
Phenotypic Modulation of Cancer-Associated Antioxidant NQO1 Activity by Post-Translational Modifications and the Natural Diversity of the Human Genome


Abstract
:1. Human NQO1: A Stress-Protein Associated with Disease
2. NQO1: A Simple Dimer with Complex Behavior
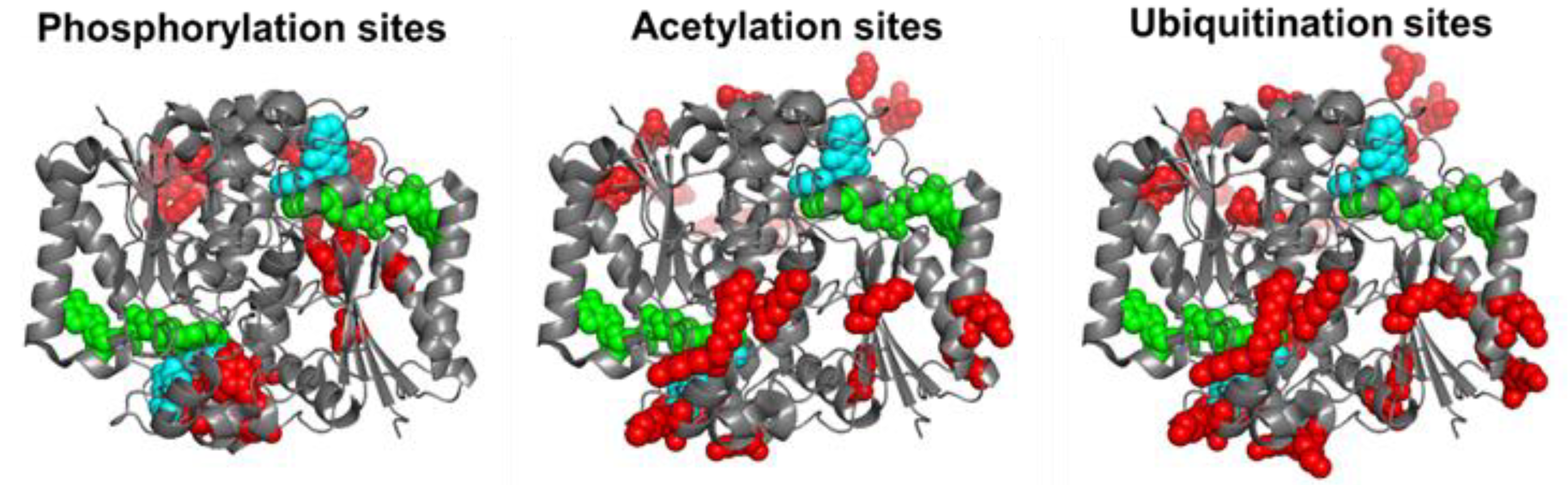
3. Post-Translational Modifications (PTMs) in hNQO1
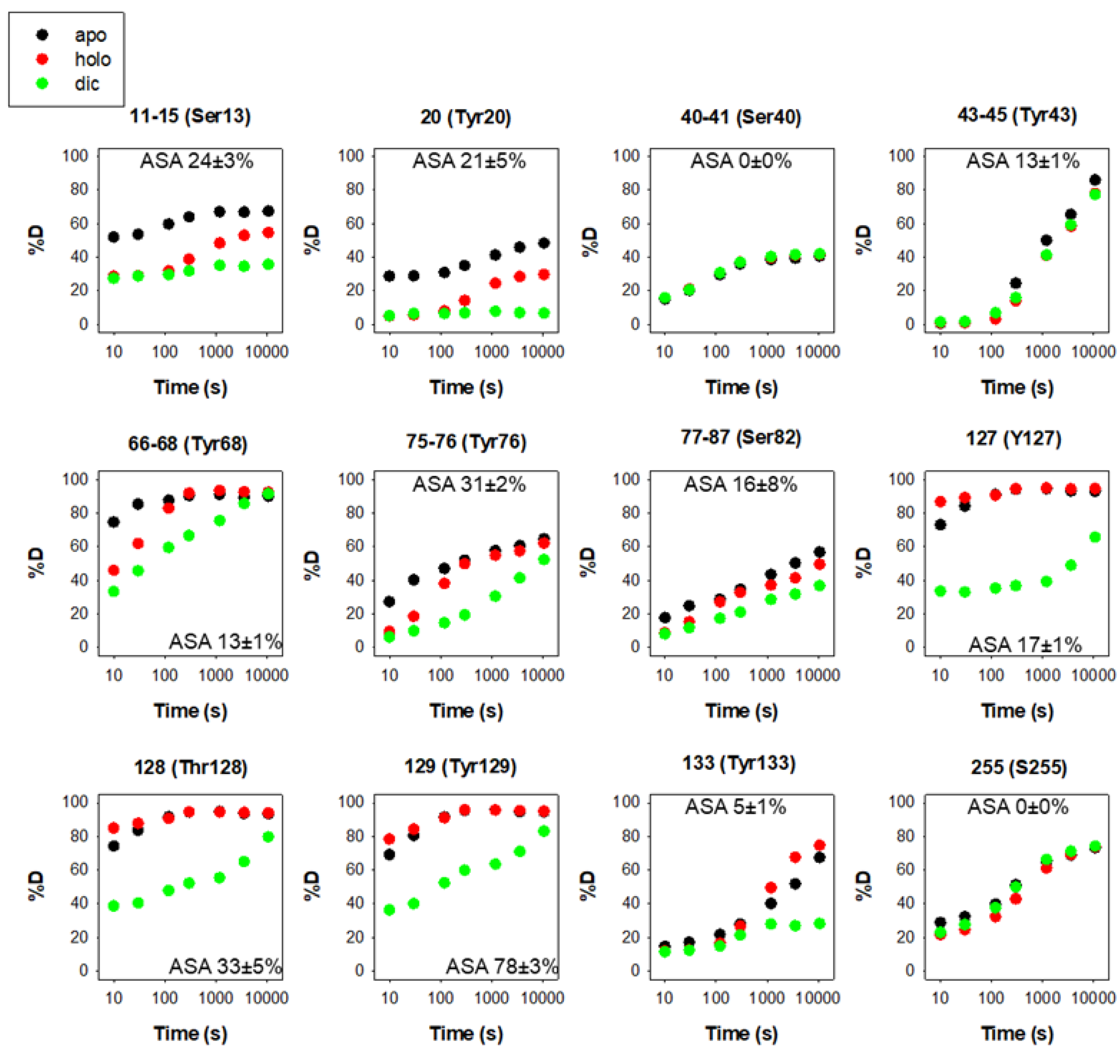
3.1. Phosphorylation
3.2. Ubiquitination
3.3. Acetylation
4. Naturally-Occurring and Artificial Mutations in NQO1: Allosteric Communication of Mutational Effects in a Multifunctional Stress Protein
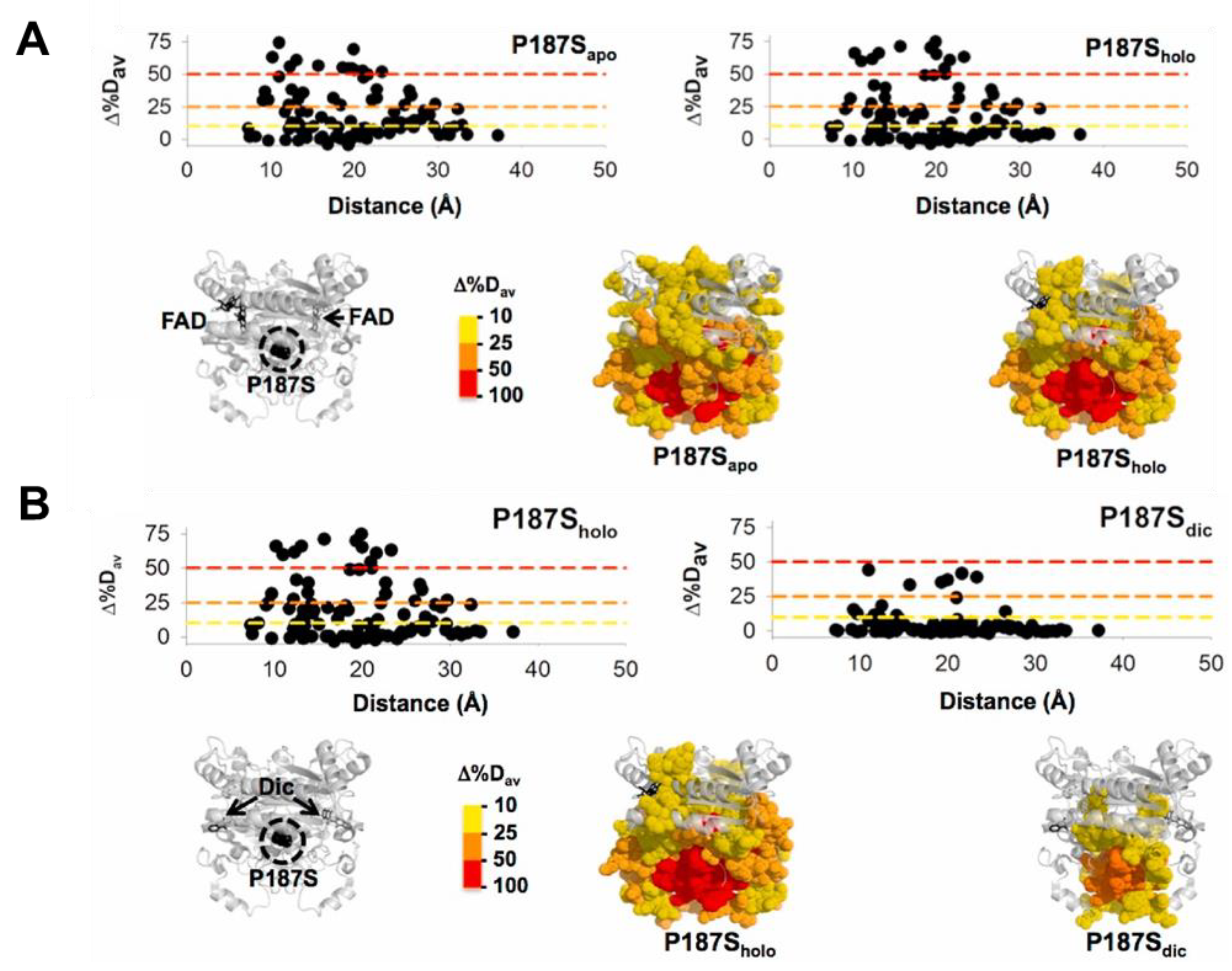
4.1. Polymorphic Variants
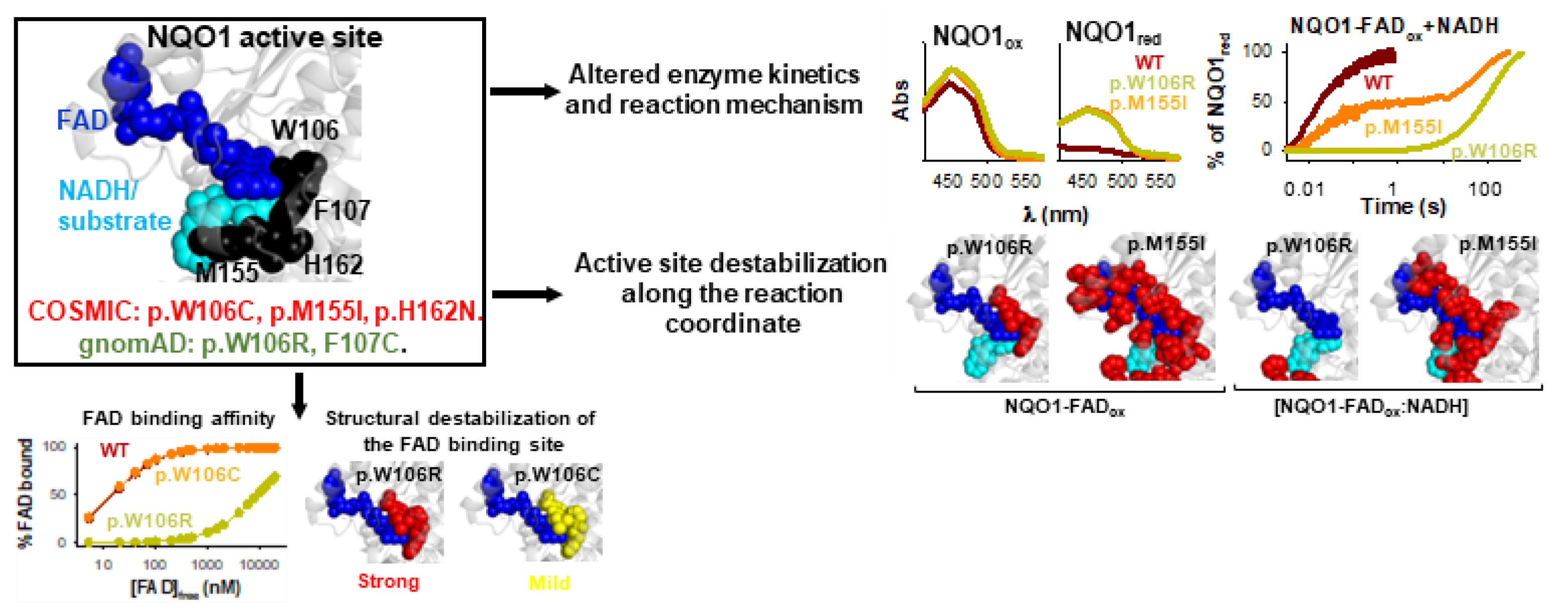
4.2. COSMIC Variants
4.3. gnomAD Variants
4.4. Artificial Variants Aimed at Evaluating the Propagation of Stability Effects and the Structural Basis of Allosterism
5. Outlook and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A target for the treatment of cancer and neurological diseases, and a model to understand loss of function disease mechanisms. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Salido, E.; Timson, D.J.; Betancor-Fernández, I.; Palomino-Morales, R.; Anoz-Carbonell, E.; Pacheco-García, J.L.; Medina, M.; Pey, A.L. Targeting HIF-1α Function in Cancer through the Chaperone Action of NQO1: Implications of Genetic Diversity of NQO1. J. Pers. Med. 2022, 12, 747. [Google Scholar] [CrossRef]
- Siegel, D.; Bersie, S.; Harris, P.; Di Francesco, A.; Armstrong, M.; Reisdorph, N.; Bernier, M.; de Cabo, R.; Fritz, K.; Ross, D. A redox-mediated conformational change in NQO1 controls binding to microtubules and α-tubulin acetylation. Redox Biol. 2021, 39, 101840. [Google Scholar] [CrossRef]
- Carbonell, E.A.; Timson, D.J.; Pey, A.L.; Medina, M. The Catalytic Cycle of the Antioxidant and Cancer-Associated Human NQO1 Enzyme: Hydride Transfer, Conformational Dynamics and Functional Cooperativity. Antioxidants 2020, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Clavería-Gimeno, R.; Velazquez-Campoy, A.; Pey, A.L. Thermodynamics of cooperative binding of FAD to human NQO1: Implications to understanding cofactor-dependent function and stability of the flavoproteome. Arch. Biochem. Biophys. 2017, 636, 17–27. [Google Scholar] [CrossRef]
