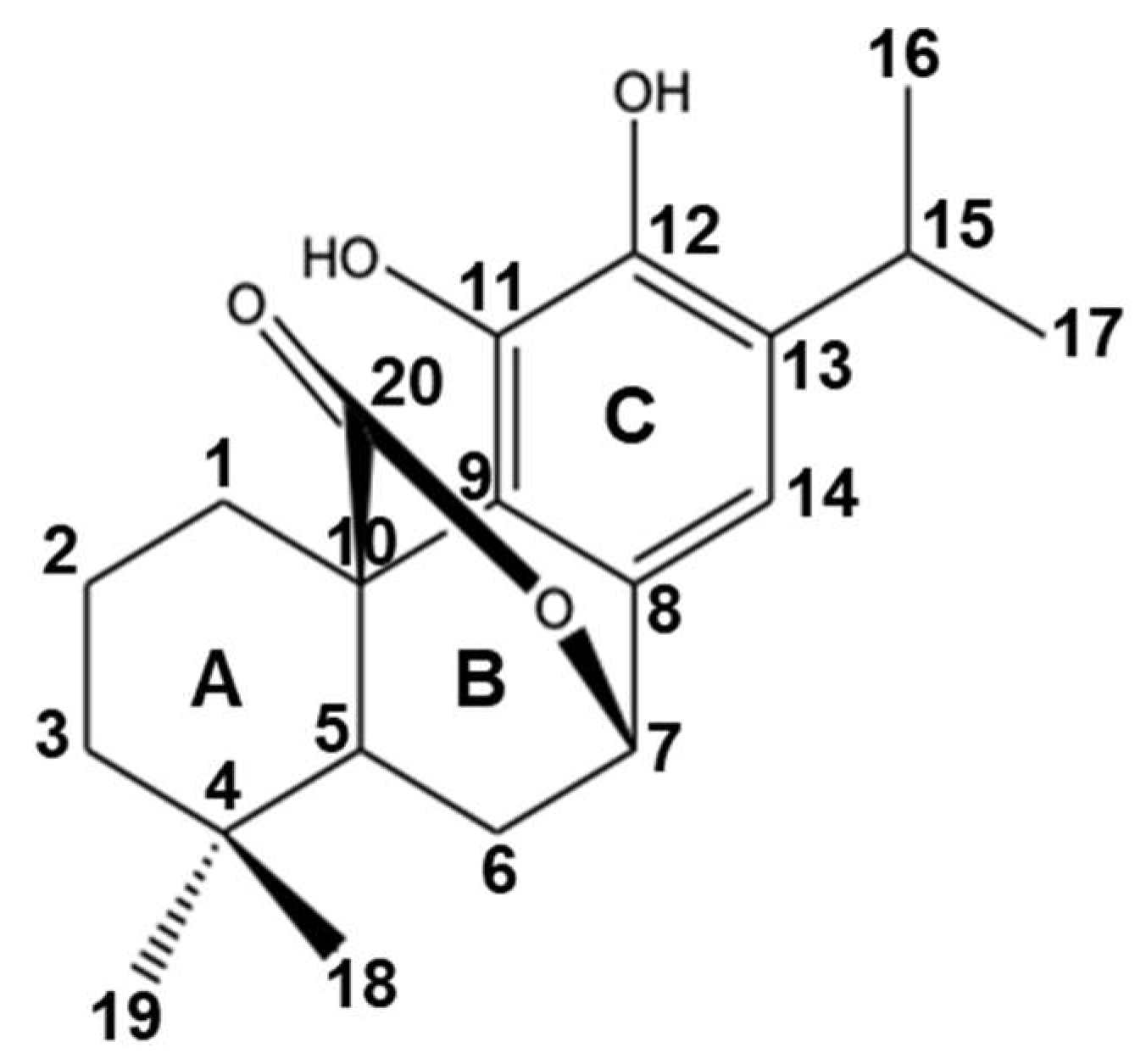
2. Carnosol: Sources, Chemistry, and Structural Characterization
3. Safety and Toxicological Studies on Carnosol
4. Carnosol as a Potent ROS Generator in Cancer Cells
5. Anticancer Activities of Carnosol against TNBC
5.1. Carnosol Decreases Cellular Viability and Induces G2 Arrest in TNBC
5.2. Carnosol Induces Programmed Cell Death (PCD) I and II in TNBC Cells via a ROS-Dependent Mechanism
5.3. Carnosol Induces ER Stress via Activation of the p38-MAPK Pathway through a ROS-Dependent Mechanism
5.4. Carnosol Inhibits p300 Acetyl Transferase
5.5. Carnosol, a Potential Targeted Protein Degradation Molecule, Targets Key Proteins Regulating Cancer Growth and Metastasis through a p38MAPK-Dependent Mechanism
5.6. Carnosol Inhibits Tumor Growth and Metastasis in TNBC
5.7. Synergetic Anticancer Effects of Carnosol in an In Vitro Combination Assay
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ritchie, H.; Roser, M. Causes of Death. Our World Data 2018. Published online at OurWorldInData.org. Available online: https://ourworldindata.org/causes-of-death (accessed on 6 June 2019).
