
TRP Channels in Tumoral Processes Mediated by Oxidative Stress and Inflammation




Abstract
1. Introduction
2. Sources of ROS and Their Targets in Tumoral Cells
3. ROS and the Redox TRP Channels
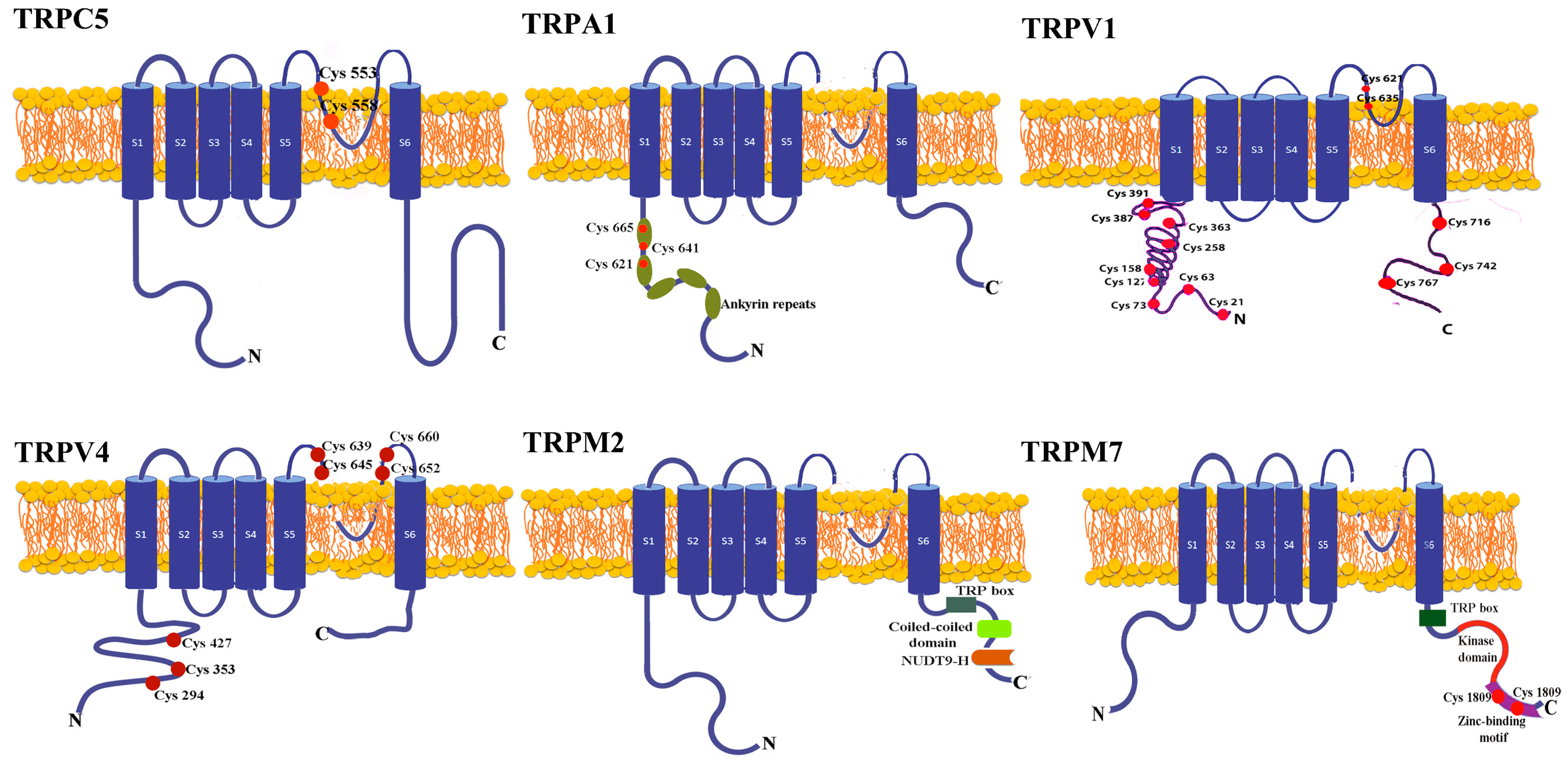
4. The Structure of the Redox TRP Channels
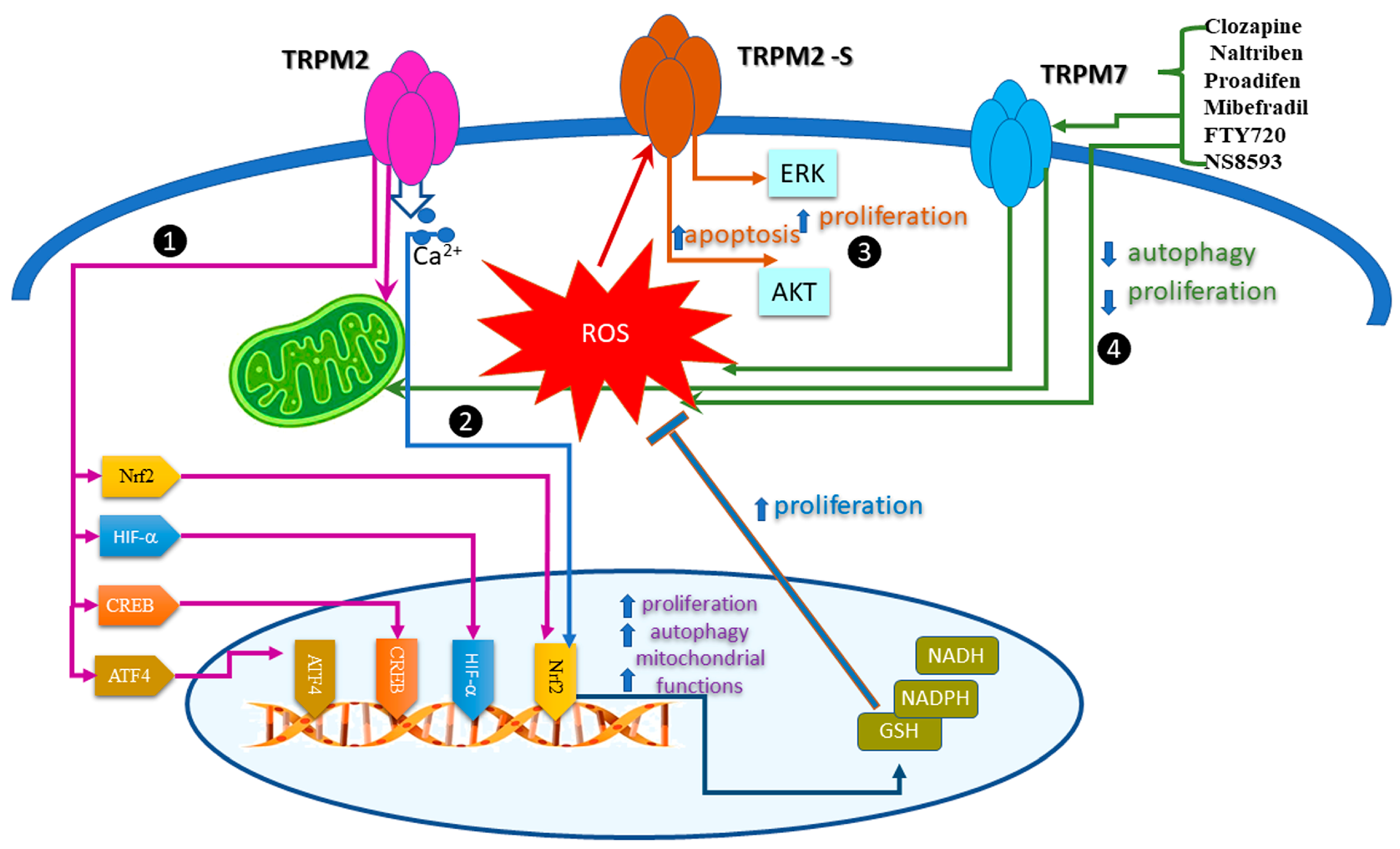
5. Regulation of the Redox TRP Channels by Oxidative Stress in Cancer Cell Proliferation
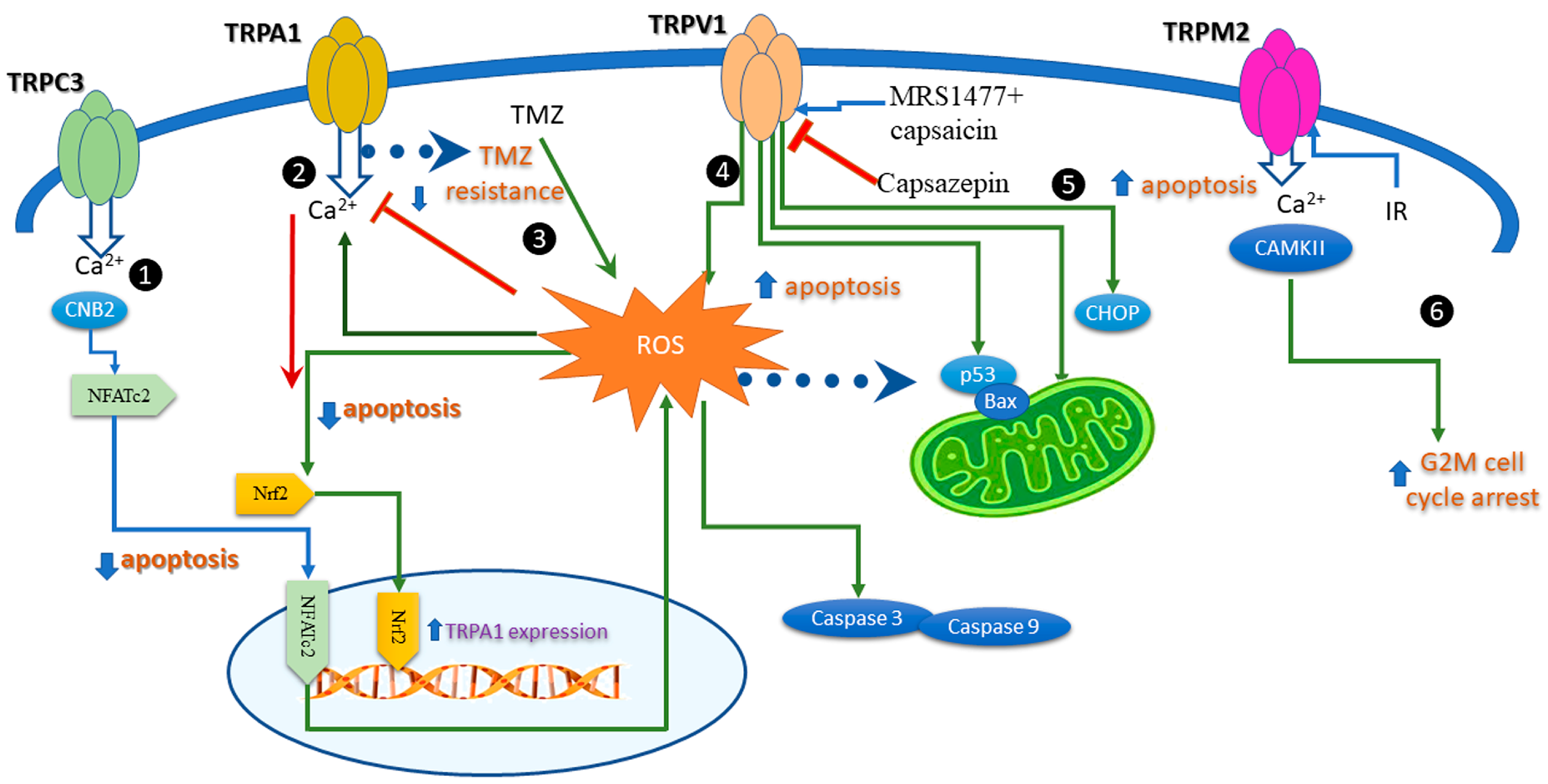
6. Apoptosis Induced by Modulation of the Redox TRP Channels
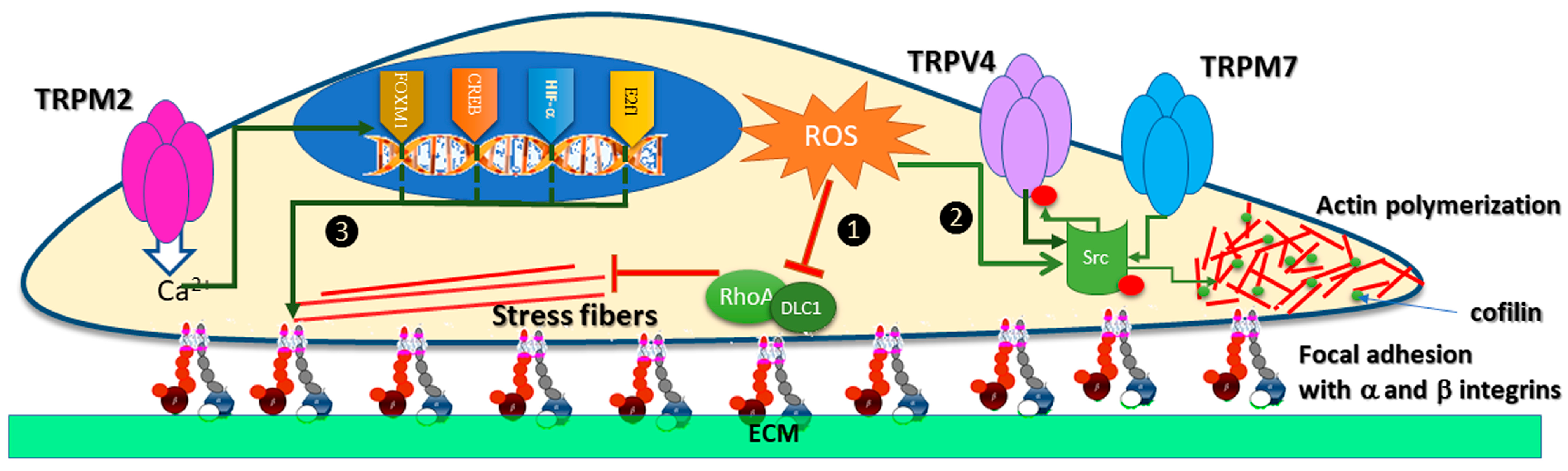
7. TRP Channels Modulation by Oxidative Stress in Cancer Cell Migration
8. Redox TRP Channels and Inflammation
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jardin, I.; Lopez, J.J.; Diez, R.; Sanchez-Collado, J.; Cantonero, C.; Albarran, L.; Woodard, G.E.; Redondo, P.C.; Salido, G.M.; Smani, T.; et al. TRPs in Pain Sensation. Front. Physiol. 2017, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef]
- Vennekens, R.; Hoenderop, J.G.; Prenen, J.; Stuiver, M.; Willems, P.H.; Droogmans, G.; Nilius, B.; Bindels, R.J. Permeation and gating properties of the novel epithelial Ca2+ channel. J. Biol. Chem. 2000, 275, 3963–3969. [Google Scholar] [CrossRef]
- Nilius, B.; Prenen, J.; Janssens, A.; Owsianik, G.; Wang, C.; Zhu, M.X.; Voets, T. The selectivity filter of the cation channel TRPM4. J. Biol. Chem. 2005, 280, 22899–22906. [Google Scholar] [CrossRef]
- Sakaguchi, R.; Mori, Y. Transient receptor potential (TRP) channels: Biosensors for redox environmental stimuli and cellular status. Free Radic. Biol. Med. 2020, 146, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef]
- Canton, M.; Sanchez-Rodriguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 2021, 12, 734229. [Google Scholar] [CrossRef] [PubMed]
- Nugud, A.; Sandeep, D.; El-Serafi, A.T. Two faces of the coin: Minireview for dissecting the role of reactive oxygen species in stem cell potency and lineage commitment. J. Adv. Res. 2018, 14, 73–79. [Google Scholar] [CrossRef]
- Vieira, H.L.; Alves, P.M.; Vercelli, A. Modulation of neuronal stem cell differentiation by hypoxia and reactive oxygen species. Prog. Neurobiol. 2011, 93, 444–455. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Catala, A.; Diaz, M. Editorial: Impact of Lipid Peroxidation on the Physiology and Pathophysiology of Cell Membranes. Front. Physiol. 2016, 7, 423. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Kang, D.H.; Park, C.K.; Chung, C.; Oh, I.J.; Kim, Y.C.; Park, D.; Kim, J.; Kwon, G.C.; Kwon, I.; Sun, P.; et al. Baseline Serum Interleukin-6 Levels Predict the Response of Patients with Advanced Non-small Cell Lung Cancer to PD-1/PD-L1 Inhibitors. Immune Netw. 2020, 20, e27. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.S.; Liu, L.; Qin, Y.W. IL-6 and TNF-alpha promote metastasis of lung cancer by inducing epithelial-mesenchymal transition. Oncol. Lett. 2017, 13, 4657–4660. [Google Scholar] [CrossRef]
- Han, F.; Li, S.; Yang, Y.; Bai, Z. Interleukin-6 promotes ferroptosis in bronchial epithelial cells by inducing reactive oxygen species-dependent lipid peroxidation and disrupting iron homeostasis. Bioengineered 2021, 12, 5279–5288. [Google Scholar] [CrossRef]
- Li, M.; Jin, S.; Zhang, Z.; Ma, H.; Yang, X. Interleukin-6 facilitates tumor progression by inducing ferroptosis resistance in head and neck squamous cell carcinoma. Cancer Lett. 2022, 527, 28–40. [Google Scholar] [CrossRef]
- Uz, U.; Eskiizmir, G. Association between Interleukin-6 and Head and Neck Squamous Cell Carcinoma: A Systematic Review. Clin. Exp. Otorhinolaryngol. 2021, 14, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Gasparovic, A.C.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. Cancer growth regulation by 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 226–234. [Google Scholar] [CrossRef] [PubMed]
