1. Introduction
Bone destruction is a major characteristic and severe consequence of multiple skeletal diseases, including osteoporosis and inflammatory arthritis. It significantly reduces the quality of life of these patients and increases their risk of disability [
1,
2]. Osteoporosis, a metabolic skeletal disease reflected by decreased bone mineral density, microarchitectural deterioration, and impaired bone strength, leads to an increased risk of fragility fracture. The development of osteoporosis and bone diseases is influenced significantly by aging, estrogen deficiency, and inflammation [
3,
4,
5]. The fundamental etiology of osteoporosis involves an imbalance in bone turnover, characterized by a higher rate of bone resorption mediated by osteoclasts compared to the rate of bone formation mediated by osteoblasts.
Osteoclasts, which originate from osteoclast/macrophage/dendritic common progenitor cells, are multinuclear giant cells responsible for bone degradation. This process is achieved through the secretion of acid and proteolytic enzymes, including cathepsin K, which effectively dissolve collagen and other matrix proteins during bone resorption [
6]. Osteoblasts secrete a receptor activator of nuclear factor-κB ligand (RANKL) that binds to the receptor activator of nuclear factor-κB (RANK) receptor on the surfaces of osteoclasts and their progenitors, thus promoting osteoclast differentiation and survival. The binding of RANKL and RANK recruits tumor necrosis factor receptor-associated factor 6 (TRAF6), which activates the downstream signaling of c-FOS and the nuclear factor of activated T cells, cytoplasmic 1 (NFATc1) [
7]. c-FOS is a constituent of the FOS gene family and, in conjunction with Jun proteins, constitutes the AP-1 family of heterodimeric transcription factors. The interaction between c-FOS and the promoter region of NFATc1 leads to the activation of the NFATc1 gene [
8], which is a key regulator of osteoclast-specific genes, including tartrate-resistant acid phosphatase (TRAP), H
+-ATPase, Atp6v0d2, Oscar, and Cathepsin K marker genes [
9]. The
c-Fos knockout mice and transgenic mice that over-express dominant-negative c-JUN exhibited severe osteopetrosis.
Reactive oxygen species (ROS) are essential secondary intracellular messengers which mediate many of the molecular signaling pathways, including apoptosis, differentiation, and the activation of cell signaling cascades [
10]. The involvement of free radicals and antioxidant systems in bone remodeling is of paramount importance [
11]. It is plausible that natural antioxidants exert a protective influence on osteoporosis through the regulation of ROS levels [
12]. Oxidative stress induced by ROS increases with age and/or estrogen deficiency in postmenopausal women, and can adversely affect bone homeostasis and lead to skeletal fragility [
13,
14]. In osteoclasts, ROS are important components that regulate the differentiation process [
15,
16]. The binding of RANKL to its receptor RANK leads to the activation of TRAF6, RAC1, and NADPH oxidase 1 (NOX1), resulting in an increase in intracellular ROS. Subsequently, the activation of MAPK, PI3K, and NF-κB signaling pathways occurs as a downstream event. ROS serves as the second messenger to facilitate the activation of these signaling pathways [
17,
18]. Mitochondrial ROS is also essential for hypoxia-induced osteoclast differentiation [
19].
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor that is expressed in various cell types, such as osteoblasts, osteocytes, and osteoclasts [
20]. It is increasingly acknowledged as a pivotal transcription factor that facilitates defense against electrophiles and oxidants, thereby promoting cell viability across multiple tissues [
21]. Nrf2 exhibits binding affinity towards antioxidant response elements, thereby promoting the transcriptional activation of antioxidant proteins [
22].
Nrf2 deficiency leads to an increase in the intracellular ROS level, as well as a defect in the production of numerous antioxidant enzymes and glutathione, in both osteoclast precursors and osteoblast progenitor cells [
23]. Exposure of RAW 264.7 macrophages to RANK ligands lowers the Nrf2/Keap1 ratio and leads to a decrease in the expression of Nrf2-dependent enzymes, which are in favor of ROS signaling [
24].