- Megarity, C.F.; Timson, D.J. Cancer-associated variants of human NQO1: Impacts on inhibitor binding and cooperativity. Biosci. Rep. 2019, 39, BSR20191874. [Google Scholar] [CrossRef] [PubMed]
- Megarity, C.F.; Bettley, H.A.; Caraher, M.C.; Scott, K.A.; Whitehead, R.C.; Jowitt, T.A.; Gutierrez, A.; Bryce, R.A.; Nolan, K.A.; Stratford, I.J.; et al. Negative Cooperativity in NAD(P)H Quinone Oxidoreductase 1 (NQO1). Chembiochem 2019, 20, 2841–2849. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Vankova, P.; Salido, E.; Timson, D.J.; Man, P.; Pey, A.L. A Dynamic Core in Human NQO1 Controls the Functional and Stability Effects of Ligand Binding and Their Communication across the Enzyme Dimer. Biomolecules 2019, 9, 728. [Google Scholar] [CrossRef]
- Pacheco-Garcia, J.L.; Anoz-Carbonell, E.; Loginov, D.S.; Vankova, P.; Salido, E.; Man, P.; Medina, M.; Palomino-Morales, R.; Pey, A.L. Different phenotypic outcome due to site-specific phosphorylation in the cancer-associated NQO1 enzyme studied by phosphomimetic mutations. Arch. Biochem. Biophys. 2022, 729, 109392. [Google Scholar] [CrossRef]
- Pacheco-Garcia, J.L.; Loginov, D.; Rizzuti, B.; Vankova, P.; Neira, J.L.; Kavan, D.; Mesa-Torres, N.; Guzzi, R.; Man, P.; Pey, A.L. A single evolutionarily divergent mutation determines the different FAD-binding affinities of human and rat NQO1 due to site-specific phosphorylation. FEBS Lett. 2022, 596, 29–41. [Google Scholar] [CrossRef]
- Pacheco-Garcia, J.L.; Loginov, D.S.; Anoz-Carbonell, E.; Vankova, P.; Palomino-Morales, R.; Salido, E.; Man, P.; Medina, M.; Naganathan, A.N.; Pey, A.L. Allosteric Communication in the Multifunctional and Redox NQO1 Protein Studied by Cavity-Making Mutations. Antioxidants 2022, 11, 1110. [Google Scholar] [CrossRef]
- Pacheco-Garcia, J.L.; Anoz-Carbonell, E.; Vankova, P.; Kannan, A.; Palomino-Morales, R.; Mesa-Torres, N.; Salido, E.; Man, P.; Medina, M.; Naganathan, A.N.; et al. Structural basis of the pleiotropic and specific phenotypic consequences of missense mutations in the multifunctional NAD(P)H:quinone oxidoreductase 1 and their pharmacological rescue. Redox Biol. 2021, 46, 102112. [Google Scholar] [CrossRef]
