
Klotho, Oxidative Stress, and Mitochondrial Damage in Kidney Disease

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Klotho Protein
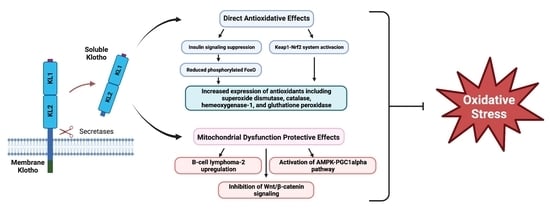
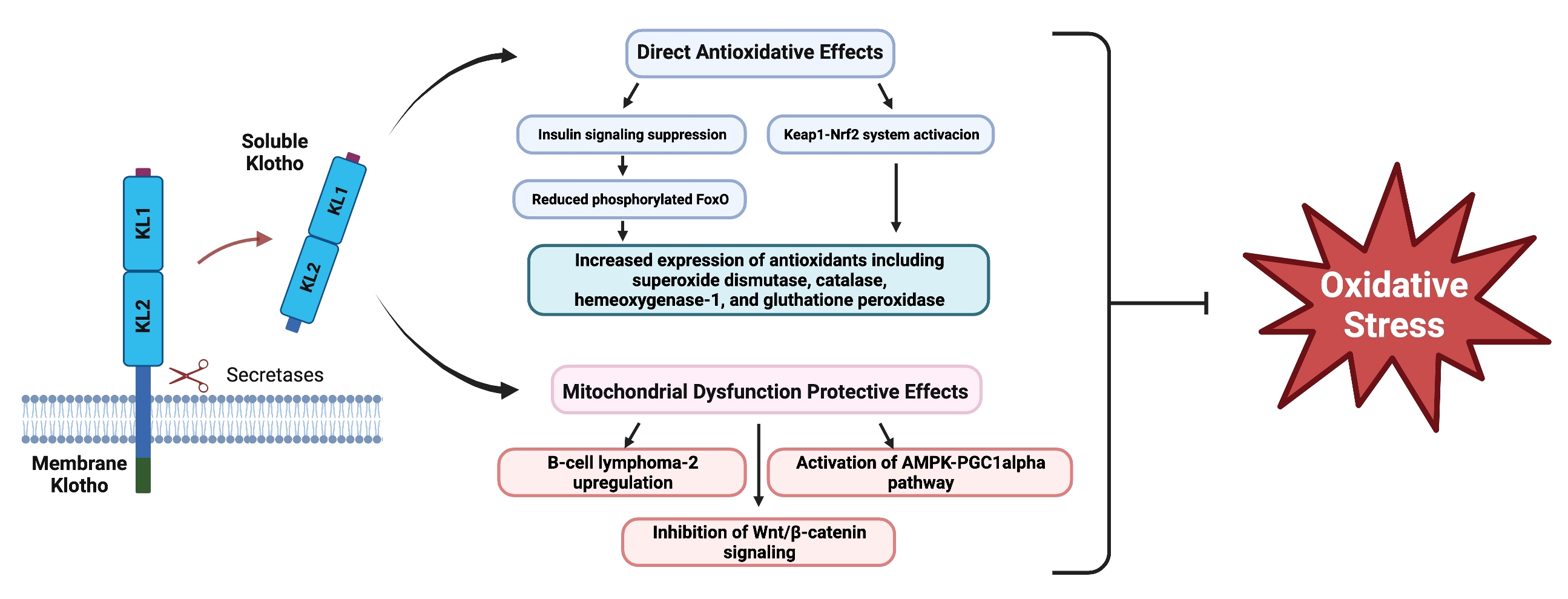
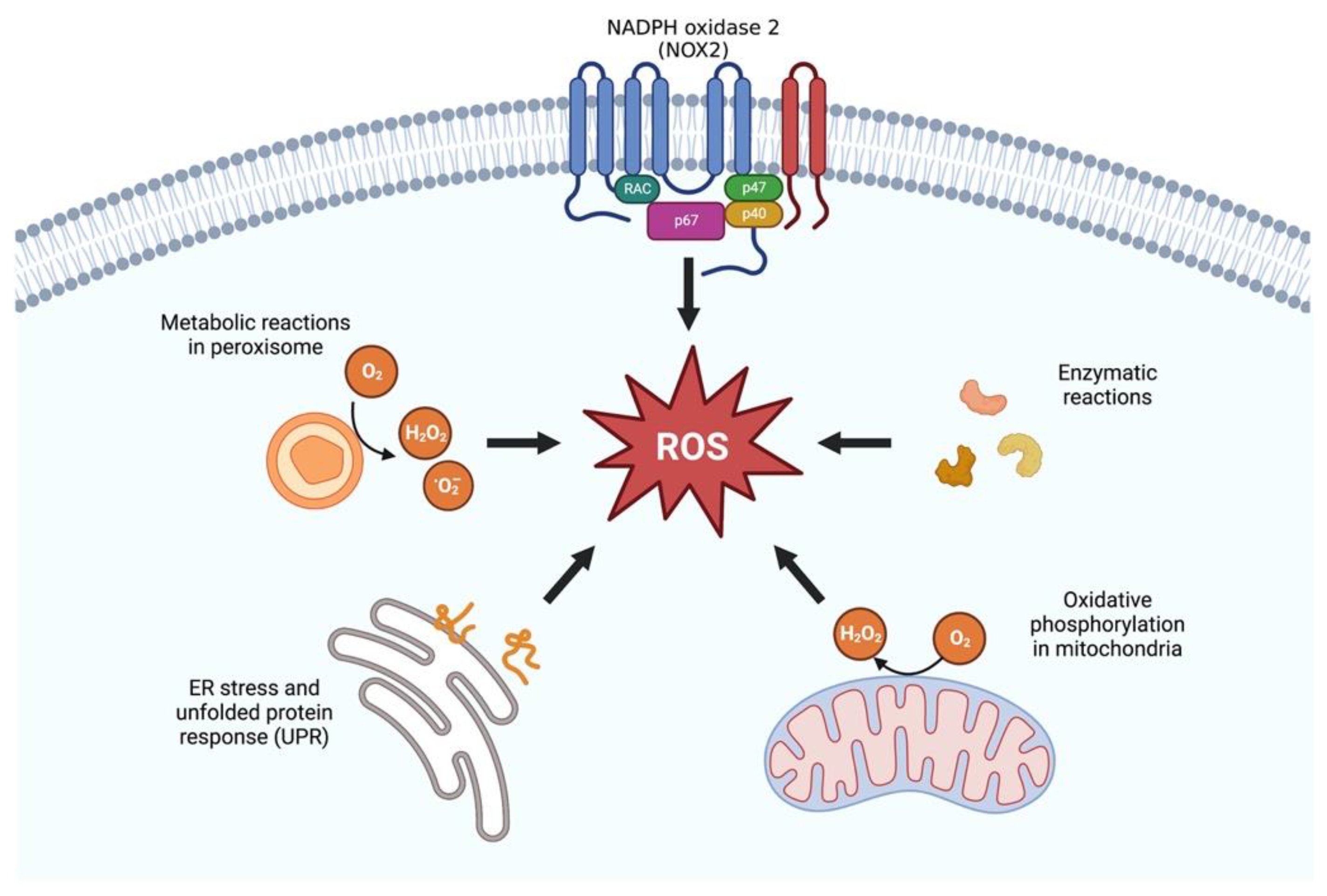
3. Antioxidative Effects of Klotho
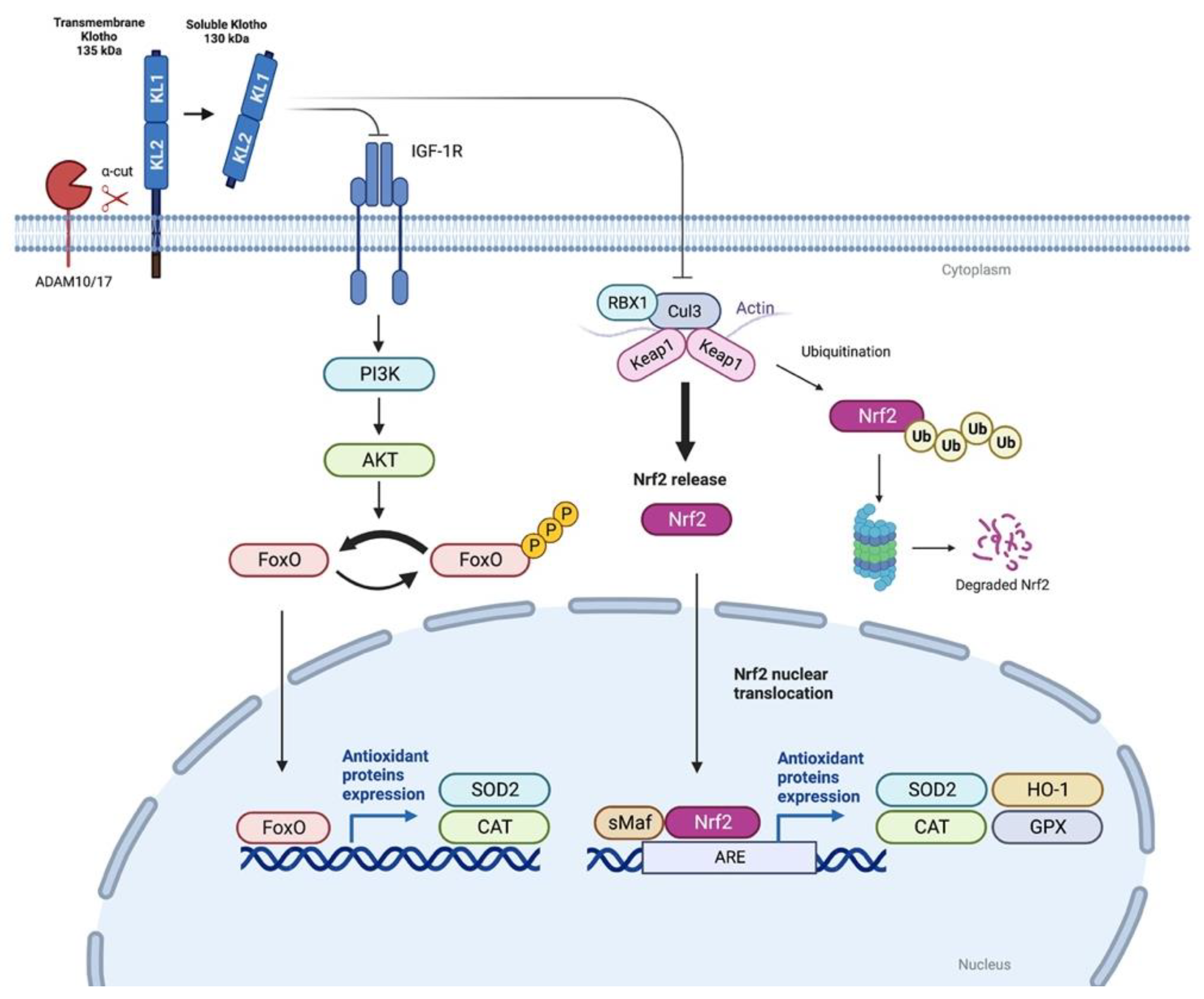
3.1. Klotho and FoxO Proteins
3.2. Nrf2
4. Klotho & Mitochondrial Dysfunction
4.1. Mitochondrial Uncoupling Protein 1 (UCP1)
4.2. B-Cell Lymphoma-2 (BCL-2)
4.3. Wnt/β-Catenin Signaling
4.4. Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha (PGC1alpha)
4.5. Transcription Factor EB (TFEB)
4.6. Peroxisome Proliferator-Activated Receptor Gamma (PPAR-γ)
5. Klotho Replacement as a Possible Antioxidant and Mitochondria-Protective Strategy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brück, K.; Stel, V.S.; Gambaro, G.; Hallan, S.; Völzke, H.; Ärnlöv, J.; Kastarinen, M.; Guessous, I.; Vinhas, J.; Stengel, B.; et al. CKD Prevalence Varies across the European General Population. J. Am. Soc. Nephrol. 2016, 27, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A. Asociacion Informacion Enfermedades Renales Geneticas EKPFFNdAplLClEdRFRIAdTRdIR. RICORS2040: The need for collaborative research in chronic kidney disease. Clin. Kidney J. 2022, 15, 372–387. [Google Scholar] [CrossRef]
- Woo, K.T.; Wong, K.S.; Choong, H.L.; Foo, M.W.; Tan, H.K.; Chan, C.M. Angiotensin receptor blocker and calcium channel blocker combination prevents cardiovascular events in CKD better than high-dose ARB alone. Kidney Int. 2013, 84, 214–215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fernandez-Fernandez, B.; Sarafidis, P.; Kanbay, M.; Navarro-González, J.F.; Soler, M.J.; Górriz, J.L.; Ortiz, A. SGLT2 inhibitors for non-diabetic kidney disease: Drugs to treat CKD that also improve glycaemia. Clin. Kidney J. 2020, 13, 728–733. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Larsson, T.E. Chronic kidney disease: A clinical model of premature aging. Am. J. Kidney Dis. 2013, 62, 339–351. [Google Scholar] [CrossRef]
- Wang, K.; Kestenbaum, B. Proximal Tubular Secretory Clearance: A Neglected Partner of Kidney Function. Clin. J. Am. Soc. Nephrol. 2018, 13, 1291–1296. [Google Scholar] [CrossRef]
- Eirin, A.; Lerman, A.; Lerman, L.O. The Emerging Role of Mitochondrial Targeting in Kidney Disease. Handb. Exp. Pharmacol. 2017, 240, 229–250. [Google Scholar] [CrossRef]
- Thérond, P.; Bonnefont-Rousselot, D.; Davit-Spraul, A.; Conti, M.; Legrand, A. Biomarkers of oxidative stress: An analytical approach. Curr. Opin. Clin. Nutr. Metab. Care 2000, 3, 373–384. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Weinberg, J.M. Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol. 2003, 14, 2199–2210. [Google Scholar] [CrossRef]
- Himmelfarb, J. Relevance of oxidative pathways in the pathophysiology of chronic kidney disease. Cardiol. Clin. 2005, 23, 319–330. [Google Scholar] [CrossRef]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.-Y.; Matsui, I.; Kitamura, H.; et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Chertow, G.M.; Devarajan, P.; Levin, A.; Andreoli, S.P.; Bangalore, S.; Warady, B.A. Chronic Inflammation in Chronic Kidney Disease Progression: Role of Nrf2. Kidney Int. Rep. 2021, 6, 1775–1787. [Google Scholar] [CrossRef]
- Chatterjee, P.K.; Cuzzocrea, S.; Brown, P.A.; Zacharowski, K.; Stewart, K.N.; Mota-Filipe, H.; Thiemermann, C. Tempol, a membrane-permeable radical scavenger, reduces oxidant stress-mediated renal dysfunction and injury in the rat. Kidney Int. 2000, 58, 658–673. [Google Scholar] [CrossRef]
- Luo, C.; Zhou, S.; Zhou, Z.; Liu, Y.; Yang, L.; Liu, J.; Zhang, Y.; Li, H.; Liu, Y.; Hou, F.F.; et al. Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells. J. Am. Soc. Nephrol. 2018, 29, 1238–1256. [Google Scholar] [CrossRef]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal. 2018, 12, 69–82. [Google Scholar] [CrossRef]
- Bonventre, J.V. Maladaptive proximal tubule repair: Cell cycle arrest. Nephron Clin. Pract. 2014, 127, 61–64. [Google Scholar] [CrossRef]
- Melk, A.; Schmidt, B.M.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520. [Google Scholar] [CrossRef]