Nrf2 deficiency promotes the RANKL-induced activation of MAPK, including c-Jun N-terminal kinase, extracellular signal-regulated kinase, p38, and the consequent induction of NFATc1, a pivotal determinant of osteoclast differentiation [
25,
26]. Our previous studies found that
Nrf2 deficiency aggravates the increase in osteoclastogenesis and bone loss induced by inorganic arsenic [
27]. Despite the extensive research conducted on the association between Nrf2 and osteoclast differentiation, further investigation is required in order to elucidate the precise mechanism by which Nrf2 influences the process of osteoclast differentiation.
While prior research suggests that ROS play a crucial role as secondary messengers in the differentiation of osteoclasts [
15,
16], there is a dearth of information concerning the impact of ROS on the population dynamics of osteoclasts in cells lacking
Nrf2, particularly with respect to the contribution of c-FOS to the augmentation of osteoclast differentiation resulting from
Nrf2 deficiency. The underlying mechanisms by which
Nrf2 deficiency enhances osteoclastogenesis have not yet been fully elucidated. In this study, we examined the role of ROS and c-FOS in osteoclastogenesis in the absence of
Nrf2 using bone-marrow-derived macrophages (BMMs) and RAW 264.7 cells, and further identified the regulatory role of Nrf2 in c-FOS and NFATc1 activation.
2. Materials and Methods
2.1. Reagent
N-acetylcysteine (NAC, HY-B0215), diphenyleneiodonium chloride (DPI, HY-100965), mitoquinone mesylate (MitoQ, HY-100116A), blastcidin (HY-103401), and puromycin (HY-B1743A) were purchased from MedChemExpress (Shanghai, China). 2′,7′-dichlorofluorescein diacetate (DCFH-DA, S0033) and a BCA protein assay kit (P0027) were purchased from Beyotime Biotechnology (Shanghai, China). MitoSOX (M36008) was purchased from Thermo Fisher Scientific (Wilmington, NC, USA). Rhodamine phalloidin (KGMP001) and DAPI (KGR0001) were purchased from KeyGEN BioTECH (Nanjing, China). The mouse leukemic monocyte/macrophage cell line RAW 264.7 was purchased from American Type Culture Collection, ATCC (Manassas, VA, USA). Dulbecco’s modified essential media (DMEM), minimum essential medium alpha medium (α-MEM), fetal bovine serum (FBS), phosphate-buffered saline (PBS, pH 7.4), and supplements for cell culture were purchased from Life Technologies (Grand Island, NY, USA). Macrophage colony-stimulating factor (M-CSF, 416-ML-010) and RANKL (462-TEC-010) were purchased from R&D Systems (Minneapolis, MN, USA). Acid phosphatase kits (387A) were purchased from Sigma (St. Louis, MO, USA). Trizol reagent, SYBR mix, and iScript cDNA Supermix were purchased from TaKaRa (Dalian, China). The primers were designed using Primer Express (Applied Biosystems, Waltham, MA, USA) and synthesized by Life Technologies (Shanghai, China). Antibodies for NFATc1 (sc-7294), β-ACTIN (sc-47778), and p-c-FOS (sc-81485) were purchased from Santa Cruz Biotechnology (Santa Clara, CA, USA). Antibodies for GCLC (12601), GCLM (14241), and HO-1 (10701) were purchased from Proteintech (Rosemont, IL, USA).
2.2. Bone-Marrow-Derived Macrophages (BMMs) Extraction
Nrf2-wildtype (
Nrf2+/+) and global
Nrf2 knockout (
Nrf2−/−) littermate mice in C57BL/6 background were generated by crossing
Nrf2-heterozygous (
Nrf2+/−) mice, which were kindly provided by Dr. Masayuki Yamamoto (Tohoku University, Japan). Genotyping was performed using genomic DNA that was isolated from tail snips, as mentioned previously [
27]. Mice were raised in a pathogen-free environment, with a diet of SPF grade and sterilized, purified drinking water. The room’s temperature was 23 ± 1 °C, the humidity was 55–70%, and a regular light/dark cycle was used (12 h day/12 h night). All animal experiments in this study were approved by the Institutional Animal Care and Use Committee of China Medical University (Shenyang, China), following all the current guidelines for animal care and welfare.