- Lajin, B.; Alachkar, A. The NQO1 polymorphism C609T (Pro187Ser) and cancer susceptibility: A comprehensive meta-analysis. Br. J. Cancer 2013, 109, 1325–1337. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Anwar, A.; Winski, S.L.; Kepa, J.K.; Zolman, K.L.; Ross, D. Rapid Polyubiquitination and Proteasomal Degradation of a Mutant Form of NAD(P)H:Quinone Oxidoreductase 1. Mol. Pharmacol. 2001, 59, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Akey, J.M. The Origins, Determinants, and Consequences of Human Mutations. Science 2015, 349, 1478–1483. [Google Scholar] [CrossRef]
- Manolio, T.A.; Fowler, D.M.; Starita, L.M.; Haendel, M.A.; MacArthur, D.G.; Biesecker, L.G.; Worthey, E.; Chisholm, R.L.; Green, E.D.; Jacob, H.J.; et al. Bedside Back to Bench: Building Bridges between Basic and Clinical Genomic Research. Cell 2017, 169, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- McInnes, G.; Sharo, A.G.; Koleske, M.L.; Brown, J.E.; Norstad, M.; Adhikari, A.N.; Wang, S.; Brenner, S.E.; Halpern, J.; Koenig, B.A.; et al. Opportunities and challenges for the computational interpretation of rare variation in clinically important genes. Am. J. Hum. Genet. 2021, 108, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Bagdasaryan, A.A.; Chubarev, V.N.; Smolyarchuk, E.A.; Drozdov, V.N.; Krasnyuk, I.I.; Liu, J.; Fan, R.; Tse, E.; Shikh, E.V.; Sukocheva, O.A. Pharmacogenetics of Drug Metabolism: The Role of Gene Polymorphism in the Regulation of Doxorubicin Safety and Efficacy. Cancers 2022, 14, 5436. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, M.; Allard, W.G.; Vossen, R.H.A.M.; Baak-Pablo, R.F.; Menafra, R.; Deiman, B.A.L.M.; Deenen, M.J.; Neven, P.; Johansson, I.; Gastaldello, S.; et al. Toward predicting CYP2D6-mediated variable drug response from CYP2D6 gene sequencing data. Sci. Transl. Med. 2021, 13, eabf3637. [Google Scholar] [CrossRef]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free. Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Fowke, J.H.; Shu, X.-O.; Dai, Q.; Jin, F.; Cai, Q.; Gao, Y.-T.; Zheng, W. Oral Contraceptive Use and Breast Cancer Risk: Modification by NAD(P)H:Quinone Oxoreductase (NQO1) Genetic Polymorphisms. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1308–1315. [Google Scholar] [CrossRef]