- Knoppert, S.N.; Valentijn, F.A.; Nguyen, T.Q.; Goldschmeding, R.; Falke, L.L. Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front. Pharmacol. 2019, 10, 770. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wan, Y.; Chen, R.; Zhang, C.; Li, X.; Meng, F.; Glaser, S.; Wu, N.; Zhou, T.; Li, S.; et al. The emerging role of cellular senescence in renal diseases. J. Cell. Mol. Med. 2020, 24, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Harper, M.-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free. Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Renal. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef]
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPARalpha and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237. [Google Scholar] [CrossRef]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Verzola, D.; Gandolfo, M.T.; Gaetani, G.; Ferraris, A.; Mangerini, R.; Ferrario, F.; Villaggio, B.; Gianiorio, F.; Tosetti, F.; Weiss, U.; et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am. J. Physiol. Physiol. 2008, 295, F1563–F1573. [Google Scholar] [CrossRef]
- Granata, S.; Zaza, G.; Simone, S.; Villani, G.; Latorre, D.; Pontrelli, P.; Carella, M.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genom. 2009, 10, 388. [Google Scholar] [CrossRef]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ren, J.; Gui, Y.; Wei, W.; Shu, B.; Lu, Q.; Xue, X.; Sun, X.; He, W.; Yang, J.; et al. Wnt/beta-Catenin-Promoted Macrophage Alternative Activation Contributes to Kidney Fibrosis. J. Am. Soc. Nephrol. 2017, 29, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.L.; Cao, W.; Xie, C.; Tian, J.; Zhou, Z.; Zhou, Q.; Zhu, P.; Li, A.; Liu, Y.; Miyata, T.; et al. The receptor of advanced glycation end products plays a central role in advanced oxidation protein products-induced podocyte apoptosis. Kidney Int. 2012, 82, 759–770. [Google Scholar] [CrossRef]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St. Calir, D.K. Manganese Superoxide Dismutase: Guardian of the Powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef]
- Theurey, P.; Pizzo, P. The Aging Mitochondria. Genes 2018, 9, 22. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Tábara, L.C.; Poveda, J.; Martin-Cleary, C.; Selgas, R.; Ortiz, A.; Sanchez-Niño, M.D. Mitochondria-targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 2014, 16, e13. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Lopez-Diaz, A.M.; Guerrero-Mauvecin, J.; Miguel, V.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Tubular Mitochondrial Dysfunction, Oxidative Stress, and Progression of Chronic Kidney Disease. Antioxidants 2022, 11, 1356. [Google Scholar] [CrossRef]
- Izbeki, F.; Asuzu, D.T.; Lorincz, A.; Bardsley, M.R.; Popko, L.N.; Choi, K.M.; Young, D.L.; Hayashi, Y.; Linden, D.R.; Kuro-o, M.; et al. Loss of Kitlow progenitors, reduced stem cell factor and high oxidative stress underlie gastric dysfunction in progeric mice. J. Physiol. 2010, 588, 3101–3117. [Google Scholar] [CrossRef]
- Mitobe, M.; Yoshida, T.; Sugiura, H.; Shirota, S.; Tsuchiya, K.; Nihei, H. Oxidative stress decreases klotho expression in a mouse kidney cell line. Nephron Exp. Nephrol. 2005, 101, e67–e74. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M. Klotho as a regulator of oxidative stress and senescence. Biol. Chem. 2008, 389, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-C.; Shi, M.; Cho, H.J.; Zhang, J.; Pavlenco, A.; Liu, S.; Sidhu, S.; Huang, L.J.-S.; Moe, O.W. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney Int. 2013, 84, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Huang, J.; Luo, C.; Ye, H.; Ling, X.; Wu, Q.; Shen, W.; Zhou, L. Klotho retards renal fibrosis through targeting mitochondrial dysfunction and cellular senescence in renal tubular cells. Physiol. Rep. 2021, 9, e14696. [Google Scholar] [CrossRef]
- Sahu, A.; Mamiya, H.; Shinde, S.N.; Cheikhi, A.; Winter, L.L.; Vo, N.V.; Stolz, D.; Roginskaya, V.; Tang, W.-Y.; St. Croix, C.; et al. Age-related declines in alpha-Klotho drive progenitor cell mitochondrial dysfunction and impaired muscle regeneration. Nat. Commun. 2018, 9, 4859. [Google Scholar] [CrossRef]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.-C.; Moe, O.W.; et al. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Kim, H.R.; Nam, B.Y.; Kim, D.W.; Kang, M.W.; Han, J.-H.; Lee, M.J.; Shin, D.H.; Doh, F.M.; Koo, H.M.; Ko, K.I.; et al. Circulating alpha-klotho levels in CKD and relationship to progression. Am. J. Kidney Dis. 2013, 61, 899–909. [Google Scholar] [CrossRef]