- de la Haba, C.; Palacio, J.R.; Martinez, P.; Morros, A. Effect of oxidative stress on plasma membrane fluidity of THP-1 induced macrophages. Biochim. Biophys. Acta 2013, 1828, 357–364. [Google Scholar] [CrossRef]
- Amiya, E.; Watanabe, M.; Takeda, N.; Saito, T.; Shiga, T.; Hosoya, Y.; Nakao, T.; Imai, Y.; Manabe, I.; Nagai, R.; et al. Angiotensin II impairs endothelial nitric-oxide synthase bioavailability under free cholesterol-enriched conditions via intracellular free cholesterol-rich membrane microdomains. J. Biol. Chem. 2013, 288, 14497–14509. [Google Scholar] [CrossRef]
- Morgan, M.J.; Kim, Y.S.; Liu, Z. Lipid rafts and oxidative stress-induced cell death. Antioxid. Redox Signal. 2007, 9, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Svobodova, B.; Groschner, K. Mechanisms of lipid regulation and lipid gating in TRPC channels. Cell Calcium 2016, 59, 271–279. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Li, M.; Zheng, J.; Wu, T.; He, Y.; Guo, J.; Xu, J.; Gao, C.; Qu, S.; Zhang, Q.; Zhao, J.; et al. Activation of TRPV4 Induces Exocytosis and Ferroptosis in Human Melanoma Cells. Int. J. Mol. Sci. 2022, 23, 4146. [Google Scholar] [CrossRef]
- Yahya, F.; Mohd Bakri, M.; Hossain, M.Z.; Syed Abdul Rahman, S.N.; Mohammed Alabsi, A.; Ramanathan, A. Combination Treatment of TRPV4 Agonist with Cisplatin Promotes Vessel Normalization in an Animal Model of Oral Squamous Cell Carcinoma. Medicina 2022, 58, 1229. [Google Scholar] [CrossRef]
- Fujii, S.; Tajiri, Y.; Hasegawa, K.; Matsumoto, S.; Yoshimoto, R.U.; Wada, H.; Kishida, S.; Kido, M.A.; Yoshikawa, H.; Ozeki, S.; et al. The TRPV4-AKT axis promotes oral squamous cell carcinoma cell proliferation via CaMKII activation. Lab. Investig. J. Technol. Methods Pathol. 2020, 100, 311–323. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, M.; Liu, J.; Ye, J.; Jiang, H.; Xu, Y.; Ye, D.; Wan, J. Inhibition of TRPA1 Attenuates Doxorubicin-Induced Acute Cardiotoxicity by Suppressing Oxidative Stress, the Inflammatory Response, and Endoplasmic Reticulum Stress. Oxidative Med. Cell. Longev. 2018, 2018, 5179468. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.K.; Chan, K.K.W.; Fong, C.H.H.; Ramanan, S.; Yap, J.L.Y.; Yin, J.N.; Yip, Y.S.; Tan, W.R.; Koh, A.P.F.; Tan, N.S.; et al. Modulated TRPC1 Expression Predicts Sensitivity of Breast Cancer to Doxorubicin and Magnetic Field Therapy: Segue Towards a Precision Medicine Approach. Front. Oncol. 2021, 11, 783803. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, K.; Numaga-Tomita, T.; Fujimoto, Y.; Tanaka, T.; Toyama, C.; Nishimura, A.; Yamashita, T.; Matsunaga, N.; Koyanagi, S.; Azuma, Y.T.; et al. Ibudilast attenuates doxorubicin-induced cytotoxicity by suppressing formation of TRPC3 channel and NADPH oxidase 2 protein complexes. Br. J. Pharmacol. 2019, 176, 3723–3738. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.M.; Feng, X.; Liu, M.; Parker, L.P.; Koh, D.W. Inhibition of the transient receptor potential melastatin-2 channel causes increased DNA damage and decreased proliferation in breast adenocarcinoma cells. Int. J. Oncol. 2015, 46, 2267–2276. [Google Scholar] [CrossRef]
- Di, Y.; Xu, T.; Tian, Y.; Ma, T.; Qu, D.; Wang, Y.; Lin, Y.; Bao, D.; Yu, L.; Liu, S.; et al. Ursolic acid protects against cisplatin-induced ototoxicity by inhibiting oxidative stress and TRPV1-mediated Ca2+-signaling. Int. J. Mol. Med. 2020, 46, 806–816. [Google Scholar] [CrossRef]
- DelloStritto, D.J.; Sinharoy, P.; Connell, P.J.; Fahmy, J.N.; Cappelli, H.C.; Thodeti, C.K.; Geldenhuys, W.J.; Damron, D.S.; Bratz, I.N. 4-Hydroxynonenal dependent alteration of TRPV1-mediated coronary microvascular signaling. Free Radic. Biol. Med. 2016, 101, 10–19. [Google Scholar] [CrossRef]
- Takahashi, N.; Mori, Y. TRP Channels as Sensors and Signal Integrators of Redox Status Changes. Front. Pharmacol. 2011, 2, 58. [Google Scholar] [CrossRef]