- Song, M. Cancer Overtakes Vascular Disease as Leading Cause of Excess Death Associated with Diabetes. Lancet Diabetes Endocrinol. 2021, 9, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Weir, H.K.; Anderson, R.N.; Coleman King, S.M.; Soman, A.; Thompson, T.D.; Hong, Y.; Moller, B.; Leadbetter, S. Heart Disease and Cancer Deaths—Trends and Projections in the United States, 1969–2020. Prev. Chronic Dis. 2016, 13, E157. [Google Scholar] [CrossRef]
- Harding, M.C.; Sloan, C.D.; Merrill, R.M.; Harding, T.M.; Thacker, B.J.; Thacker, E.L. Transitions From Heart Disease to Cancer as the Leading Cause of Death in US States, 1999–2016. Prev. Chronic Dis. 2018, 15, E158. [Google Scholar] [CrossRef] [PubMed]
- Definition of Cancer—NCI Dictionary of Cancer Terms—National Cancer Institute. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/cancer (accessed on 8 April 2021).
- Cancer Today—Population Fact Sheets. International Agency for Research on Cancer. Available online: http://gco.iarc.fr/today/home (accessed on 8 April 2021).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L. Breast Cancer: Challenges, Controversies, Breakthroughs. Nat. Rev. Clin. Oncol. 2010, 7, 669–670. [Google Scholar] [CrossRef]
- Greenwalt, I.; Zaza, N.; Das, S.; Li, B.D. Precision Medicine and Targeted Therapies in Breast Cancer. Surg. Oncol. Clin. N. Am. 2020, 29, 51–62. [Google Scholar] [CrossRef]
- Harris, E.E.R. Precision Medicine for Breast Cancer: The Paths to Truly Individualized Diagnosis and Treatment. Int. J. Breast Cancer 2018, 2018, 4809183. [Google Scholar] [CrossRef]
- Maisonneuve, P.; Disalvatore, D.; Rotmensz, N.; Curigliano, G.; Colleoni, M.; Dellapasqua, S.; Pruneri, G.; Mastropasqua, M.G.; Luini, A.; Bassi, F.; et al. Proposed New Clinicopathological Surrogate Definitions of Luminal A and Luminal B (HER2-Negative) Intrinsic Breast Cancer Subtypes. Breast Cancer Res. 2014, 16, R65. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene Expression Patterns of Breast Carcinomas Distinguish Tumor Subclasses with Clinical Implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Chodosh, L.A. Breast Cancer: Current State and Future Promise. Breast Cancer Res. 2011, 13, 113. [Google Scholar] [CrossRef]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in Breast Carcinoma Characteristics in Newly Diagnosed African-American and Caucasian Patients: A Single-Institution Compilation Compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results Database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Provenzano, E.; Caldas, C. Triple Negative Breast Cancers: Clinical and Prognostic Implications. Eur. J. Cancer 2009, 45 (Suppl. S1), 27–40. [Google Scholar] [CrossRef] [PubMed]
- Irvin, W.J.; Carey, L.A. What Is Triple-Negative Breast Cancer? Eur. J. Cancer 2008, 44, 2799–2805. [Google Scholar] [CrossRef] [PubMed]
- Collett, K.; Stefansson, I.M.; Eide, J.; Braaten, A.; Wang, H.; Eide, G.E.; Thoresen, S.Ø.; Foulkes, W.D.; Akslen, L.A. A Basal Epithelial Phenotype Is More Frequent in Interval Breast Cancers Compared with Screen Detected Tumors. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Fulford, L.G.; Reis-Filho, J.S.; Ryder, K.; Jones, C.; Gillett, C.E.; Hanby, A.; Easton, D.; Lakhani, S.R. Basal-like Grade III Invasive Ductal Carcinoma of the Breast: Patterns of Metastasis and Long-Term Survival. Breast Cancer Res. 2007, 9, R4. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13 Pt 1, 4429–4434. [Google Scholar] [CrossRef]