- Wang, Q.; Ren, D.; Li, Y.; Xu, G. Klotho attenuates diabetic nephropathy in db/db mice and ameliorates high glucose-induced injury of human renal glomerular endothelial cells. Cell Cycle 2019, 18, 696–707. [Google Scholar] [CrossRef]
- Silva, A.P.; Mendes, F.; Carias, E.; Gonçalves, R.B.; Fragoso, A.; Dias, C.; Tavares, N.; Café, H.M.; Santos, N.; Rato, F.; et al. Plasmatic Klotho and FGF23 Levels as Biomarkers of CKD-Associated Cardiac Disease in Type 2 Diabetic Patients. Int. J. Mol. Sci. 2019, 20, 1536. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, J.; Chae, D.-W.; Lee, K.-B.; Sung, S.A.; Yoo, T.-H.; Han, S.H.; Ahn, C.; Oh, K.-H. Serum klotho is inversely associated with metabolic syndrome in chronic kidney disease: Results from the KNOW-CKD study. BMC Nephrol. 2019, 20, 119. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Huang, X.; Fu, C.; Wu, X.; Peng, Y.; Lin, X.; Wang, Y. Recombinant Klotho Protects Human Periodontal Ligament Stem Cells by Regulating Mitochondrial Function and the Antioxidant System during H(2)O(2)-Induced Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019, 9261565. [Google Scholar] [CrossRef]
- Ikushima, M.; Rakugi, H.; Ishikawa, K.; Maekawa, Y.; Yamamoto, K.; Ohta, J.; Chihara, Y.; Kida, I.; Ogihara, T. Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem. Biophys. Res. Commun. 2006, 339, 827–832. [Google Scholar] [CrossRef]
- Lim, S.W.; Jin, L.; Luo, K.; Jin, J.; Shin, Y.J.; Hong, S.Y.; Yang, C.W. Klotho enhances FoxO3-mediated manganese superoxide dismutase expression by negatively regulating PI3K/AKT pathway during tacrolimus-induced oxidative stress. Cell Death Dis. 2017, 8, e2972. [Google Scholar] [CrossRef]
- Hsieh, C.-C.; Kuro-O, M.; Rosenblatt, K.P.; Brobey, R.; Papaconstantinou, J. The ASK1-Signalosome regulates p38 MAPK activity in response to levels of endogenous oxidative stress in the Klotho mouse models of aging. Aging 2010, 2, 597–611. [Google Scholar] [CrossRef]
- Maltese, G.; Psefteli, P.-M.; Rizzo, B.; Srivastava, S.; Gnudi, L.; Mann, G.E.; Siow, R.C. The anti-ageing hormone klotho induces Nrf2-mediated antioxidant defences in human aortic smooth muscle cells. J. Cell. Mol. Med. 2017, 21, 621–627. [Google Scholar] [CrossRef]
- Zhu, H.; Gao, Y.; Zhu, S.; Cui, Q.; Du, J. Klotho Improves Cardiac Function by Suppressing Reactive Oxygen Species (ROS) Mediated Apoptosis by Modulating Mapks/Nrf2 Signaling in Doxorubicin-Induced Cardiotoxicity. Med. Sci. Monit. 2017, 23, 5283–5293. [Google Scholar] [CrossRef]
- Xing, L.; Guo, H.; Meng, S.; Zhu, B.; Fang, J.; Huang, J.; Chen, J.; Wang, Y.; Wang, L.; Yao, X.; et al. Klotho ameliorates diabetic nephropathy by activating Nrf2 signaling pathway in podocytes. Biochem. Biophys. Res. Commun. 2021, 534, 450–456. [Google Scholar] [CrossRef]
- Xiang, T.; Luo, X.; Ye, L.; Huang, H.; Wu, Y. Klotho alleviates NLRP3 inflammasome-mediated neuroinflammation in a temporal lobe epilepsy rat model by activating the Nrf2 signaling pathway. Epilepsy Behav. 2022, 128, 108509. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Chikuda, H.; Suga, T.; Kawaguchi, H.; Kuro-O, M. Regulation of multiple ageing-like phenotypes by inducible klotho gene expression in klotho mutant mice. Mech. Ageing Dev. 2005, 126, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Groen, A.J.; Molostvov, G.; Lu, T.; Lilley, K.S.; Snead, D.; James, S.; Wilkinson, I.B.; Ting, S.; Hsiao, L.-L.; et al. alpha-Klotho Expression in Human Tissues. J. Clin. Endocrinol. Metab. 2015, 100, E1308–E1318. [Google Scholar] [CrossRef] [PubMed]
- Mencke, R.; Harms, G.; Moser, J.; Van Meurs, M.; Diepstra, A.; Leuvenink, H.; Hillebrands, J.-L. Human alternative Klotho mRNA is a nonsense-mediated mRNA decay target inefficiently spliced in renal disease. JCI Insight 2017, 2, e94375. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Kuro-O, M.; Moe, O.W. The emerging role of Klotho in clinical nephrology. Nephrol. Dial. Transplant. 2012, 27, 2650–2657. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, K.; Lei, H.; Sun, Z. Klotho gene deficiency causes salt-sensitive hypertension via monocyte chemotactic protein-1/CC chemokine receptor 2-mediated inflammation. J. Am. Soc. Nephrol. 2014, 26, 121–132. [Google Scholar] [CrossRef]