- Yoshida, T.; Inoue, R.; Morii, T.; Takahashi, N.; Yamamoto, S.; Hara, Y.; Tominaga, M.; Shimizu, S.; Sato, Y.; Mori, Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2006, 2, 596–607. [Google Scholar] [CrossRef]
- Ogawa, N.; Kurokawa, T.; Fujiwara, K.; Polat, O.K.; Badr, H.; Takahashi, N.; Mori, Y. Functional and Structural Divergence in Human TRPV1 Channel Subunits by Oxidative Cysteine Modification. J. Biol. Chem. 2016, 291, 4197–4210. [Google Scholar] [CrossRef]
- Vellino, S.; Oddou, C.; Rivier, P.; Boyault, C.; Hiriart-Bryant, E.; Kraut, A.; Martin, R.; Coute, Y.; Knolker, H.J.; Valverde, M.A.; et al. Cross-talk between the calcium channel TRPV4 and reactive oxygen species interlocks adhesive and degradative functions of invadosomes. J. Cell Biol. 2021, 220, e201910079. [Google Scholar] [CrossRef]
- Talavera, K.; Startek, J.B.; Alvarez-Collazo, J.; Boonen, B.; Alpizar, Y.A.; Sanchez, A.; Naert, R.; Nilius, B. Mammalian Transient Receptor Potential TRPA1 Channels: From Structure to Disease. Physiol. Rev. 2020, 100, 725–803. [Google Scholar] [CrossRef] [PubMed]
- Poteser, M.; Graziani, A.; Rosker, C.; Eder, P.; Derler, I.; Kahr, H.; Zhu, M.X.; Romanin, C.; Groschner, K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol. Chem. 2006, 281, 13588–13595. [Google Scholar] [CrossRef] [PubMed]
- Balzer, M.; Lintschinger, B.; Groschner, K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc. Res. 1999, 42, 543–549. [Google Scholar] [CrossRef][Green Version]
- Kitajima, N.; Numaga-Tomita, T.; Watanabe, M.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; et al. TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci. Rep. 2016, 6, 37001. [Google Scholar] [CrossRef]
- Graziani, A.; Rosker, C.; Kohlwein, S.D.; Zhu, M.X.; Romanin, C.; Sattler, W.; Groschner, K.; Poteser, M. Cellular cholesterol controls TRPC3 function: Evidence from a novel dominant-negative knockdown strategy. Biochem. J. 2006, 396, 147–155. [Google Scholar] [CrossRef]
- Ding, R.; Yin, Y.L.; Jiang, L.H. Reactive Oxygen Species-Induced TRPM2-Mediated Ca2+ Signalling in Endothelial Cells. Antioxidants 2021, 10, 718. [Google Scholar] [CrossRef] [PubMed]
- Malko, P.; Jiang, L.H. TRPM2 channel-mediated cell death: An important mechanism linking oxidative stress-inducing pathological factors to associated pathological conditions. Redox Biol. 2020, 37, 101755. [Google Scholar] [CrossRef]
- Inoue, H.; Murayama, T.; Tashiro, M.; Sakurai, T.; Konishi, M. Mg2+- and ATP-dependent inhibition of transient receptor potential melastatin 7 by oxidative stress. Free Radic. Biol. Med. 2014, 72, 257–266. [Google Scholar] [CrossRef]
- Inoue, H.; Murayama, T.; Kobayashi, T.; Konishi, M.; Yokoyama, U. The zinc-binding motif of TRPM7 acts as an oxidative stress sensor to regulate its channel activity. J. Gen. Physiol. 2021, 153, e202012708. [Google Scholar] [CrossRef]
- Fricke, T.C.; Echtermeyer, F.; Zielke, J.; de la Roche, J.; Filipovic, M.R.; Claverol, S.; Herzog, C.; Tominaga, M.; Pumroy, R.A.; Moiseenkova-Bell, V.Y.; et al. Oxidation of methionine residues activates the high-threshold heat-sensitive ion channel TRPV2. Proc. Natl. Acad. Sci. USA 2019, 116, 24359–24365. [Google Scholar] [CrossRef]
- Chen, S.J.; Bao, L.; Keefer, K.; Shanmughapriya, S.; Chen, L.; Lee, J.; Wang, J.; Zhang, X.Q.; Hirschler-Laszkiewicz, I.; Merali, S.; et al. Transient receptor potential ion channel TRPM2 promotes AML proliferation and survival through modulation of mitochondrial function, ROS, and autophagy. Cell Death Dis. 2020, 11, 247. [Google Scholar] [CrossRef]