- Hamachi, T.; Tajima, O.; Uezono, K.; Tabata, S.; Abe, H.; Ohnaka, K.; Kono, S. CYP1A1, GSTM1, GSTT1 and NQO1polymorphisms and colorectal adenomas in Japanese men. World J. Gastroenterol. 2013, 19, 4023–4030. [Google Scholar] [CrossRef]
- Lienhart, W.; Gudipati, V.; Uhl, M.K.; Binter, A.; Pulido, S.A.; Saf, R.; Zangger, K.; Gruber, K.; Macheroux, P. Collapse of the native structure caused by a single amino acid exchange in humanNAD(P)H:quinone oxidoreductase1. FEBS J. 2014, 281, 4691–4704. [Google Scholar] [CrossRef]
- Lienhart, W.; Strandback, E.; Gudipati, V.; Koch, K.; Binter, A.; Uhl, M.K.; Rantasa, D.M.; Bourgeois, B.; Madl, T.; Zangger, K.; et al. Catalytic competence, structure and stability of the cancer-associated R139W variant of the human NAD (P)H:quinone oxidoreductase 1 (NQO 1). FEBS J. 2017, 284, 1233–1245. [Google Scholar] [CrossRef]
- Pacheco-Garcia, J.L.; Cagiada, M.; Tienne-Matos, K.; Salido, E.; Lindorff-Larsen, K.; Pey, A.L. Effect of naturally-occurring mutations on the stability and function of cancer-associated NQO1: Comparison of experiments and computation. Front. Mol. Biosci. 2022, 9, 1063620. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Neira, J.L.; Salido, E.; Fuchs, J.E.; Palomino-Morales, R.; Timson, D.J.; Pey, A.L. Site-to-site interdomain communication may mediate different loss-of-function mechanisms in a cancer-associated NQO1 polymorphism. Sci. Rep. 2017, 7, srep44532. [Google Scholar] [CrossRef]
- Carmona, E.M.; Betancor-Fernández, I.; Santos, J.; Mesa-Torres, N.; Grottelli, S.; Batlle, C.; Naganathan, A.N.; Oppici, E.; Cellini, B.; Ventura, S.; et al. Insight into the specificity and severity of pathogenic mechanisms associated with missense mutations through experimental and structural perturbation analyses. Hum. Mol. Genet. 2019, 28, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The Crystal Structure of NAD(P)H Quinone Oxidoreductase 1 in Complex with Its Potent Inhibitor Dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Rizzuti, B.; Martín-Escolano, R.; Pacheco-García, J.L.; Mesa-Torres, N.; Neira, J.L.; Guzzi, R.; Pey, A.L. Phosphorylation compromises FAD binding and intracellular stability of wild-type and cancer-associated NQO1: Insights into flavo-proteome stability. Int. J. Biol. Macromol. 