- Shi, M.; Flores, B.; Gillings, N.; Bian, A.; Cho, H.J.; Yan, S.; Liu, Y.; Levine, B.; Moe, O.W.; Hu, M.C. alphaKlotho Mitigates Progression of AKI to CKD through Activation of Autophagy. J. Am. Soc. Nephrol. 2016, 27, 2331–2345. [Google Scholar] [CrossRef]
- Wang, Y.; Kuro-O, M.; Sun, Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell 2012, 11, 410–417. [Google Scholar] [CrossRef]
- Lee, J.; Tsogbadrakh, B.; Yang, S.; Ryu, H.; Kang, E.; Kang, M.; Kang, H.G.; Ahn, C.; Oh, K.-H. Klotho ameliorates diabetic nephropathy via LKB1-AMPK-PGC1alpha-mediated renal mitochondrial protection. Biochem. Biophys. Res. Commun. 2021, 534, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.G.; Kang, S.H.; Lim, S.W.; Chung, B.H.; Doh, K.C.; Heo, S.B.; Jin, L.; Li, C.; Yang, C.W. Influence of N-acetylcysteine on Klotho expression and its signaling pathway in experimental model of chronic cyclosporine nephropathy in mice. Transplantation 2013, 96, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.E.; Choi, B.S. The renin-angiotensin system and aging in the kidney. Korean J. Intern. Med. 2014, 29, 291–295. [Google Scholar] [CrossRef]
- Yoon, H.E.; Lim, S.W.; Piao, S.G.; Song, J.-H.; Kim, J.; Yang, C.W. Statin upregulates the expression of klotho, an anti-aging gene, in experimental cyclosporine nephropathy. Nephron Exp. Nephrol. 2012, 120, e123–e133. [Google Scholar] [CrossRef]
- Yoon, H.E.; Ghee, J.Y.; Piao, S.; Song, J.-H.; Han, D.H.; Kim, S.; Ohashi, N.; Kobori, H.; Kuro-O, M.; Yang, C.W. Angiotensin II blockade upregulates the expression of Klotho, the anti-ageing gene, in an experimental model of chronic cyclosporine nephropathy. Nephrol. Dial. Transplant. 2011, 26, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Haruna, Y.; Kashihara, N.; Satoh, M.; Tomita, N.; Namikoshi, T.; Sasaki, T.; Fujimori, T.; Xie, P.; Kanwar, Y.S. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc. Natl. Acad. Sci. USA 2007, 104, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Harris, R.C. Renal endothelial dysfunction in diabetic nephropathy. Cardiovasc. Hematol. Disord. Targets 2014, 14, 22–33. [Google Scholar] [CrossRef]
- Buendía, P.; Carracedo, J.; Soriano, S.; Madueño, J.A.; Ortiz, A.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Klotho Prevents NFkappaB Translocation and Protects Endothelial Cell From Senescence Induced by Uremia. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1198–1209. [Google Scholar] [CrossRef]
- Yang, K.; Nie, L.; Huang, Y.; Zhang, J.; Xiao, T.; Guan, X.; Zhao, J. Amelioration of uremic toxin indoxyl sulfate-induced endothelial cell dysfunction by Klotho protein. Toxicol. Lett. 2012, 215, 77–83. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.-H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef]
- Carracedo, J.; Buendía, P.; Merino, A.; Madueño, J.A.; Peralbo, E.; Ortiz, A.; Martín-Malo, A.; Aljama, P.; Rodríguez, M.; Ramírez, R. Klotho modulates the stress response in human senescent endothelial cells. Mech. Ageing Dev. 2012, 133, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, S.; Ren, H.; Gu, J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat. Cell Biol. 2011, 13, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; de Mattos, S.F.; van der Horst, A.; Klompmaker, R.; Kops, G.J.P.L.; Lam, E.W.-F.; Burgering, B.M.T.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [PubMed]
- Burgering, B.M.; Kops, G.J. Cell cycle and death control: Long live Forkheads. Trends Biochem. Sci. 2002, 27, 352–360. [Google Scholar] [CrossRef]
- Barthel, A.; Schmoll, D.; Unterman, T.G. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 2005, 16, 183–189. [Google Scholar] [CrossRef]
- Tatar, M.; Bartke, A.; Antebi, A. The endocrine regulation of aging by insulin-like signals. Science 2003, 299, 1346–1351. [Google Scholar] [CrossRef]
- Jin, J.; Jin, L.; Lim, S.W.; Yang, C.W. Klotho Deficiency Aggravates Tacrolimus-Induced Renal Injury via the Phosphatidylinositol 3-Kinase-Akt-Forkhead Box Protein O Pathway. Am. J. Nephrol. 2016, 43, 357–365. [Google Scholar] [CrossRef]