- Bao, L.; Festa, F.; Freet, C.S.; Lee, J.P.; Hirschler-Laszkiewicz, I.M.; Chen, S.J.; Keefer, K.A.; Wang, H.G.; Patterson, A.D.; Cheung, J.Y.; et al. The Human Transient Receptor Potential Melastatin 2 Ion Channel Modulates ROS Through Nrf2. Sci. Rep. 2019, 9, 14132. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bu, F.; Ma, S.; Cananzi, F.; Zhao, Y.; Xiao, M.; Min, L.; Luo, C. The Janus-faced role of TRPM2-S in retroperitoneal liposarcoma via increasing ROS levels. Cell Commun. Signal. CCS 2022, 20, 128. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Wei, X.; Wang, M.M.; Liu, Y.; Sui, Z.; Wang, X.; Zhang, Y.; Fei, Y.H.; Jiang, Y.; Lu, C.; et al. Stimulating TRPM7 suppresses cancer cell proliferation and metastasis by inhibiting autophagy. Cancer Lett. 2022, 525, 179–197. [Google Scholar] [CrossRef]
- Berrout, J.; Kyriakopoulou, E.; Moparthi, L.; Hogea, A.S.; Berrout, L.; Ivan, C.; Lorger, M.; Boyle, J.; Peers, C.; Muench, S.; et al. TRPA1-FGFR2 binding event is a regulatory oncogenic driver modulated by miRNA-142-3p. Nat. Commun. 2017, 8, 947. [Google Scholar] [CrossRef]
- Xu, C.; Luo, J.; He, L.; Montell, C.; Perrimon, N. Oxidative stress induces stem cell proliferation via TRPA1/RyR-mediated Ca2+ signaling in the Drosophila midgut. eLife 2017, 6, e22441. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Feng, F.; Luo, W.; Sanders, A.J.; Zhang, Y.; Liang, J.; Chen, C.; Feng, W.; Gu, W.; Liao, W.; et al. Overexpressed transient receptor potential vanilloid 1 (TRPV1) in lung adenocarcinoma harbours a new opportunity for therapeutic targeting. Cancer Gene Ther. 2022, 29, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, C.; Chiang, C.; Xiao, T.; Chen, Y.; Zhao, Y.; Zheng, D. The Impact of TRPV1 on Cancer Pathogenesis and Therapy: A Systematic Review. Int. J. Biol. Sci. 2021, 17, 2034–2049. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Moreno-Celis, U.; Garcia-Gasca, T.; Mejia, C. Apoptosis-Induced Compensatory Proliferation in Cancer. In Metastasis; Sergi, C.M., Ed.; Exon Publications: Brisbane, Australia, 2022. [Google Scholar]
- Diwanji, N.; Bergmann, A. An unexpected friend—ROS in apoptosis-induced compensatory proliferation: Implications for regeneration and cancer. Semin. Cell Dev. Biol. 2018, 80, 74–82. [Google Scholar] [CrossRef]
- Yang, D.; Kim, J. Emerging role of transient receptor potential (TRP) channels in cancer progression. BMB Rep. 2020, 53, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Zheng, S.Y.; Zhang, Z.G.; Luo, J.H.; Zhu, Z.L.; Li, L.; Chen, L.S.; Lin, X.; Sham, J.S.K.; Lin, M.J.; et al. TRPC3 promotes tumorigenesis of gastric cancer via the CNB2/GSK3beta/NFATc2 signaling pathway. Cancer Lett. 2021, 519, 211–225. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018, 33, 985–1003.e1007. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, F.; Selescu, T.; Domocos, D.; Marutescu, L.; Chiritoiu, G.; Chelaru, N.R.; Dima, S.; Mihailescu, D.; Babes, A.; Cucu, D. Publisher Correction: Functional expression of the transient receptor potential ankyrin type 1 channel in pancreatic adenocarcinoma cells. Sci. Rep. 2021, 11, 8853. [Google Scholar] [CrossRef]
- Chen, H.; Li, C.; Hu, H.; Zhang, B. Activated TRPA1 plays a therapeutic role in TMZ resistance in glioblastoma by altering mitochondrial dynamics. BMC Mol. Cell Biol. 2022, 23, 38. [Google Scholar] [CrossRef] [PubMed]
- Scala, R.; Maqoud, F.; Angelelli, M.; Latorre, R.; Perrone, M.G.; Scilimati, A.; Tricarico, D. Zoledronic Acid Modulation of TRPV1 Channel Currents in Osteoblast Cell Line and Native Rat and Mouse Bone Marrow-Derived Osteoblasts: Cell Proliferation and Mineralization Effect. Cancers 2019, 11, 206. [Google Scholar] [CrossRef]
- Scala, R.; Maqoud, F.; Antonacci, M.; Dibenedetto, J.R.; Perrone, M.G.; Scilimati, A.; Castillo, K.; Latorre, R.; Conte, D.; Bendahhou, S.; et al. Bisphosphonates Targeting Ion Channels and Musculoskeletal Effects. Front. Pharmacol. 2022, 13, 837534. [Google Scholar] [CrossRef]
- Naziroglu, M.; Cig, B.; Blum, W.; Vizler, C.; Buhala, A.; Marton, A.; Katona, R.; Josvay, K.; Schwaller, B.; Olah, Z.; et al. Targeting breast cancer cells by MRS1477, a positive allosteric modulator of TRPV1 channels. PLoS ONE 2017, 12, e0179950. [Google Scholar] [CrossRef]