2019, 125, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Carmona, E.M.; Fuchs, J.E.; Gavira, J.A.; Mesa-Torres, N.; Neira, J.L.; Salido, E.; Palomino-Morales, R.; Burgos, M.; Timson, D.; Pey, A.L. Enhanced vulnerability of human proteins towards disease-associated inactivation through divergent evolution. Hum. Mol. Genet. 2017, 26, 3531–3544. [Google Scholar] [CrossRef]
- Luo, S.; Kang, S.S.; Wang, Z.-H.; Liu, X.; Day, J.X.; Wu, Z.; Peng, J.; Xiang, D.; Springer, W.; Ye, K. Akt Phosphorylates NQO1 and Triggers its Degradation, Abolishing Its Antioxidative Activities in Parkinson’s Disease. J. Neurosci. 2019, 39, 7291–7305. [Google Scholar] [CrossRef]
- Martínez-Limón, A.; Alriquet, M.; Lang, W.-H.; Calloni, G.; Wittig, I.; Vabulas, R.M. Recognition of enzymes lacking bound cofactor by protein quality control. Proc. Natl. Acad. Sci. USA 2016, 113, 12156–12161. [Google Scholar] [CrossRef]
- Muñoz, I.G.; Morel, B.; Medina-Carmona, E.; Pey, A.L. A mechanism for cancer-associated inactivation of NQO1 due to P187S and its reactivation by the consensus mutation H80R. FEBS Lett. 2017, 591, 2826–2835. [Google Scholar] [CrossRef]
- Siegel, D.; Harris, P.S.; Michel, C.R.; de Cabo, R.; Fritz, K.S.; Ross, D. Redox state and the sirtuin deacetylases are major factors that regulate the acetylation status of the stress protein NQO1. Front. Pharmacol. 2022, 13, 1015642. [Google Scholar] [CrossRef]
- Pan, S.S.; Forrest, G.L.; Akman, S.A.; Hu, L.T. NAD(P)H:quinone oxidoreductase expression and mitomycin C resistance developed by human colon cancer HCT 116 cells. Cancer Res. 1995, 55, 330–335. [Google Scholar]
- Traver, R.D.; Siegel, D.; Beall, H.D.; Phillips, R.M.; Gibson, N.W.; Franklin, W.A.; Ross, D.T. Characterization of a polymorphism in NAD(P)H: Quinone oxidoreductase (DT-diaphorase). Br. J. Cancer 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Traver, R.D.; Horikoshi, T.; Danenberg, K.D.; Stadlbauer, T.H.; Danenberg, P.V.; Ross, D.; Gibson, N.W. NAD(P)H:quinone oxidoreductase gene expression in human colon carcinoma cells: Characterization of a mutation which modulates DT-diaphorase activity and mitomycin sensitivity. Cancer Res 1992, 52, 797–802. [Google Scholar]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. FAD binding overcomes defects in activity and stability displayed by cancer-associated variants of human NQO1. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 2163–2173. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-García, J.; Cano-Muñoz, M.; Sánchez-Ramos, I.; Salido, E.; Pey, A. Naturally-Occurring Rare Mutations Cause Mild to Catastrophic Effects in the Multifunctional and Cancer-Associated NQO1 Protein. J. Pers. Med. 2020, 10, 207. [Google Scholar] [CrossRef]
- Zhou, H.; Wan, H.; Zhu, L.; Mi, Y. Research on the effects of rs1800566 C/T polymorphism of NAD(P)H quinone oxidoreductase 1 gene on cancer risk involves analysis of 43,736 cancer cases and 56,173 controls. Front. Oncol. 2022, 12, 980897. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; McGuinness, S.M.; Winski, S.L.; Ross, D. Genotype-phenotype relationships in studies of a polymorphism in NAD(P)H. Pharmacogenetics 1999, 9, 113–122. [Google Scholar] [CrossRef]
- Pey, A.L. Biophysical and functional perturbation analyses at cancer-associated P187 and K240 sites of the multifunctional NADP(H):quinone oxidoreductase 1. Int. J. Biol. Macromol. 2018, 118, 1912–1923. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Palomino-Morales, R.J.; Fuchs, J.E.; Padín-Gonzalez, E.; Mesa-Torres, N.; Salido, E.; Timson, D.J.; Pey, A.L. Erratum: Conformational Dynamics Is Key to Understanding Loss-of-Function of NQO1 Cancer-Associated Polymorphisms and Its Correction by Pharmacological Ligands. Sci. Rep. 2016, 6, 21939. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Limón, A.; Calloni, G.; Ernst, R.; Vabulas, R.M. Flavin dependency undermines proteome stability, lipid metabolism and cellular proliferation during vitamin B2 deficiency. Cell Death Dis. 2020, 11, 725. [Google Scholar] [CrossRef]
- Eguchi-Ishimae, M.; Eguchi, M.; Ishii, E.; Knight, D.; Sadakane, Y.; Isoyama, K.; Yabe, H.; Mizutani, S.; Greaves, M. The association of a distinctive allele of NAD(P)H:quinone oxidoreductase with pediatric acute lymphoblastic leukemias with MLL fusion genes in Japan. Haematologica 2005, 90, 1511–1515. [Google Scholar]