- Wolf, I.; Levanon-Cohen, S.; Bose, S.; Ligumsky, H.; Sredni, B.; Kanety, H.; Kuro-O, M.; Karlan, B.; Kaufman, B.; Koeffler, H.P.; et al. Klotho: A tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 2008, 27, 7094–7105. [Google Scholar] [CrossRef]
- Yoon, H.E.; Yang, C.W. Established and newly proposed mechanisms of chronic cyclosporine nephropathy. Korean J. Intern. Med. 2009, 24, 81–92. [Google Scholar] [CrossRef]
- Kuang, X.; Du, J.R.; Chen, Y.S.; Wang, J.; Wang, Y.N. Protective effect of Z-ligustilide against amyloid beta-induced neurotoxicity is associated with decreased pro-inflammatory markers in rat brains. Pharmacol. Biochem. Behav. 2009, 92, 635–641. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Manolopoulos, K.N.; Klotz, L.-O.; Korsten, P.; Bornstein, S.R.; Barthel, A. Linking Alzheimer’s disease to insulin resistance: The FoxO response to oxidative stress. Mol. Psychiatry 2010, 15, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Zeldich, E.; Chen, C.-D.; Colvin, T.A.; Bove-Fenderson, E.A.; Liang, J.; Zhou, T.B.T.; Harris, D.; Abraham, C.R. The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J. Biol. Chem. 2014, 289, 24700–24715. [Google Scholar] [CrossRef]
- Kuang, X.; Chen, Y.S.; Wang, L.F.; Li, Y.J.; Liu, K.; Zhang, M.X.; Li, L.-J.; Chen, C.; He, Q.; Wang, Y.; et al. Klotho upregulation contributes to the neuroprotection of ligustilide in an Alzheimer’s disease mouse model. Neurobiol. Aging. 2014, 35, 169–178. [Google Scholar] [CrossRef]
- Yu, Y.; Du, J.-R.; Wang, C.-Y.; Qian, Z.-M. Protection against hydrogen peroxide-induced injury by Z-ligustilide in PC12 cells. Exp. Brain Res. 2008, 184, 307–312. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Panda, H.; Wen, H.; Suzuki, M.; Yamamoto, M. Multifaceted Roles of the KEAP1-NRF2 System in Cancer and Inflammatory Disease Milieu. Antioxidants 2022, 11, 538. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M. Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found Symp. 2007, 287, 60–63, discussion 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ucar, B.; Ucar, G.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Pharmacological Protection against Ischemia-Reperfusion Injury by Regulating the Nrf2-Keap1-ARE Signaling Pathway. Antioxidants 2021, 10, 823. [Google Scholar] [CrossRef]
- González-Bosch, C.; Boorman, E.; Zunszain, P.A.; Mann, G.E. Short-chain fatty acids as modulators of redox signaling in health and disease. Redox Biol. 2021, 47, 102165. [Google Scholar] [CrossRef]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Min, X.; Xu, X.; Du, B.; Luo, P. Role of Nuclear Factor Erythroid 2-Related Factor 2 in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 3797802. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, L.; Wang, F.; Shi, Y.; Ren, Y.; Liu, Q.; Cao, Y.; Duan, H. Attenuation of glomerular injury in diabetic mice with tert-butylhydroquinone through nuclear factor erythroid 2-related factor 2-dependent antioxidant gene activation. Am. J. Nephrol. 2011, 33, 289–297. [Google Scholar] [CrossRef]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, S.; Sun, Q.W.; Zhang, B.; Ullah, M.F.; Sun, Z. Klotho Deficiency Causes Heart Aging via Impairing the Nrf2-GR Pathway. Circ. Res. 2021, 128, 492–507. [Google Scholar] [CrossRef]
- Zhao, M.; Murakami, S.; Matsumaru, D.; Kawauchi, T.; Nabeshima, Y.-I.; Motohashi, H. NRF2 pathway activation attenuates ageing-related renal phenotypes due to alpha-klotho deficiency. J. Biochem. 2022, 171, 579–589. [Google Scholar] [CrossRef]
- Guo, Y.; Zhuang, X.; Zou, J.; Yang, D.; Hu, X.; Du, Z.; Wang, L.; Liao, X. Klotho protects the heart from hyperglycemia-induced injury by inactivating ROS and NF-kappaB-mediated inflammation both in vitro and in vivo. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 238–251. [Google Scholar] [CrossRef]
- Romero, A.; San Hipolito-Luengo, A.; Villalobos, L.A.; Vallejo, S.; Valencia, I.; Michalska, P.; Pajuelo-Lozano, N.; Sánchez-Pérez, I.; León, R.; Bartha, J.L.; et al. The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell 2019, 18, e12913. [Google Scholar] [CrossRef]
- Cui, W.; Leng, B.; Wang, G. Klotho protein inhibits H(2)O(2)-induced oxidative injury in endothelial cells via regulation of PI3K/AKT/Nrf2/HO-1 pathways. Can. J. Physiol. Pharmacol. 2019, 97, 370–376. [Google Scholar] [CrossRef]