- Mahtani, R.; Kittaneh, M.; Kalinsky, K.; Mamounas, E.; Badve, S.; Vogel, C.; Lower, E.; Schwartzberg, L.; Pegram, M.; Breast Cancer Therapy Expert Group (BCTEG). Advances in Therapeutic Approaches for Triple-Negative Breast Cancer. Clin. Breast Cancer 2021, 21, 383–390. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef]
- Jhan, J.-R.; Andrechek, E.R. Triple-Negative Breast Cancer and the Potential for Targeted Therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef]
- Kim, H.; Samuel, S.L.; Zhai, G.; Rana, S.; Taylor, M.; Umphrey, H.R.; Oelschlager, D.K.; Buchsbaum, D.J.; Zinn, K.R. Combination Therapy with Anti-DR5 Antibody and Tamoxifen for Triple Negative Breast Cancer. Cancer Biol. Ther. 2014, 15, 1053–1060. [Google Scholar] [CrossRef]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 Promotes Anthracycline Resistance and Poor Prognosis in Triple Negative Breast Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, S.; Akhtar, N.; Khan, M.S.; Hameed, A.; Irfan, M.; Arshad, M.A.; Ali, S.; Asrar, M. Plant Derived Anticancer Agents: A Green Approach towards Skin Cancers. Biomed Pharm. 2018, 103, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Kapinova, A.; Stefanicka, P.; Kubatka, P.; Zubor, P.; Uramova, S.; Kello, M.; Mojzis, J.; Blahutova, D.; Qaradakhi, T.; Zulli, A.; et al. Are Plant-Based Functional Foods Better Choice against Cancer than Single Phytochemicals? A Critical Review of Current Breast Cancer Research. Biomed Pharm. 2017, 96, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.L.; Nasrollahi, S.; Shah, K.N.; Soltisz, A.; Paruchuri, S.; Yun, Y.H.; Luker, G.D.; Bishayee, A.; Tavana, H. Phytochemicals Potently Inhibit Migration of Metastatic Breast Cancer Cells. Integr. Biol. 2015, 7, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, S.; Moradi, S.Z.; Yarmohammadi, A.; Narimani, F.; Wallace, C.E.; Bishayee, A. Modulation of TLR/NF-ΚB/NLRP Signaling by Bioactive Phytocompounds: A Promising Strategy to Augment Cancer Chemotherapy and Immunotherapy. Front. Oncol. 2022, 12, 834072. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, X.; Li, H.; Zhuang, J.; Feng, F.; Liu, L.; Liu, C.; Sun, C. Identifying the Effect of Ursolic Acid Against Triple-Negative Breast Cancer: Coupling Network Pharmacology With Experiments Verification. Front. Pharm. 2021, 12, 685773. [Google Scholar] [CrossRef]
- Wei, C.; Khan, M.A.; Du, J.; Cheng, J.; Tania, M.; Leung, E.L.-H.; Fu, J. Cordycepin Inhibits Triple-Negative Breast Cancer Cell Migration and Invasion by Regulating EMT-TFs SLUG, TWIST1, SNAIL1, and ZEB1. Front. Oncol. 2022, 12, 898583. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, Z.; Ma, M.; Zhao, Y.; Zhang, C.; Qian, Z.; Wang, B. The Role Played by Ailanthone in Inhibiting Bone Metastasis of Breast Cancer by Regulating Tumor-Bone Microenvironment through the RANKL-Dependent Pathway. Front. Pharm. 2022, 13, 1081978. [Google Scholar] [CrossRef]
- Dandawate, P.; Padhye, S.; Ahmad, A.; Sarkar, F.H. Novel Strategies Targeting Cancer Stem Cells through Phytochemicals and Their Analogs. Drug Deliv. Transl. Res. 2013, 3, 165–182. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Cole, P.A.; Dannenberg, A.J. Retinoids and Carnosol Suppress Cyclooxygenase-2 Transcription by CREB-Binding Protein/P300-Dependent and -Independent Mechanisms. Cancer Res. 2002, 62, 2522–2530. [Google Scholar]