- Sung, B.; Prasad, S.; Ravindran, J.; Yadav, V.R.; Aggarwal, B.B. Capsazepine, a TRPV1 antagonist, sensitizes colorectal cancer cells to apoptosis by TRAIL through ROS-JNK-CHOP-mediated upregulation of death receptors. Free Radic. Biol. Med. 2012, 53, 1977–1987. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, J.; Liu, X. TRPV4 induces apoptosis via p38 MAPK in human lung cancer cells. Braz. J. Med. Biol. Res. 2021, 54, e10867. [Google Scholar] [CrossRef]
- Zhao, L.Y.; Xu, W.L.; Xu, Z.Q.; Qi, C.; Li, Y.; Cheng, J.; Liu, L.K.; Wu, Y.N.; Gao, J.; Ye, J.H. The overexpressed functional transient receptor potential channel TRPM2 in oral squamous cell carcinoma. Sci. Rep. 2016, 6, 38471. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, D.; Misovic, M.; Szteyn, K.; Shumilina, E.; Rudner, J.; Huber, S.M. Targeting TRPM2 Channels Impairs Radiation-Induced Cell Cycle Arrest and Fosters Cell Death of T Cell Leukemia Cells in a Bcl-2-Dependent Manner. Oxidative Med. Cell. Longev. 2016, 2016, 8026702. [Google Scholar] [CrossRef]
- Akpinar, O.; Ozsimsek, A.; Guzel, M.; Naziroglu, M. Clostridium botulinum neurotoxin A induces apoptosis and mitochondrial oxidative stress via activation of TRPM2 channel signaling pathway in neuroblastoma and glioblastoma tumor cells. J. Recept. Signal Transduct. Res. 2020, 40, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Meyskens, F.L., Jr.; McNulty, S.E.; Buckmeier, J.A.; Tohidian, N.B.; Spillane, T.J.; Kahlon, R.S.; Gonzalez, R.I. Aberrant redox regulation in human metastatic melanoma cells compared to normal melanocytes. Free Radic. Biol. Med. 2001, 31, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhu, W.Z.; Liu, T.T.; Fu, H.L.; Liu, Z.J.; Yang, B.W.; Song, T.Y.; Li, G.R. H2O2 inhibits proliferation and mediates suppression of migration via DLC1/RhoA signaling in cancer cells. Asian Pac. J. Cancer Prev. APJCP 2015, 16, 1637–1642. [Google Scholar] [CrossRef]
- Savino, L.; Di Marcantonio, M.C.; Moscatello, C.; Cotellese, R.; Centurione, L.; Muraro, R.; Aceto, G.M.; Mincione, G. Effects of H2O2 Treatment Combined with PI3K Inhibitor and MEK Inhibitor in AGS Cells: Oxidative Stress Outcomes in a Model of Gastric Cancer. Front. Oncol. 2022, 12, 860760. [Google Scholar] [CrossRef]
- Chen, Y.H.; Hsu, J.Y.; Chu, C.T.; Chang, Y.W.; Fan, J.R.; Yang, M.H.; Chen, H.C. Loss of cell-cell adhesion triggers cell migration through Rac1-dependent ROS generation. Life Sci. Alliance 2023, 6. [Google Scholar] [CrossRef]
- Yao, X.; Kwan, H.Y.; Huang, Y. Regulation of TRP channels by phosphorylation. Neuro-Signals 2005, 14, 273–280. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, H.; Tian, W.; Yoshida, K.; Roullet, J.B.; Cohen, D.M. Regulation of a transient receptor potential (TRP) channel by tyrosine phosphorylation. SRC family kinase-dependent tyrosine phosphorylation of TRPV4 on TYR-253 mediates its response to hypotonic stress. J. Biol. Chem. 2003, 278, 11520–11527. [Google Scholar] [CrossRef]
- Huang, S.; Yu, S.; Deng, R.; Liu, H.; Ding, Y.; Sun, Y.; Chen, W.; Wang, A.; Wei, Z.; Lu, Y. TRPV4 Promotes Metastasis in Melanoma by Regulating Cell Motility through Cytoskeletal Rearrangement. Int. J. Mol. Sci. 2022, 23, 5155. [Google Scholar] [CrossRef]
- Li, X.; Cheng, Y.; Wang, Z.; Zhou, J.; Jia, Y.; He, X.; Zhao, L.; Dong, Y.; Fan, Y.; Yang, X.; et al. Calcium and TRPV4 promote metastasis by regulating cytoskeleton through the RhoA/ROCK1 pathway in endometrial cancer. Cell Death Dis. 2020, 11, 1009. [Google Scholar] [CrossRef] [PubMed]