- Ravikumar, P.; Ye, J.; Zhang, J.; Pinch, S.N.; Hu, M.C.; Kuro-O, M.; Hsia, C.C.W.; Moe, O.W. α-Klotho protects against oxidative damage in pulmonary epithelia. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L566–L575. [Google Scholar] [CrossRef]
- Marquez-Exposito, L.; Tejedor-Santamaria, L.; Valentijn, F.A.; Tejera-Muñoz, A.; Rayego-Mateos, S.; Marchant, V.; Rodrigues-Diez, R.R.; Rubio-Soto, I.; Knoppert, S.N.; Ortiz, A.; et al. Oxidative Stress and Cellular Senescence Are Involved in the Aging Kidney. Antioxidants 2022, 11, 301. [Google Scholar] [CrossRef] [PubMed]
- Ahmatjan, B.; Ruotian, L.; Rahman, A.; Bin, M.; Heng, D.; Yi, H.; Tao, C.; Le, G.; Mahmut, M. Klotho inhibits the formation of calcium oxalate stones by regulating the Keap1-Nrf2-ARE signaling pathway. Int. Urol. Nephrol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Panesso, M.C.; Shi, M.; Cho, H.J.; Paek, J.; Ye, J.; Moe, O.W.; Hu, M.C. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney Int. 2014, 85, 855–870. [Google Scholar] [CrossRef]
- Kawai, M.; Kinoshita, S.; Ozono, K.; Michigami, T. Inorganic Phosphate Activates the AKT/mTORC1 Pathway and Shortens the Life Span of an alpha-Klotho-Deficient Model. J. Am. Soc. Nephrol. 2016, 27, 2810–2824. [Google Scholar] [CrossRef]
- Skulachev, V.P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996, 29, 169–202. [Google Scholar] [CrossRef] [PubMed]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Wu, X.; Pan, T.; Xu, S.; Hu, J.; Ding, X. Uncoupling protein 1 inhibits mitochondrial reactive oxygen species generation and alleviates acute kidney injury. EBioMedicine 2019, 49, 331–340. [Google Scholar] [CrossRef]
- Li, P.; Shi, M.; Maique, J.; Shaffer, J.; Yan, S.; Moe, O.W.; Hu, M.C. Beclin 1/Bcl-2 complex-dependent autophagy activity modulates renal susceptibility to ischemia-reperfusion injury and mediates renoprotection by Klotho. Am. J. Physiol. Physiol. 2020, 318, F772–F792. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Miao, J.; Liu, J.; Niu, J.; Zhang, Y.; Shen, W.; Luo, C.; Liu, Y.; Li, C.; Li, H.; Yang, P.; et al. Wnt/beta-catenin/RAS signaling mediates age-related renal fibrosis and is associated with mitochondrial dysfunction. Aging Cell 2019, 18, e13004. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Zhou, D.; Tan, R.J.; Liu, Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J. Am. Soc. Nephrol. 2013, 24, 771–785. [Google Scholar] [CrossRef]
- He, W.; Dai, C.; Li, Y.; Zeng, G.; Monga, S.P.; Liu, Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2009, 20, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Y.; Hao, S.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Multiple genes of the renin-angiotensin system are novel targets of Wnt/beta-catenin signaling. J. Am. Soc. Nephrol. 2015, 26, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Mo, H.; Miao, J.; Zhou, D.; Tan, R.J.; Hou, F.F.; Liu, Y. Klotho Ameliorates Kidney Injury and Fibrosis and Normalizes Blood Pressure by Targeting the Renin-Angiotensin System. Am. J. Pathol. 2015, 185, 3211–3223. [Google Scholar] [CrossRef]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef]
- Dugan, L.L.; You, Y.-H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.-E.; Andreyev, A.Y.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Investig. 2013, 123, 4888–4899. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef]
- Bulluck, H.; Hausenloy, D.J. Modulating NAD(+) metabolism to prevent acute kidney injury. Nat. Med. 2018, 24, 1306–1307. [Google Scholar] [CrossRef]
- Lim, S.W.; Shin, Y.J.; Luo, K.; Quan, Y.; Ko, E.J.; Chung, B.H.; Yang, C.W. Effect of Klotho on autophagy clearance in tacrolimus-induced renal injury. FASEB J. 2019, 33, 2694–2706. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Y.; Li, X.; Zhang, W.; Liu, Z.; Wu, M.; Pan, Q.; Liu, H. Emerging role of transcription factor EB in mitochondrial quality control. Biomed. Pharmacother. 2020, 128, 110272. [Google Scholar] [CrossRef] [PubMed]
- Ivankovic, D.; Chau, K.; Schapira, A.H.V.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 2015, 136, 388–402. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Zheng, F.; Guan, Y. PPARs and the kidney in metabolic syndrome. Am. J. Physiol. Physiol. 2008, 294, F1032–F1047. [Google Scholar] [CrossRef] [PubMed]