- Satoh, T.; Izumi, M.; Inukai, Y.; Tsutsumi, Y.; Nakayama, N.; Kosaka, K.; Shimojo, Y.; Kitajima, C.; Itoh, K.; Yokoi, T.; et al. Carnosic Acid Protects Neuronal HT22 Cells through Activation of the Antioxidant-Responsive Element in Free Carboxylic Acid- and Catechol Hydroxyl Moieties-Dependent Manners. Neurosci. Lett. 2008, 434, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Kim, J.-S.; Cho, H.-S.; Lee, H.J.; Kim, S.Y.; Kim, S.; Lee, S.-Y.; Chun, H.S. Carnosol, a Component of Rosemary (Rosmarinus Officinalis L.) Protects Nigral Dopaminergic Neuronal Cells. Neuroreport 2006, 17, 1729–1733. [Google Scholar] [CrossRef] [PubMed]
- Weckesser, S.; Engel, K.; Simon-Haarhaus, B.; Wittmer, A.; Pelz, K.; Schempp, C.M. Screening of Plant Extracts for Antimicrobial Activity against Bacteria and Yeasts with Dermatological Relevance. Phytomedicine 2007, 14, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.J.; Syed, D.N.; Suh, Y.; Heren, C.R.; Saleem, M.; Siddiqui, I.A.; Mukhtar, H. Disruption of Androgen and Estrogen Receptor Activity in Prostate Cancer by a Novel Dietary Diterpene Carnosol: Implications for Chemoprevention. Cancer Prev. Res. 2010, 3, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Al Dhaheri, Y.; Attoub, S.; Ramadan, G.; Arafat, K.; Bajbouj, K.; Karuvantevida, N.; AbuQamar, S.; Eid, A.; Iratni, R. Carnosol Induces ROS-Mediated Beclin1-Independent Autophagy and Apoptosis in Triple Negative Breast Cancer. PLoS ONE 2014, 9, e109630. [Google Scholar] [CrossRef]
- Wang, X.Z.; Ron, D. Stress-Induced Phosphorylation and Activation of the Transcription Factor CHOP (GADD153) by P38 MAP Kinase. Science 1996, 272, 1347–1349. [Google Scholar] [CrossRef]
- Yan, M.; Vemu, B.; Veenstra, J.; Petiwala, S.M.; Johnson, J.J. Carnosol, a Dietary Diterpene from Rosemary (Rosmarinus officinalis) Activates Nrf2 Leading to Sestrin 2 Induction in Colon Cells. Integr. Mol. Med. 2018, 5, 1–7. [Google Scholar] [CrossRef]
- O’Neill, E.J.; Hartogh, D.J.D.; Azizi, K.; Tsiani, E. Anticancer Properties of Carnosol: A Summary of in Vitro and In Vivo Evidence. Antioxidants 2020, 9, 961. [Google Scholar] [CrossRef]
- Brieskorn, C.H.; Fuchs, A.; Bredenberg, J.B.; McChesney, J.D.; Wenkert, E. The Structure of Carnosol. J. Org. Chem. 1964, 29, 2293–2298. [Google Scholar] [CrossRef]
- Gajhede, M.; Anthoni, U.; Per Nielsen, H.; Pedersen, E.J.; Christophersen, C. Carnosol. Crystal Structure, Absolute Configuration, and Spectroscopic Properties of a Diterpene. J. Crystallogr. Spectrosc. Res. 1990, 20, 165–171. [Google Scholar] [CrossRef]
- Birtić, S.; Dussort, P.; Pierre, F.-X.; Bily, A.C.; Roller, M. Carnosic Acid. Phytochemistry 2015, 115, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Singletary, K.; MacDonald, C.; Wallig, M. Inhibition by Rosemary and Carnosol of 7,12-Dimethylbenz[a]Anthracene (DMBA)-Induced Rat Mammary Tumorigenesis and in Vivo DMBA-DNA Adduct Formation. Cancer Lett. 1996, 104, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Phipps, K.R.; Lozon, D.; Baldwin, N. Genotoxicity and Subchronic Toxicity Studies of Supercritical Carbon Dioxide and Acetone Extracts of Rosemary. Regul. Toxicol. Pharm. 2021, 119, 104826. [Google Scholar] [CrossRef] [PubMed]
- Use of Rosemary Extracts as a Food Additive—Scientific Opinion of the Panel on Food Additives, Flavourings, Processing Aids and Materials in Contact with Food|EFSA. Available online: https://www.efsa.europa.eu/en/efsajournal/pub/721 (accessed on 17 April 2023).