- Pan, S.-S.; Han, Y.; Farabaugh, P.; Xia, H. Implication of alternative splicing for expression of a variant NAD(P)H:quinone oxidoreductase-1 with a single nucleotide polymorphism at 465C>T. Pharmacogenetics 2002, 12, 479–488. [Google Scholar] [CrossRef]
- Pacheco-García, J.L.; Anoz-Carbonell, E.; Loginov, D.S.; Kavan, D.; Salido, E.; Man, P.; Medina, M.; Pey, A.L. Counterintuitive structural and functional effects due to naturally occurring mutations targeting the active site of the disease-associated NQO1 enzyme. FEBS J. 2022. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. What’s next for AlphaFold and the AI protein-folding revolution. Nature 2022, 604, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A structural biology community assessment of AlphaFold2 applications. Nat. Struct. Mol. Biol. 2022, 29, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Høie, M.H.; Cagiada, M.; Frederiksen, A.H.B.; Stein, A.; Lindorff-Larsen, K. Predicting and interpreting large-scale mutagenesis data using analyses of protein stability and conservation. Cell Rep. 2022, 38, 110207. [Google Scholar] [CrossRef]
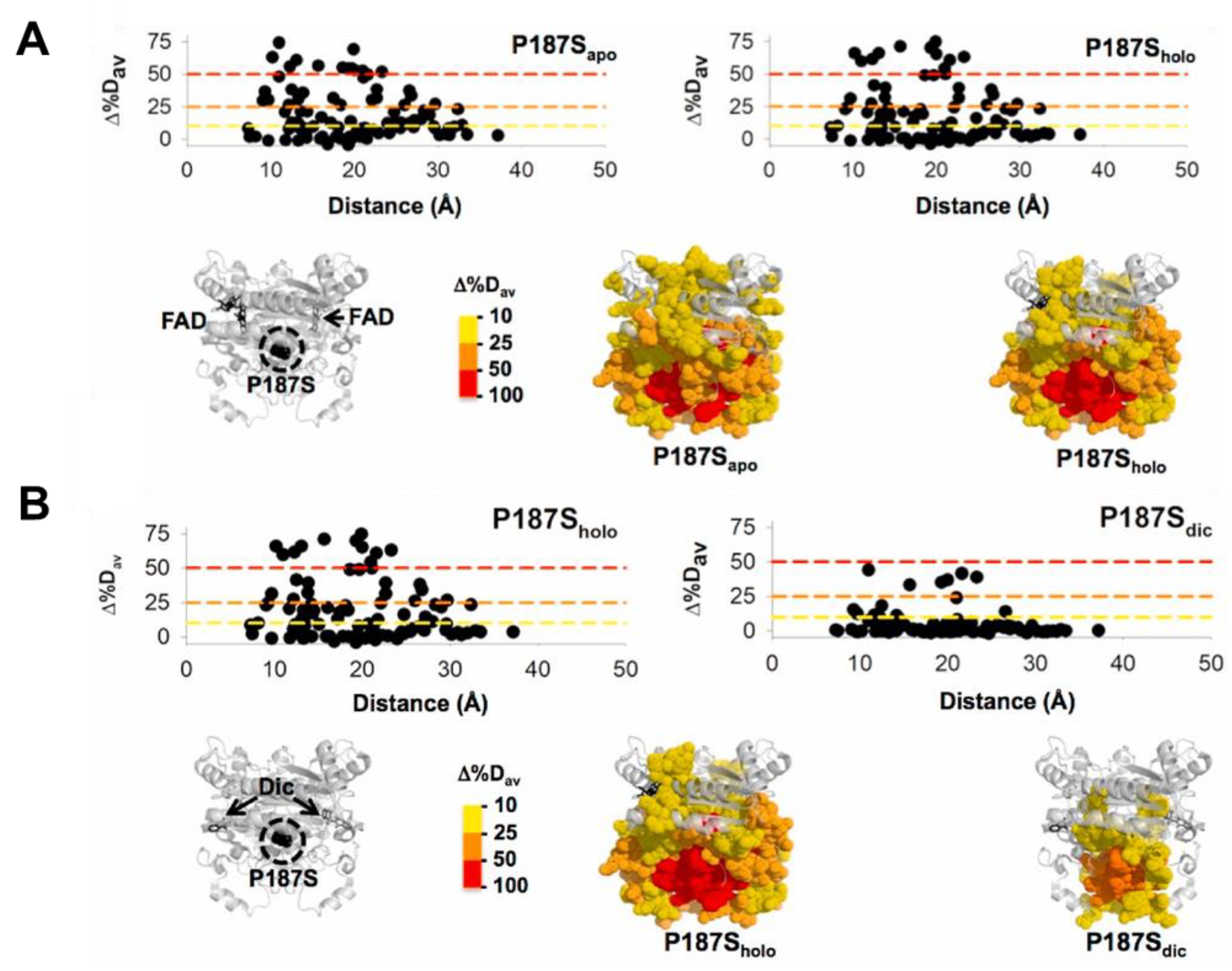


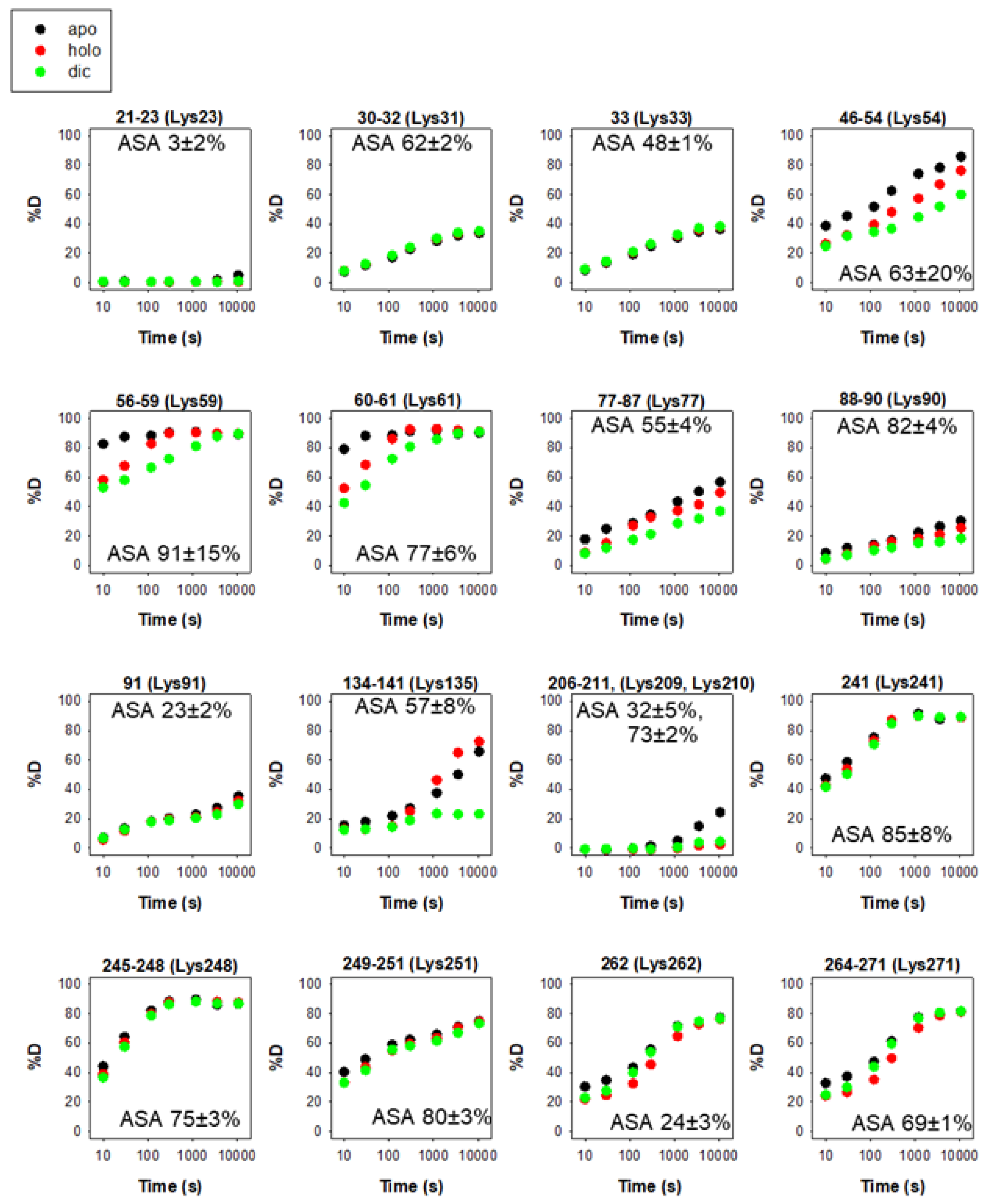

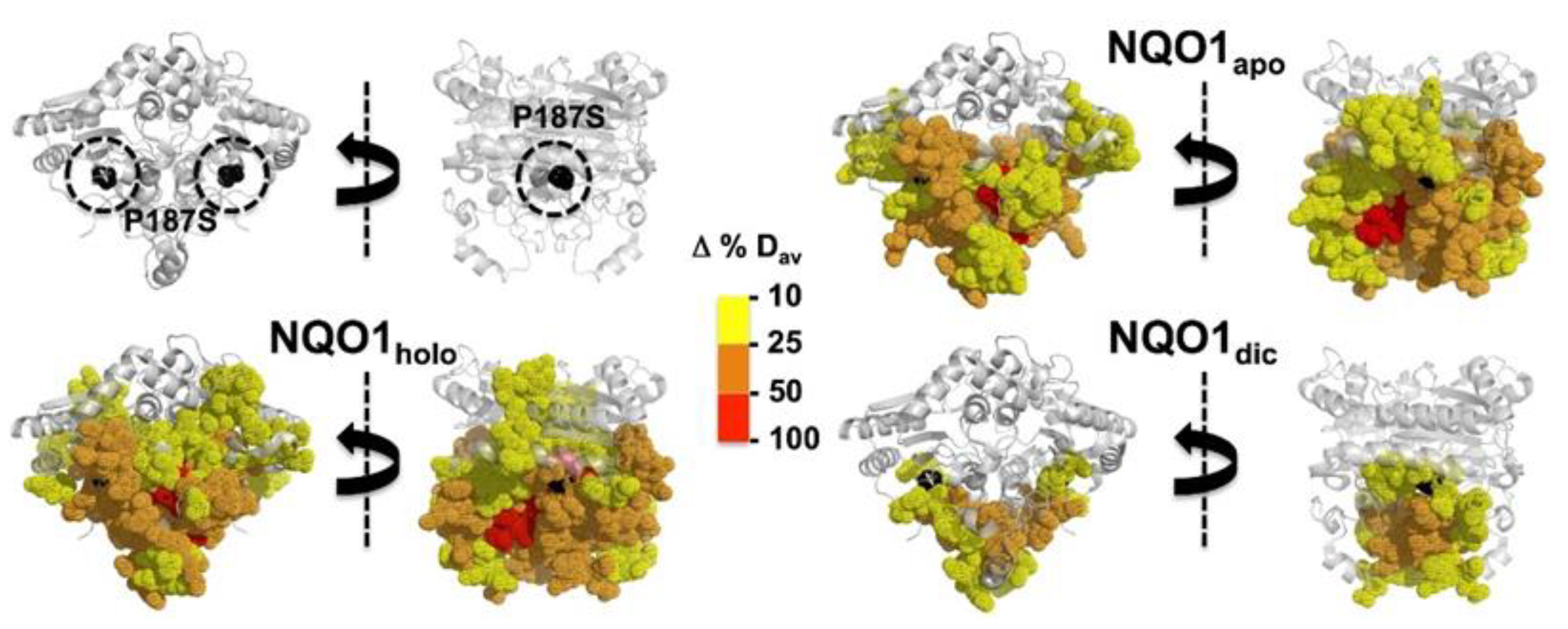
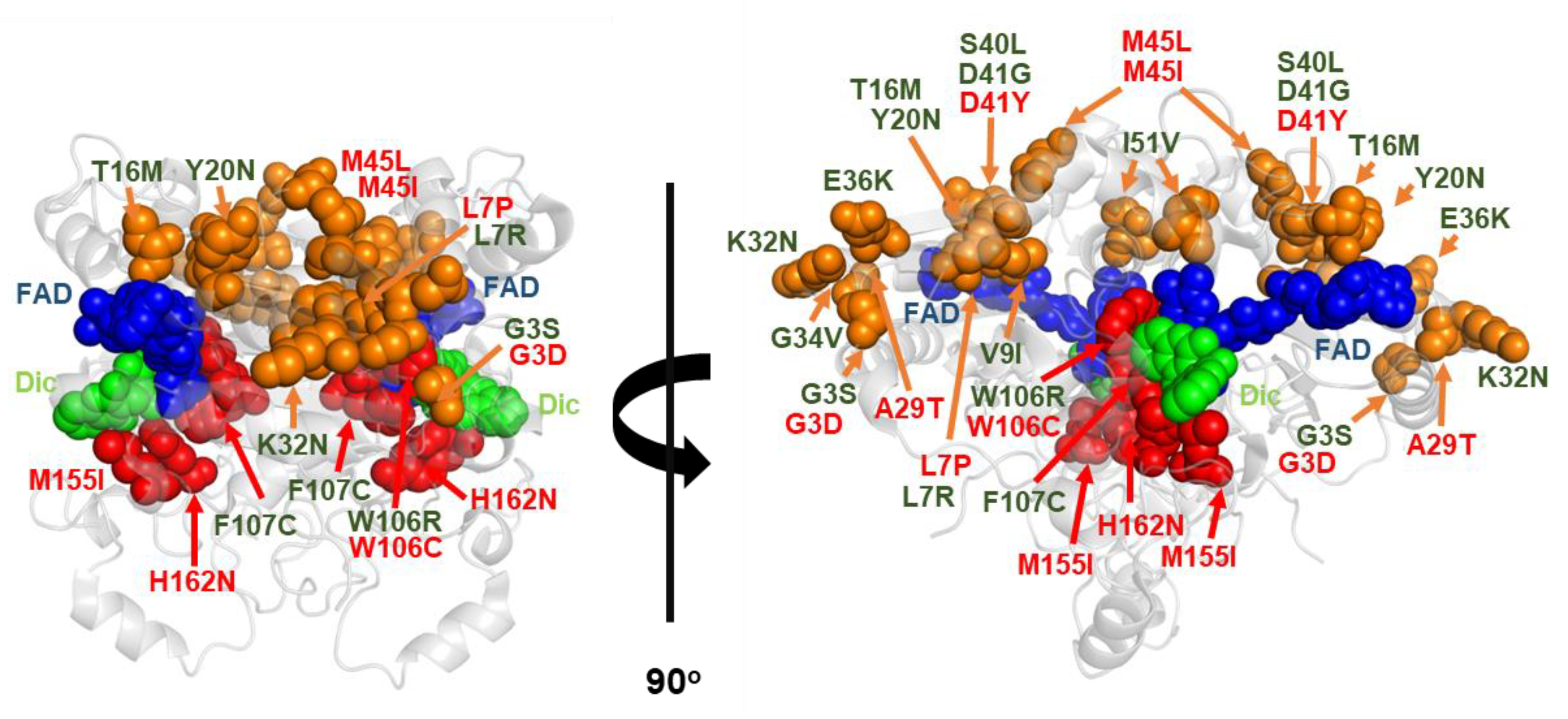


{kind=link}
{kind=link}
{kind=link}
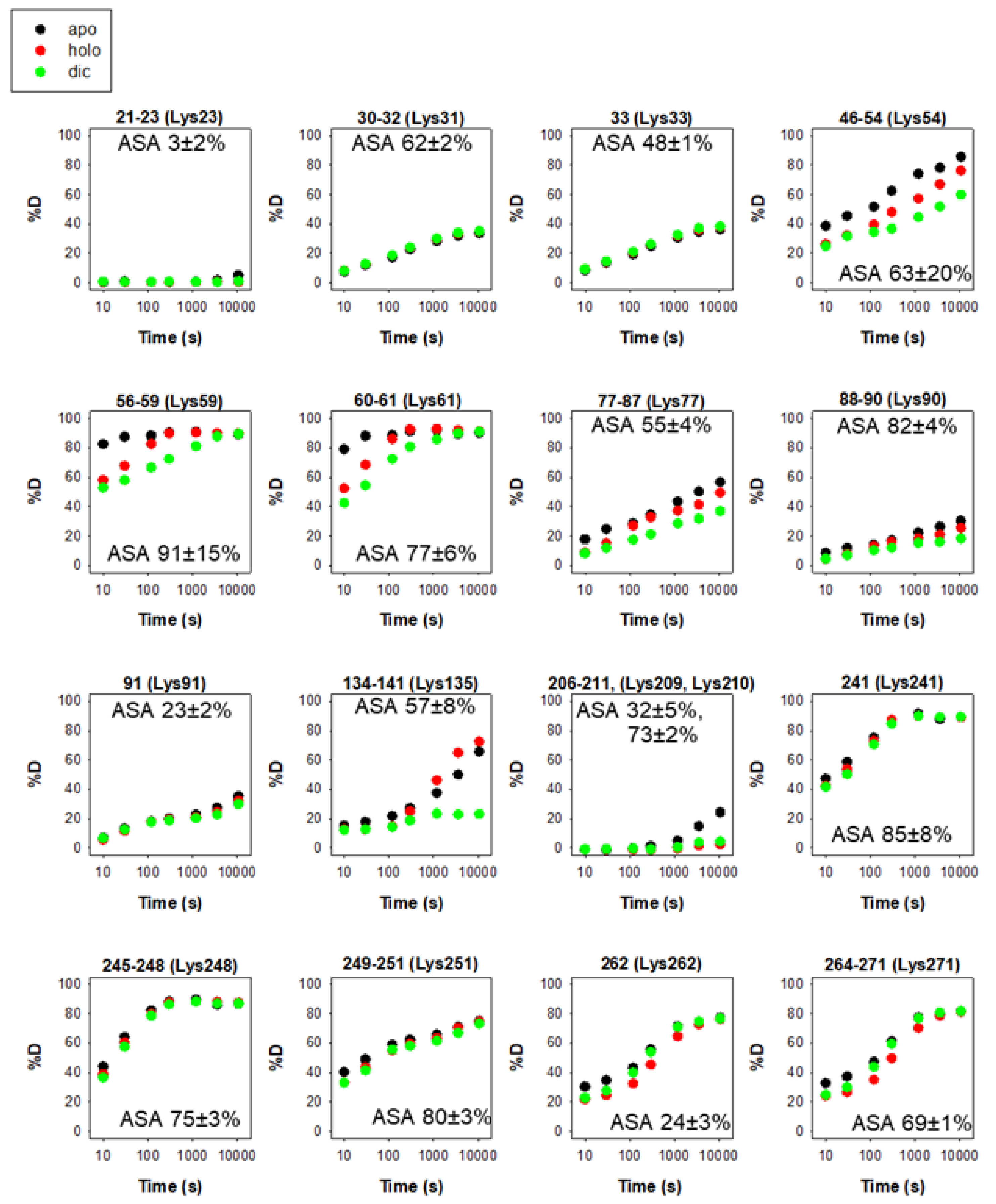{kind=link}
{kind=link}
{kind=link}
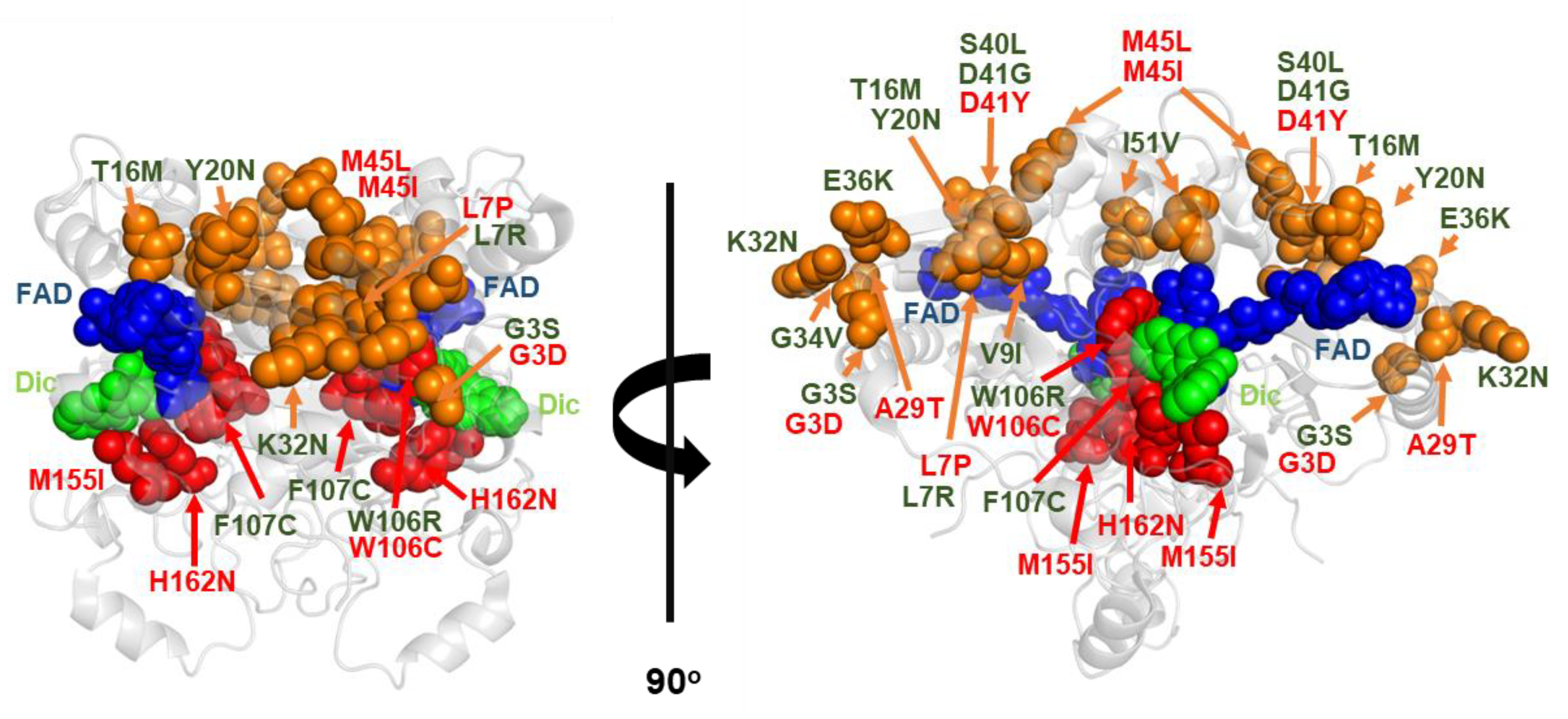{kind=link}
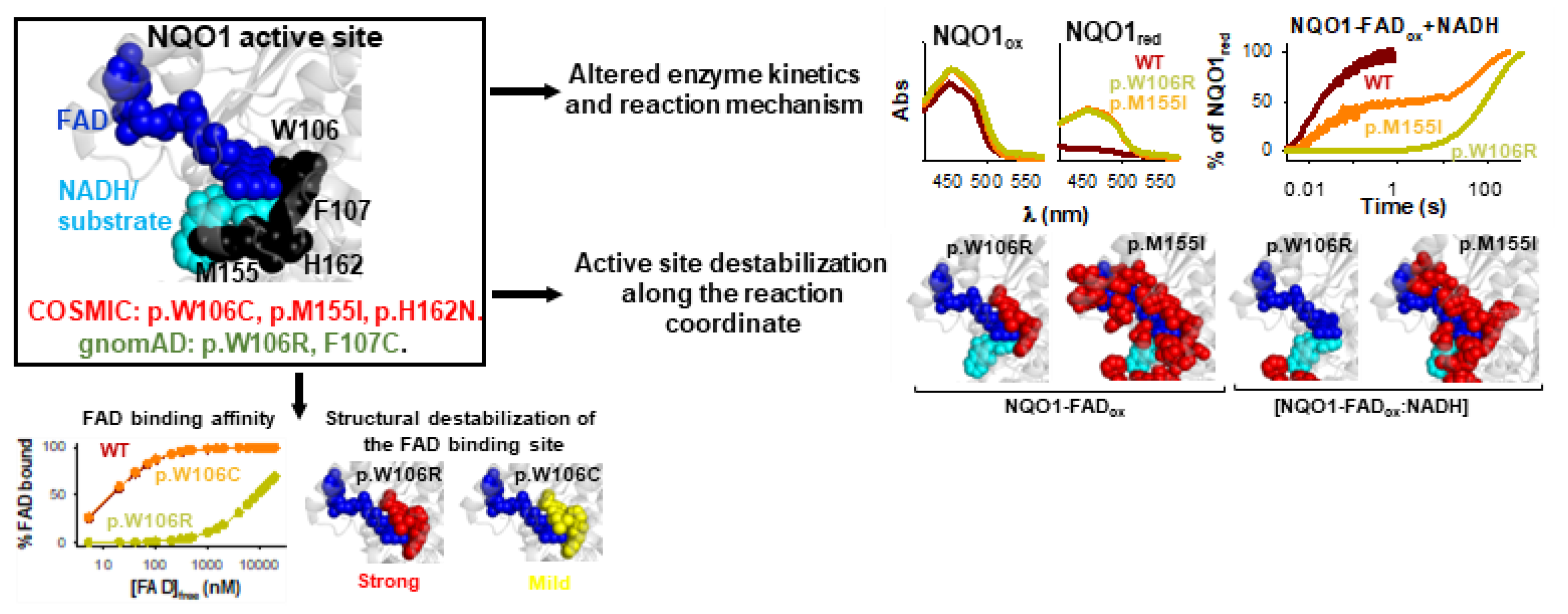{kind=link}
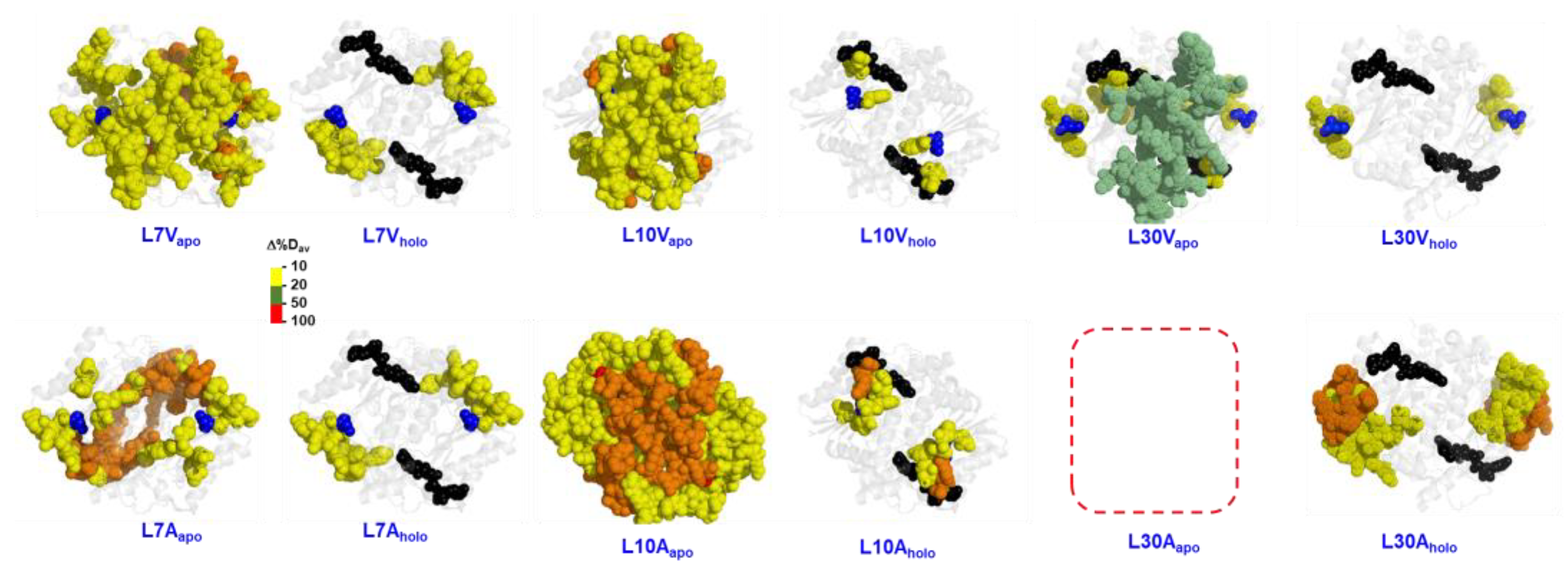{kind=link}
| NQO1 Variant | kcat (s−1) | KM (NADH) (mM) | kcat/KM (s−1·mM−1) |
|---|---|---|---|
| WT | 50 ± 4 | 0.54 ± 0.10 | 91 ± 18 |
| K31Q | 36 ± 4 | 0.44 ± 0.10 | 82 ± 20 |
| K31E | 32 ± 5 | 0.38 ± 0.08 | 86 ± 19 |
| K209Q | 43 ± 6 | 0.29 ± 0.10 | 147 ± 50 |
| K209E | 42 ± 5 | 0.30 ± 0.08 | 142 ± 40 |
| NQO1 Variant | Ligation State 1 | Tm (°C) 2 |
|---|---|---|
| WT | Holo | 55.8 ± 0.5 |
| Apo | 52.0 ± 0.6 | |
| K31Q | Holo | 54.5 ± 0.1 |
| Apo | 50.5 ± 0.7 | |
| K31E | Holo | 53.3 ± 0.1 |
| Apo | 49.6 ± 0.6 | |
| K209Q | Holo | 55.4 ± 0.1 |