- Panchapakesan, U.; Pollock, C.A.; Chen, X.M. The effect of high glucose and PPAR-gamma agonists on PPAR-gamma expression and function in HK-2 cells. Am. J. Physiol. Physiol. 2004, 287, F528–F534. [Google Scholar] [CrossRef]
- Schneider, C.A.; Ferrannini, E.; DeFronzo, R.; Schernthaner, G.; Yates, J.; Erdmann, E. Effect of pioglitazone on cardiovascular outcome in diabetes and chronic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 182–187. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Fan, Y.; Wu, J.; Zhao, B.; Guan, Y.; Chien, S.; Wang, N. Klotho is a target gene of PPAR-gamma. Kidney Int. 2008, 74, 732–739. [Google Scholar] [CrossRef]
- Maquigussa, E.; Paterno, J.C.; Pokorny, G.H.D.O.; Perez, M.D.S.; Varela, V.A.; Novaes, A.D.S.; Schor, N.; Boim, M.A. Klotho and PPAR Gamma Activation Mediate the Renoprotective Effect of Losartan in the 5/6 Nephrectomy Model. Front. Physiol. 2018, 9, 1033. [Google Scholar] [CrossRef]
- Ragab, D.; Abdallah, D.M.; El-Abhar, H.S. Cilostazol renoprotective effect: Modulation of PPAR-gamma, NGAL, KIM-1 and IL-18 underlies its novel effect in a model of ischemia-reperfusion. PLoS ONE 2014, 9, e95313. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-C.; Shi, M.; Zhang, J.; Quiñones, H.; Kuro-O, M.; Moe, O.W. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010, 78, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Shiraki-Iida, T.; Iida, A.; Nabeshima, Y.; Anazawa, H.; Nishikawa, S.; Noda, M.; Kuro-O, M.; Nabeshima, Y. Improvement of multiple pathophysiological phenotypes of klotho (kl/kl) mice by adenovirus-mediated expression of the klotho gene. J. Gene Med. 2000, 2, 233–242. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Z. Antiaging gene Klotho regulates endothelin-1 levels and endothelin receptor subtype B expression in kidneys of spontaneously hypertensive rats. J. Hypertens. 2014, 32, 1629–1636, discussion 36. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension 2009, 54, 810–817. [Google Scholar] [CrossRef]
- Mitani, H.; Ishizaka, N.; Aizawa, T.; Ohno, M.; Usui, S.-I.; Suzuki, T.; Amaki, T.; Mori, I.; Nakamura, Y.; Sato, M.; et al. In vivo klotho gene transfer ameliorates angiotensin II-induced renal damage. Hypertension 2002, 39, 838–843. [Google Scholar] [CrossRef]
- Saito, Y.; Nakamura, T.; Ohyama, Y.; Suzuki, T.; Iida, A.; Shiraki-Iida, T.; Kuro-O, M.; Nabeshima, Y.-I.; Kurabayashi, M.; Nagai, R. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem. Biophys. Res. Commun. 2000, 276, 767–772. [Google Scholar] [CrossRef]
- Xie, J.; Yoon, J.; An, S.-W.; Kuro, M.; Kuro-O, M.; Huang, C.-L. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. J. Am. Soc. Nephrol. 2015, 26, 1150–1160. [Google Scholar] [CrossRef]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef]
- Chen, T.-H.; Kuro-O, M.; Chen, C.-H.; Sue, Y.-M.; Chen, Y.-C.; Wu, H.-H.; Cheng, C.-Y. The secreted Klotho protein restores phosphate retention and suppresses accelerated aging in Klotho mutant mice. Eur. J. Pharmacol. 2013, 698, 67–73. [Google Scholar] [CrossRef]
- Yang, H.-C.; Deleuze, S.; Zuo, Y.; Potthoff, S.A.; Ma, L.-J.; Fogo, A.B. The PPARgamma agonist pioglitazone ameliorates aging-related progressive renal injury. J. Am. Soc. Nephrol. 2009, 20, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-J.; Cheng, M.-F.; Ku, P.-M.; Lin, J.-W. Rosiglitazone increases cerebral klotho expression to reverse baroreflex in type 1-like diabetic rats. BioMed Res. Int. 2014, 2014, 309151. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Lin, S.; Tang, R.; Veeraragoo, P.; Peng, W.; Wu, R. Role of Fosinopril and Valsartan on Klotho Gene Expression Induced by Angiotensin II in Rat Renal Tubular Epithelial Cells. Kidney Blood Press. Res. 2010, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Karalliedde, J.; Maltese, G.; Hill, B.; Viberti, G.; Gnudi, L. Effect of renin-angiotensin system blockade on soluble Klotho in patients with type 2 diabetes, systolic hypertension, and albuminuria. Clin. J. Am. Soc. Nephrol. 2013, 8, 1899–1905. [Google Scholar] [CrossRef]