- Nakamura, H.; Takada, K. Reactive Oxygen Species in Cancer: Current Findings and Future Directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Alsamri, H.; El Hasasna, H.; Al Dhaheri, Y.; Eid, A.H.; Attoub, S.; Iratni, R. Carnosol, a Natural Polyphenol, Inhibits Migration, Metastasis, and Tumor Growth of Breast Cancer via a ROS-Dependent Proteasome Degradation of STAT3. Front. Oncol. 2019, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-W.; Kundu, J.; Chae, I.-G.; Kim, D.-H.; Yu, M.-H.; Kundu, J.K.; Chun, K.-S. Carnosol Induces Apoptosis through Generation of ROS and Inactivation of STAT3 Signaling in Human Colon Cancer HCT116 Cells. Int. J. Oncol. 2014, 44, 1309–1315. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Lin, Y.-C.; Huang, Y.-F.; Hsieh, C.-P.; Wu, C.-C.; Chang, I.-L.; Chen, C.-L.; Cheng, C.-H.; Chen, H.-Y. Carnosol-Induced ROS Inhibits Cell Viability of Human Osteosarcoma by Apoptosis and Autophagy. Am. J. Chin. Med. 2017, 45, 1761–1772. [Google Scholar] [CrossRef]
- Alsamri, H.; Alneyadi, A.; Muhammad, K.; Ayoub, M.A.; Eid, A.; Iratni, R. Carnosol Induces P38-Mediated ER Stress Response and Autophagy in Human Breast Cancer Cells. Front. Oncol. 2022, 12, 911615. [Google Scholar] [CrossRef]
- Vergara, D.; Simeone, P.; Bettini, S.; Tinelli, A.; Valli, L.; Storelli, C.; Leo, S.; Santino, A.; Maffia, M. Antitumor Activity of the Dietary Diterpene Carnosol against a Panel of Human Cancer Cell Lines. Food Funct. 2014, 5, 1261–1269. [Google Scholar] [CrossRef]
- Ling, T.; Tran, M.; González, M.A.; Gautam, L.N.; Connelly, M.; Wood, R.K.; Fatima, I.; Miranda-Carboni, G.; Rivas, F. (+)-Dehydroabietylamine Derivatives Target Triple-Negative Breast Cancer. Eur. J. Med. Chem. 2015, 102, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Alsamri, H.; Hasasna, H.E.; Baby, B.; Alneyadi, A.; Dhaheri, Y.A.; Ayoub, M.A.; Eid, A.H.; Vijayan, R.; Iratni, R. Carnosol Is a Novel Inhibitor of P300 Acetyltransferase in Breast Cancer. Front. Oncol. 2021, 11, 664403. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell Cycle Proteins as Promising Targets in Cancer Therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Dembic, Z. Antitumor Drugs and Their Targets. Molecules 2020, 25, 5776. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, D.; Mittal, S.; Sak, K.; Singhal, P.; Tuli, H.S. Molecular Mechanisms of Action of Quercetin in Cancer: Recent Advances. Tumor Biol. 2016, 37, 12927–12939. [Google Scholar] [CrossRef] [PubMed]
- Visanji, J.M.; Thompson, D.G.; Padfield, P.J. Induction of G2/M Phase Cell Cycle Arrest by Carnosol and Carnosic Acid Is Associated with Alteration of Cyclin A and Cyclin B1 Levels. Cancer Lett. 2006, 237, 130–136. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.-Y.; Lin, L.-T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad Targeting of Resistance to Apoptosis in Cancer. Semin Cancer Biol. 2015, 35 (Suppl.), S78–S103. [Google Scholar] [CrossRef]
- Plati, J.; Bucur, O.; Khosravi-Far, R. Dysregulation of Apoptotic Signaling in Cancer: Molecular Mechanisms and Therapeutic Opportunities. J. Cell. Biochem. 2008, 104, 1124–1149. [Google Scholar] [CrossRef]
- Carafa, V.; Altucci, L. Deregulation of Cell Death in Cancer: Recent Highlights. Cancers 2020, 12, 3517. [Google Scholar] [CrossRef]
- Tamm, I.; Schriever, F.; Dörken, B. Apoptosis: Implications of Basic Research for Clinical Oncology. Lancet Oncol. 2001, 2, 33–42. [Google Scholar] [CrossRef] [PubMed]