- Cai, N.; Bai, Z.; Nanda, V.; Runnels, L.W. Mass Spectrometric Analysis of TRPM6 and TRPM7 Phosphorylation Reveals Regulatory Mechanisms of the Channel-Kinases. Sci. Rep. 2017, 7, 42739. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Chun, S.Y.; Kim, B.; Yoon, B.H.; Lee, J.N.; Kim, B.S.; Yoo, E.S.; Lee, S.; Song, P.H.; Kwon, T.G.; et al. Knockdown of TRPM7 prevents tumor growth, migration, and invasion through the Src, Akt, and JNK pathway in bladder cancer. BMC Urol. 2020, 20, 145. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Middelbeek, J.; Morrice, N.A.; Figdor, C.G.; Lasonder, E.; van Leeuwen, F.N. Massive autophosphorylation of the Ser/Thr-rich domain controls protein kinase activity of TRPM6 and TRPM7. PLoS ONE 2008, 3, e1876. [Google Scholar] [CrossRef]
- Zhang, W.; Tong, Q.; Conrad, K.; Wozney, J.; Cheung, J.Y.; Miller, B.A. Regulation of TRP channel TRPM2 by the tyrosine phosphatase PTPL1. Am. J. Physiol. Cell Physiol. 2007, 292, C1746–C1758. [Google Scholar] [CrossRef]
- Almasi, S.; Sterea, A.M.; Fernando, W.; Clements, D.R.; Marcato, P.; Hoskin, D.W.; Gujar, S.; El Hiani, Y. TRPM2 ion channel promotes gastric cancer migration, invasion and tumor growth through the AKT signaling pathway. Sci. Rep. 2019, 9, 4182. [Google Scholar] [CrossRef]
- Bao, L.; Festa, F.; Hirschler-Laszkiewicz, I.; Keefer, K.; Wang, H.G.; Cheung, J.Y.; Miller, B.A. The human ion channel TRPM2 modulates migration and invasion in neuroblastoma through regulation of integrin expression. Sci. Rep. 2022, 12, 20544. [Google Scholar] [CrossRef]
- Manolache, A.; Selescu, T.; Maier, G.L.; Mentel, M.; Ionescu, A.E.; Neacsu, C.; Babes, A.; Szedlacsek, S.E. Regulation of TRPM8 channel activity by Src-mediated tyrosine phosphorylation. J. Cell. Physiol. 2020, 235, 5192–5203. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Tan, Z.; Xue, H.; Sun, Y.; Zhang, C.; Song, Y.; Qi, Y. The Role of Tumor Inflammatory Microenvironment in Lung Cancer. Front. Pharmacol. 2021, 12, 688625. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Dajee, M.; Lazarov, M.; Zhang, J.Y.; Cai, T.; Green, C.L.; Russell, A.J.; Marinkovich, M.P.; Tao, S.; Lin, Q.; Kubo, Y.; et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 2003, 421, 639–643. [Google Scholar] [CrossRef]
- Ralph, S.J.; Pritchard, R.; Rodriguez-Enriquez, S.; Moreno-Sanchez, R.; Ralph, R.K. Hitting the Bull’s-Eye in Metastatic Cancers-NSAIDs Elevate ROS in Mitochondria, Inducing Malignant Cell Death. Pharmaceuticals 2015, 8, 62–106. [Google Scholar] [CrossRef] [PubMed]
- Sevinç, S.K.; Orun, O.; Tiber, P.M.; Çıkla-Süzgün, P.; Küçükgüzel, Ş.G. Anti-Cancer Acitivity of Etodolac and Its Derivatives on Prostate and Colorectal Cancer Cell Lines. Proceedings 2018, 2, 1573. [Google Scholar]
- Tsagareli, M.G.; Nozadze, I.; Tsiklauri, N.; Gurtskaia, G. Non-steroidal anti-inflammatory drugs attenuate agonist-evoked activation of transient receptor potential channels. Biomed. Pharmacother. 2018, 97, 745–751. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2018, 11, 487. [Google Scholar] [CrossRef]
- Li, D.; Ilnytskyy, Y.; Ghasemi Gojani, E.; Kovalchuk, O.; Kovalchuk, I. Analysis of Anti-Cancer and Anti-Inflammatory Properties of 25 High-THC Cannabis Extracts. Molecules 2022, 27, 6057. [Google Scholar] [CrossRef]
- Dogru, A.; Naziroglu, M.; Cig, B. Modulator role of infliximab and methotrexate through the transient receptor potential melastatin 2 (TRPM2) channel in neutrophils of patients with rheumatoid arthritis: A pilot study. Arch. Med. Sci. AMS 2019, 15, 1415–1424. [Google Scholar] [CrossRef]
- Sharan, M.; Jha, M.; Chandel, R.; Syeda, S.; Mathur, R.; Jha, N.K.; Jha, S.K.; Goel, H.; Shrivastava, A.; Chauhan, S.; et al. Demethylation of CADM1 and SOCS1 using capsaicin in cervical cancer cell line. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Pawar, J.S.; Mustafa, S.; Ghosh, I. Chrysin and Capsaicin induces premature senescence and apoptosis via mitochondrial dysfunction and p53 elevation in Cervical cancer cells. Saudi J. Biol. Sci. 2022, 29, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dong, J.; Tian, W.; Qiao, S.; Wang, H. Role of TRPV1 ion channel in cervical squamous cell carcinoma genesis. Front. Mol. Biosci. 2022, 9, 980262. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Jung, S.H.; Sethi, G.; Ahn, K.S. Pleiotropic Pharmacological Actions of Capsazepine, a Synthetic Analogue of Capsaicin, against Various Cancers and Inflammatory Diseases. Molecules 2019, 24, 995. [Google Scholar] [CrossRef]