- De Borst, M.; Vervloet, M.G.; Ter Wee, P.M.; Navis, G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1603–1609. [Google Scholar] [CrossRef]
- Lim, K.; Lu, T.-S.; Molostvov, G.; Lee, C.; Lam, F.; Zehnder, D.; Hsiao, L.-L. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef]
- Lau, W.L.; Leaf, E.M.; Hu, M.C.; Takeno, M.M.; Kuro-O, M.; Moe, O.W.; Giachelli, C.M. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012, 82, 1261–1270. [Google Scholar] [CrossRef]
- Forster, R.E.; Jurutka, P.W.; Hsieh, J.-C.; Haussler, C.A.; Lowmiller, C.L.; Kaneko, I.; Haussler, M.R.; Whitfield, G.K. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochem. Biophys. Res. Commun. 2011, 414, 557–562. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Sánchez-Niño, M.D.; Donate-Correa, J.; Martín-Núñez, E.; Ferri, C.; Pérez-Delgado, N.; Górriz, J.L.; Martínez-Castelao, A.; Ortiz, A.; Mora-Fernández, C. Effects of Pentoxifylline on Soluble Klotho Concentrations and Renal Tubular Cell Expression in Diabetic Kidney Disease. Diabetes Care 2018, 41, 1817–1820. [Google Scholar] [CrossRef]
- Kang, W.-L.; Xu, G.-S. Atrasentan increased the expression of klotho by mediating miR-199b-5p and prevented renal tubular injury in diabetic nephropathy. Sci. Rep. 2016, 6, 19979. [Google Scholar] [CrossRef]
- Cho, N.-J.; Han, D.-J.; Lee, J.-H.; Jang, S.-H.; Kang, J.S.; Gil, H.-W.; Park, S.; Lee, E.Y. Soluble klotho as a marker of renal fibrosis and podocyte injuries in human kidneys. PLoS ONE 2018, 13, e0194617. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Luo, Y.; Li, Y.; Yang, Q.; Deng, X.; Wu, P.; Ma, H. Klotho gene delivery ameliorates renal hypertrophy and fibrosis in streptozotocin-induced diabetic rats by suppressing the Rho-associated coiled-coil kinase signaling pathway. Mol. Med. Rep. 2015, 12, 45–54. [Google Scholar] [CrossRef]
- Neyra, J.A.; Hu, M.C. Potential application of klotho in human chronic kidney disease. Bone 2017, 100, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Afsar, B.; Afsar, R.E. Sodium-glucose cotransporter inhibitors and kidney fibrosis: Review of the current evidence and related mechanisms. Pharmacol. Rep. 2022, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Zhou, R.; Sun, Z.F.; Long, J.W.; Gong, Y.Q. Novel Insights into the Roles and Mechanisms of GLP-1 Receptor Agonists against Aging-Related Diseases. Aging Dis. 2022, 13, 468–490. [Google Scholar] [CrossRef]
- Peng, X.; Chen, S.; Wang, Y.; Jin, M.; Mei, F.; Bao, Y.; Liao, X.; Chen, Y.; Gong, W. SGLT2i reduces renal injury by improving mitochondrial metabolism and biogenesis. Mol. Metab. 2022, 101613. [Google Scholar] [CrossRef]
- Lau, H.K.; Son, D.O.; Jin, T.; Yang, Y.; Zhang, Z.; Li, Y.; Prud’homme, G.J.; Wang, G. Combined use of GABA and sitagliptin promotes human beta-cell proliferation and reduces apoptosis. J. Endocrinol. 2021, 248, 133–143. [Google Scholar] [CrossRef]
- Mora-Fernández, C.; Sánchez-Niño, M.D.; Donate-Correa, J.; Martín-Núñez, E.; Pérez-Delgado, N.; Valiño-Rivas, L.; Fernández-Fernández, B.; Ortiz, A.; Navarro-González, J.F. Sodium-glucose co-transporter-2 inhibitors increase Klotho in patients with diabetic kidney disease: A clinical and experimental study. Biomed. Pharmacother. 2022, 154, 113677. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donate-Correa, J.; Martín-Carro, B.; Cannata-Andía, J.B.; Mora-Fernández, C.; Navarro-González, J.F. Klotho, Oxidative Stress, and Mitochondrial Damage in Kidney Disease. Antioxidants 2023, 12, 239. https://doi.org/10.3390/antiox12020239
Donate-Correa J, Martín-Carro B, Cannata-Andía JB, Mora-Fernández C, Navarro-González JF. Klotho, Oxidative Stress, and Mitochondrial Damage in Kidney Disease. Antioxidants. 2023; 12(2):239. https://doi.org/10.3390/antiox12020239
Chicago/Turabian StyleDonate-Correa, Javier, Beatriz Martín-Carro, Jorge B. Cannata-Andía, Carmen Mora-Fernández, and Juan F. Navarro-González. 2023. "Klotho, Oxidative Stress, and Mitochondrial Damage in Kidney Disease" Antioxidants 12, no. 2: 239. https://doi.org/10.3390/antiox12020239
APA StyleDonate-Correa, J., Martín-Carro, B., Cannata-Andía, J. B., Mora-Fernández, C., & Navarro-González, J. F. (2023). Klotho, Oxidative Stress, and Mitochondrial Damage in Kidney Disease. Antioxidants, 12(2), 239. https://doi.org/10.3390/antiox12020239