- González-Cardenete, M.A.; González-Zapata, N.; Boyd, L.; Rivas, F. Discovery of Novel Bioactive Tanshinones and Carnosol Analogues against Breast Cancer. Cancers 2023, 15, 1318. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of Autophagy in Cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yu, Q.; Zhang, R.; Liu, B. Core Signaling Pathways of Survival/Death in Autophagy-Related Cancer Networks. Int. J. Biochem. Cell Biol. 2011, 43, 1263–1266. [Google Scholar] [CrossRef]
- Li, X.; Wu, W.K.K.; Sun, B.; Cui, M.; Liu, S.; Gao, J.; Lou, H. Dihydroptychantol A, a Macrocyclic Bisbibenzyl Derivative, Induces Autophagy and Following Apoptosis Associated with P53 Pathway in Human Osteosarcoma U2OS Cells. Toxicol. Appl. Pharm. 2011, 251, 146–154. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-Eating and Self-Killing: Crosstalk between Autophagy and Apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Wang, W.; Fan, H.; Zhou, Y.; Duan, P.; Zhao, G.; Wu, G. Knockdown of Autophagy-Related Gene BECLIN1 Promotes Cell Growth and Inhibits Apoptosis in the A549 Human Lung Cancer Cell Line. Mol. Med. Rep. 2013, 7, 1501–1505. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. A Trip to the ER: Coping with Stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The Role of the Unfolded Protein Response in Cancer Progression: From Oncogenesis to Chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Moretti, R.M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Montagnani Marelli, M. Role of Endoplasmic Reticulum Stress in the Anticancer Activity of Natural Compounds. Int. J. Mol. Sci. 2019, 20, 961. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. P38(MAPK): Stress Responses from Molecular Mechanisms to Therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. P38 MAPK-Mediated Regulation of Xbp1s Is Crucial for Glucose Homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Darling, N.J.; Cook, S.J. The Role of MAPK Signalling Pathways in the Response to Endoplasmic Reticulum Stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal Integration by JNK and P38 MAPK Pathways in Cancer Development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Koeberle, A.; Pergola, C.; Shindou, H.; Koeberle, S.C.; Shimizu, T.; Laufer, S.A.; Werz, O. Role of P38 Mitogen-Activated Protein Kinase in Linking Stearoyl-CoA Desaturase-1 Activity with Endoplasmic Reticulum Homeostasis. FASEB J. 2015, 29, 2439–2449. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell Death and Endoplasmic Reticulum Stress: Disease Relevance and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Qu, M.; Liu, Y.; Xu, K.; Wang, D. Activation of P38 MAPK Signaling-Mediated Endoplasmic Reticulum Unfolded Protein Response by Nanopolystyrene Particles. Adv. Biosyst. 2019, 3, e1800325. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Caldas, C. Chromatin Modifier Enzymes, the Histone Code and Cancer. Eur. J. Cancer 2005, 41, 2381–2402. [Google Scholar] [CrossRef]
- Glass, C.K.; Rosenfeld, M.G. The Coregulator Exchange in Transcriptional Functions of Nuclear Receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-Dependent Acetylation of P53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Zhou, M.-M. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and Mechanisms of Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.L.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP Mutations in Relapsed Acute Lymphoblastic Leukaemia. Nature 2011, 471, 235–239. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating Mutations of Acetyltransferase Genes in B-Cell Lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef]