- Bautista, D.M.; Pellegrino, M.; Tsunozaki, M. TRPA1: A gatekeeper for inflammation. Annu. Rev. Physiol. 2013, 75, 181–200. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, C.; Liu, Z.; Sun, Y.; Chen, M.; Guo, Y.; Liu, W.; Zhang, C.; Chen, W.; Sun, J.; et al. Cancer cells co-opt nociceptive nerves to thrive in nutrient-poor environments and upon nutrient-starvation therapies. Cell Metab. 2022, 34, 1999–2017.e10. [Google Scholar] [CrossRef]
- De Logu, F.; Souza Monteiro de Araujo, D.; Ugolini, F.; Iannone, L.F.; Vannucchi, M.; Portelli, F.; Landini, L.; Titiz, M.; De Giorgi, V.; Geppetti, P.; et al. The TRPA1 Channel Amplifies the Oxidative Stress Signal in Melanoma. Cells 2021, 10, 3131. [Google Scholar] [CrossRef]
- Yao, K.; Dou, B.; Zhang, Y.; Chen, Z.; Li, Y.; Fan, Z.; Ma, Y.; Du, S.; Wang, J.; Xu, Z.; et al. Inflammation-the role of TRPA1 channel. Front. Physiol. 2023, 14, 1093925. [Google Scholar] [CrossRef]
- Lee, K.I.; Lee, H.T.; Lin, H.C.; Tsay, H.J.; Tsai, F.C.; Shyue, S.K.; Lee, T.S. Role of transient receptor potential ankyrin 1 channels in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 92. [Google Scholar] [CrossRef]
- Maki-Opas, I.; Hamalainen, M.; Moilanen, L.J.; Haavikko, R.; Ahonen, T.J.; Alakurtti, S.; Moreira, V.M.; Muraki, K.; Yli-Kauhaluoma, J.; Moilanen, E. Pyrazine-Fused Triterpenoids Block the TRPA1 Ion Channel in Vitro and Inhibit TRPA1-Mediated Acute Inflammation in Vivo. ACS Chem. Neurosci. 2019, 10, 2848–2857. [Google Scholar] [CrossRef]
- Vinuesa, A.G.; Sancho, R.; Garcia-Limones, C.; Behrens, A.; ten Dijke, P.; Calzado, M.A.; Munoz, E. Vanilloid receptor-1 regulates neurogenic inflammation in colon and protects mice from colon cancer. Cancer Res. 2012, 72, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, W.; Yang, H.; Shao, D.; Zhao, X.; Zhang, G. Intraperitoneal injection of 4-hydroxynonenal (4-HNE), a lipid peroxidation product, exacerbates colonic inflammation through activation of Toll-like receptor 4 signaling. Free Radic. Biol. Med. 2019, 131, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Janion, K.; Szczepanska, E.; Nowakowska-Zajdel, E.; Walkiewicz, K.; Strzelczyk, J. Lipid peroxidation and total oxidant/antioxidant status in colorectal cancer patients. J. Biol. Regul. Homeost. Agents 2020, 34, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhu, Y.; Dong, Y.; Zhang, P.; Han, X.; Jin, J.; Ma, X. Overexpression of TrpC5 promotes tumor metastasis via the HIF-1alpha-Twist signaling pathway in colon cancer. Clin. Sci. 2017, 131, 2439–2450. [Google Scholar] [CrossRef]
- Park, M.K.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, D.H.; Kang, B.S.; Suh, S.W. Effects of Transient Receptor Potential Cation 5 (TRPC5) Inhibitor, NU6027, on Hippocampal Neuronal Death after Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 8256. [Google Scholar] [CrossRef]
- Chen, Y.; Li, J.; Jin, L.; Lei, K.; Liu, H.; Yang, Y. Fibulin-5 contributes to colorectal cancer cell apoptosis via the ROS/MAPK and Akt signal pathways by downregulating transient receptor potential cation channel subfamily V member 1. J. Cell. Biochem. 2019, 120, 17838–17846. [Google Scholar] [CrossRef]
- Wang, Z.; Li, S.; Cao, Y.; Tian, X.; Zeng, R.; Liao, D.F.; Cao, D. Oxidative Stress and Carbonyl Lesions in Ulcerative Colitis and Associated Colorectal Cancer. Oxidative Med. Cell. Longev. 2016, 2016, 9875298. [Google Scholar] [CrossRef]