| Apo | 51.9 ± 0.3 | |
| K209E | Holo | 53.5 ± 0.1 |
| Apo | 50.8 ± 0.6 |
| NQO1 Variant | Soluble Protein Levels (vs. WT) | ΔTm (vs. WT) (°C) | Kd (FAD) (WT-Fold) |
|---|---|---|---|
| WT | 1.0 ± 0.1 | 0.0 ± 0.6 | 1.0 ± 0.2 |
| G3S | 2.0 ± 0.3 | −0.4 ± 0.8 | 1.4 ± 0.3 |
| G3D | 3.8 ± 1.1 | −1.5 ± 0.7 | 0.9 ± 0.4 |
| L7P | 0.5 ± 0.1 | N.Det. | N.Det. |
| L7R | >>0.1 | N.Det. | N.Det. |
| V9I | 0.8 ± 0.6 | −1.7 ± 0.6 | 1.4 ± 0.3 |
| T16M | 0.4 ± 0.2 | −4.3 ± 0.7 | 10.8 ± 0.2 |
| Y20N | 0.6 ± 0.1 | −5.1 ± 0.6 | 2.9 ± 0.5 |
| A29T | 1.3 ± 0.3 | 0.1 ± 0.7 | 5.0 ± 0.4 |
| K32N | 0.8 ± 0.2 | −0.2 ± 0.6 | 0.8 ± 0.4 |
| G34V | >>0.1 | N.Det. | N.Det. |
| E36K | 0.9 ± 0.2 | 0.0 ± 0.8 | 0.8 ± 0.5 |
| S40L | >>0.1 | N.Det. | N.Det. |
| D41G | >>0.1 | −7.9 ± 0.5 | N.Det. |
| D41Y | >>0.1 | −9.7 ± 0.6 | N.Det. |
| M45L | 0.4 ± 0.5 | −3.7 ± 0.5 | 0.5 ± 1.0 |
| M45I | 0.2 ± 0.4 | −3.4 ± 0.6 | 0.6 ± 0.8 |
| I51V | 0.4 ± 0.3 | −5.0 ± 0.5 | 8.9 ± 0.3 |
| W106R | >0.1 | −6.4 ± 0.7 | ~ 500 |
| W106C | 0.2 ± 0.5 | −2.5 ± 0.7 | 0.9 ± 0.4 |
| F107C | 0.1 ± 0.7 | 0.6 ± 0.7 | ~ 0.2 |
| M155I | >0.1 | −0.9 ± 0.6 | 45 ± 1 |
| H162N | 0.3 ± 0.9 | −0.7 ± 1.0 | 27 ± 1 |
| K240Q | 1.3 ± 0.3 | 0.5 ± 0.6 | 2.8 ± 0.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pey, A.L. Phenotypic Modulation of Cancer-Associated Antioxidant NQO1 Activity by Post-Translational Modifications and the Natural Diversity of the Human Genome. Antioxidants 2023, 12, 379. https://doi.org/10.3390/antiox12020379
Pey AL. Phenotypic Modulation of Cancer-Associated Antioxidant NQO1 Activity by Post-Translational Modifications and the Natural Diversity of the Human Genome. Antioxidants. 2023; 12(2):379. https://doi.org/10.3390/antiox12020379
Chicago/Turabian StylePey, Angel L. 2023. "Phenotypic Modulation of Cancer-Associated Antioxidant NQO1 Activity by Post-Translational Modifications and the Natural Diversity of the Human Genome" Antioxidants 12, no. 2: 379. https://doi.org/10.3390/antiox12020379
APA StylePey, A. L. (2023). Phenotypic Modulation of Cancer-Associated Antioxidant NQO1 Activity by Post-Translational Modifications and the Natural Diversity of the Human Genome. Antioxidants, 12(2), 379. https://doi.org/10.3390/antiox12020379