- Zhao, D.; Zou, S.-W.; Liu, Y.; Zhou, X.; Mo, Y.; Wang, P.; Xu, Y.-H.; Dong, B.; Xiong, Y.; Lei, Q.-Y.; et al. Lysine-5 Acetylation Negatively Regulates Lactate Dehydrogenase A and Is Decreased in Pancreatic Cancer. Cancer Cell 2013, 23, 464–476. [Google Scholar] [CrossRef]
- Debes, J.D.; Sebo, T.J.; Lohse, C.M.; Murphy, L.M.; Haugen, D.A.L.; Tindall, D.J. P300 in Prostate Cancer Proliferation and Progression. Cancer Res. 2003, 63, 7638–7640. [Google Scholar]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the Interaction of BRD4 with Diacetylated Twist Suppresses Tumorigenesis in Basal-like Breast Cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone Deacetylases and Their Inhibitors in Cancer, Neurological Diseases and Immune Disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Dwarakanath, B.S.; Verma, A.; Bhatt, A.N.; Parmar, V.S.; Raj, H.G. Targeting Protein Acetylation for Improving Cancer Therapy. Indian J. Med. Res. 2008, 128, 13–21. [Google Scholar] [PubMed]
- Gujral, P.; Mahajan, V.; Lissaman, A.C.; Ponnampalam, A.P. Histone Acetylation and the Role of Histone Deacetylases in Normal Cyclic Endometrium. Reprod. Biol. Endocrinol. 2020, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Dekker, F.J.; Haisma, H.J. Histone Acetyl Transferases as Emerging Drug Targets. Drug Discov. Today 2009, 14, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.G.; Xian, J.; Chin, S.-F.; Bannister, A.J.; Daigo, Y.; Aparicio, S.; Kouzarides, T.; Caldas, C. P300 Is Required for Orderly G1/S Transition in Human Cancer Cells. Oncogene 2007, 26, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Ding, L.; Bohrer, L.R.; Pan, Y.; Liu, P.; Zhang, J.; Sebo, T.J.; Karnes, R.J.; Tindall, D.J.; van Deursen, J.; et al. P300 Acetyltransferase Regulates Androgen Receptor Degradation and PTEN-Deficient Prostate Tumorigenesis. Cancer Res. 2014, 74, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gural, A.; Sun, X.-J.; Zhao, X.; Perna, F.; Huang, G.; Hatlen, M.A.; Vu, L.; Liu, F.; Xu, H.; et al. The Leukemogenicity of AML1-ETO Is Dependent on Site-Specific Lysine Acetylation. Science 2011, 333, 765–769. [Google Scholar] [CrossRef]
- Gojis, O.; Rudraraju, B.; Gudi, M.; Hogben, K.; Sousha, S.; Coombes, R.C.; Cleator, S.; Palmieri, C. The Role of SRC-3 in Human Breast Cancer. Nat. Rev. Clin. Oncol. 2010, 7, 83–89. [Google Scholar] [CrossRef]
- Nandy, D.; Rajam, S.M.; Dutta, D. A Three Layered Histone Epigenetics in Breast Cancer Metastasis. Cell Biosci. 2020, 10, 52. [Google Scholar] [CrossRef]
- Xiao, X.-S.; Cai, M.-Y.; Chen, J.-W.; Guan, X.-Y.; Kung, H.-F.; Zeng, Y.-X.; Xie, D. High Expression of P300 in Human Breast Cancer Correlates with Tumor Recurrence and Predicts Adverse Prognosis. Chin. J. Cancer Res. 2011, 23, 201–207. [Google Scholar] [CrossRef]
- Yokomizo, C.; Yamaguchi, K.; Itoh, Y.; Nishimura, T.; Umemura, A.; Minami, M.; Yasui, K.; Mitsuyoshi, H.; Fujii, H.; Tochiki, N.; et al. High Expression of P300 in HCC Predicts Shortened Overall Survival in Association with Enhanced Epithelial Mesenchymal Transition of HCC Cells. Cancer Lett. 2011, 310, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted Protein Degradation: Mechanisms, Strategies and Application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Targeted Protein Degraders Crowd into the Clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250. [Google Scholar] [CrossRef]
- Li, H.; Qiu, Z.; Li, F.; Wang, C. The Relationship between MMP-2 and MMP-9 Expression Levels with Breast Cancer Incidence and Prognosis. Oncol. Lett. 2017, 14, 5865–5870. [Google Scholar] [CrossRef]
- Li, C.; Guo, S.; Shi, T. Role of NF-ΚB Activation in Matrix Metalloproteinase 9, Vascular Endothelial Growth Factor and Interleukin 8 Expression and Secretion in Human Breast Cancer Cells. Cell Biochem. Funct. 2013, 31, 263–268. [Google Scholar] [CrossRef]