- Matsumoto, K.; Deguchi, A.; Motoyoshi, A.; Morita, A.; Maebashi, U.; Nakamoto, T.; Kawanishi, S.; Sueyoshi, M.; Nishimura, K.; Takata, K.; et al. Role of transient receptor potential vanilloid subtype 4 in the regulation of azoymethane/dextran sulphate sodium-induced colitis-associated cancer in mice. Eur. J. Pharmacol. 2020, 867, 172853. [Google Scholar] [CrossRef]
- Kaya, İ.; Dağ, S.; Kaya, M.M.; Tanrıverdi, E.A.; Beşeren, H.; Aşasın, G. Modulatory effect of pomegranate extract on TRPA1, TRPM2 and caspase-3 expressions in colorectal cancer induction of mice. Turk. J. Biochem. 2022, 47, 612–619. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Wu, K.; Liu, Z. Expression of 4-hydroxynonenal in esophageal squamous cell carcinoma. Oncol. Lett. 2017, 14, 35–40. [Google Scholar] [CrossRef]
- Nakashima, S.; Shiozaki, A.; Ichikawa, D.; Hikami, S.; Kosuga, T.; Konishi, H.; Komatsu, S.; Fujiwara, H.; Okamoto, K.; Kishimoto, M.; et al. Transient Receptor Potential Melastatin 7 as an Independent Prognostic Factor in Human Esophageal Squamous Cell Carcinoma. Anticancer Res. 2017, 37, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevic, A.; Zarkovic, K.; Jakovcevic, D.; Rakusic, Z.; Prgomet, D.; Waeg, G.; Sunjic, S.B.; Zarkovic, N. The Appearance of 4-Hydroxy-2-Nonenal (HNE) in Squamous Cell Carcinoma of the Oropharynx. Molecules 2020, 25, 868. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, Y.; Tian, Y.; Cao, R.; Shang, D. Pan-Cancer Analysis of the TRP Family, Especially TRPV4 and TRPC4, and Its Expression Correlated with Prognosis, Tumor Microenvironment, and Treatment Sensitivity. Biomolecules 2023, 13, 282. [Google Scholar] [CrossRef]
- Yasuda, T.; Baba, H.; Ishimoto, T. Cellular senescence in the tumor microenvironment and context-specific cancer treatment strategies. FEBS J. 2023, 290, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Farfariello, V.; Gordienko, D.V.; Mesilmany, L.; Touil, Y.; Germain, E.; Fliniaux, I.; Desruelles, E.; Gkika, D.; Roudbaraki, M.; Shapovalov, G.; et al. TRPC3 shapes the ER-mitochondria Ca2+ transfer characterizing tumour-promoting senescence. Nat. Commun. 2022, 13, 956. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-Inflammation Drug | TRP Channel Affected | Role in Tumorigenesis | Ref. |
|---|---|---|---|
| Nonsteroidal anti-inflammatory drugs (NSAIDs), e.g., aspirin and cyclooxygenase | Inhibition of TRPC4/C5 | reduces the risk of breast cancer | [48,52,96] |
| Etodolac (Cox-2 inhibitor) | Desensitization of TRPA1 | Inhibition of prostate and colorectal carcinoma cells proliferation | [97,98] |
| Cannabinoids and Cannabis Extracts | Activation of TRPV4 and TRPA1 | Inhibits growth of breast cancer cells | [99,100] |
| Methotrexate | Inhibition of TRPM2 | Chemotherapy of breast cancer, head, and neck cancer, lymphoma | [101] |
| Capsaicin | Activation of TRPV1 | Senescence and Apoptosis in cervical cancer cells | [102,103,104] |
| Capsazepine | Inhibition of TRPV1 | Inhibits growth and survival of prostate cancer, breast cancer, colorectal cancer, oral cancer, and osteosarcoma | [105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piciu, F.; Balas, M.; Badea, M.A.; Cucu, D. TRP Channels in Tumoral Processes Mediated by Oxidative Stress and Inflammation. Antioxidants 2023, 12, 1327. https://doi.org/10.3390/antiox12071327
Piciu F, Balas M, Badea MA, Cucu D. TRP Channels in Tumoral Processes Mediated by Oxidative Stress and Inflammation. Antioxidants. 2023; 12(7):1327. https://doi.org/10.3390/antiox12071327
Chicago/Turabian StylePiciu, Florentina, Mihaela Balas, Madalina Andreea Badea, and Dana Cucu. 2023. "TRP Channels in Tumoral Processes Mediated by Oxidative Stress and Inflammation" Antioxidants 12, no. 7: 1327. https://doi.org/10.3390/antiox12071327
APA StylePiciu, F., Balas, M., Badea, M. A., & Cucu, D. (2023). TRP Channels in Tumoral Processes Mediated by Oxidative Stress and Inflammation. Antioxidants, 12(7), 1327. https://doi.org/10.3390/antiox12071327