- Cancemi, P.; Buttacavoli, M.; Roz, E.; Feo, S. Expression of Alpha-Enolase (ENO1), Myc Promoter-Binding Protein-1 (MBP-1) and Matrix Metalloproteinases (MMP-2 and MMP-9) Reflect the Nature and Aggressiveness of Breast Tumors. Int. J. Mol. Sci. 2019, 20, 3952. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-C.; Ho, C.-T.; Lin-Shiau, S.-Y.; Lin, J.-K. Carnosol Inhibits the Invasion of B16/F10 Mouse Melanoma Cells by Suppressing Metalloproteinase-9 through down-Regulating Nuclear Factor-Kappa B and c-Jun. Biochem. Pharm. 2005, 69, 221–232. [Google Scholar] [CrossRef]
- Yanagimichi, M.; Nishino, K.; Sakamoto, A.; Kurodai, R.; Kojima, K.; Eto, N.; Isoda, H.; Ksouri, R.; Irie, K.; Kambe, T.; et al. Analyses of Putative Anti-Cancer Potential of Three STAT3 Signaling Inhibitory Compounds Derived from Salvia officinalis. Biochem. Biophys. Rep. 2021, 25, 100882. [Google Scholar] [CrossRef]
- Şakalar, Ç.; İzgi, K.; İskender, B.; Sezen, S.; Aksu, H.; Çakır, M.; Kurt, B.; Turan, A.; Canatan, H. The Combination of Thymoquinone and Paclitaxel Shows Anti-Tumor Activity through the Interplay with Apoptosis Network in Triple-Negative Breast Cancer. Tumor Biol. 2016, 37, 4467–4477. [Google Scholar] [CrossRef]
- Jafri, S.H.; Glass, J.; Shi, R.; Zhang, S.; Prince, M.; Kleiner-Hancock, H. Thymoquinone and Cisplatin as a Therapeutic Combination in Lung Cancer: In Vitro and in Vivo. J. Exp. Clin. Cancer Res. 2010, 29, 87. [Google Scholar] [CrossRef]
- Islam, M.T. Diterpenes and Their Derivatives as Potential Anticancer Agents. Phytother. Res. 2017, 31, 691–712. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell Line | Carnosol | Experimental Model | Targeted Proteins | Reference | ||||
|---|---|---|---|---|---|---|---|---|
| Alone | In Combination | In Vitro | In Vivo | In Silico | Upregulated | Downregulated | ||
| HBL-100 MDA-231 MDA-435 | 12.5–200 μM; 4 h, 24 h, 48 h and 72 h | Carnosol 50 μM + Curcumin 70 μM; 4 h | ✓ | - | - | p27 | Cyclin D1 Bcl2 Survivin | [54] |
| MDA-MB-231 | 25–100 μM; 24 h and 48 h | - | ✓ | - | - | p21/WAF1 Bax LC3II pERK1/2 γH2AX | p27 PARP Cleaved Caspases 3,8,9 Bcl2 p62 (SQSTM1) | [38] |
| MDA-MB-231 | EC50 < 9 μM | - | ✓ | - | - | [55] | ||
| MDA-MB-231 Hs578T | 25–100 μM | - | ✓ | ✓ | - | MMP-9 STAT3 | [50] | |
| MDA-MB-231 Hs578T | 25–100 μM; 24 h | - | ✓ | - | ✓ | Ac-H3K14 Ac-H3K56 Ac-H4K9 Ac-H4K16 Ac-H4K5 P300 PCAF | [56] | |
| MDA-MB-231 Hs578T | 50 and 100 μM; 24 h | - | ✓ | - | - | PARP LC3 I/II IRE1α XBP-1s Cl. ATF6 eIF2α ATF4 CHOP pP38 Ubiquitination | mTOR PDI Ero1α PCAF STAT3 | [53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsamri, H.; Al Dhaheri, Y.; Iratni, R. Targeting Triple-Negative Breast Cancer by the Phytopolyphenol Carnosol: ROS-Dependent Mechanisms. Antioxidants 2023, 12, 1349. https://doi.org/10.3390/antiox12071349
Alsamri H, Al Dhaheri Y, Iratni R. Targeting Triple-Negative Breast Cancer by the Phytopolyphenol Carnosol: ROS-Dependent Mechanisms. Antioxidants. 2023; 12(7):1349. https://doi.org/10.3390/antiox12071349
Chicago/Turabian StyleAlsamri, Halima, Yusra Al Dhaheri, and Rabah Iratni. 2023. "Targeting Triple-Negative Breast Cancer by the Phytopolyphenol Carnosol: ROS-Dependent Mechanisms" Antioxidants 12, no. 7: 1349. https://doi.org/10.3390/antiox12071349
APA StyleAlsamri, H., Al Dhaheri, Y., & Iratni, R. (2023). Targeting Triple-Negative Breast Cancer by the Phytopolyphenol Carnosol: ROS-Dependent Mechanisms. Antioxidants, 12(7), 1349. https://doi.org/10.3390/antiox12071349