Cervical dislocation was used to euthanize the mice (8 weeks old, male), and they were immersed in 75% alcohol for 5 min for disinfection and sterilization. The bilateral femur and tibia were removed and collected. The long shaft was placed in a 4 mL centrifuge tube and transferred to a ventilation cabinet hood to ensure sterile operation for the remaining experiments. After the muscle and other tissues attached to long bones were carefully removed, epiphyses were cut off. Bone marrow was flushed out repeatedly using a 1 mL sterile syringe containing α-MEM culture medium until bone tissue turned white. The collected bone marrow was filtered using a 70 μm filter screen to remove the bone and muscle tissue residue in the culture medium. The sample was then centrifuged at 800 rpm for 5 min, and the supernatant was discarded. The bone marrow was re-suspended in 3 mL of culture medium and cultured in a 6 cm dish at 37 °C for 24 h. Then, the suspended cells were harvested and further cultured with 30 ng/mL M-CSF in α-MEM containing 10% (v/v) of heat-inactivated fetal bovine serum (HI-FBS). After 3 days of culture, the adherent cells were used for osteoclast differentiation studies.
2.3. Cell Culture
The mouse leukemic monocyte/macrophage cell line, RAW 264.7, was cultured in DMEM supplemented with 10% HI-FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 10 mmol/L HEPES at 37 °C with 5% CO
2. Scramble (Scr) and
Nrf2-knockdown (
Nrf2-KD) RAW 264.7 cells were established via MISSION shRNA lentiviral transduction (mouse Nrf2 (SHVRSNM_008686, Sigma, USA) or scrambled nontarget negative control (SHC002V, Sigma, USA) as described in [
27]. The lentiviral transfer vectors encoding mouse
Nrf2 were developed as detailed previously [
28]. The lentiviral transfer vectors encoding mouse
Nrf2 were constructed by cloning PCR-generated fragments into the lentiviral vector pLVX-IRES-NEO (PP2374, St. Louis, MO, USA) and pTK642, a gift from Dr. Tal Kafri of the University of North Carolina at Chapel Hill. Following transfection, the
Nrf2 overexpression
(Nrf2-OE) cells were selected and maintained in culture medium containing 2 μg/mL blastcidin. In the current study, we define cells transfected with negative control lentiviral vectors as controls (Cont) in contrast to the
Nrf2-OE.
2.4. TRAP Staining
Bone-marrow-derived OPCs (osteoclast precursor cells) and osteoclasts derived from RAW 264.7 cells were stained for the activity of TRAP to confirm the osteoclasts. The RAW 264.7 cell line was cultured in 24-well plates at a density of 1.5 × 104 cells per well, and primary bone marrow hematopoietic stem cells were evenly distributed in 24-well plates at a density of 8 × 104 cells per well. The culture medium, supplemented with 50 ng/mL RANKL and 30 ng/mL M-CSF, was refreshed every alternate day for a duration of 5 days. TRAP staining was then performed using an acid phosphatase kit according to the manufacturer’s instructions. The images of TRAP staining were captured using a microscope (DMi8, Lecia, Wetzlar, Germany). Analyses of the osteoclasts were implemented using ImageJ software (1.50i, NIH, Bethesda, MD, USA).
2.5. Quantitative Real-Time RT-PCR
Total RNA was extracted using the Trizol reagent following the manufacturer’s recommendations. Concentration of RNA was quantified by Nanodrop 2000 (Thermo, Wilmington, DE, USA). Reverse-transcription to cDNA (50 ng per sample) was carried out with iScript cDNA Supermix. Quantitative RT-PCR was performed using a reaction mixture containing SYBR mix, and real-time fluorescence was detected by QuantStudio 6 Flex (ABI, Foster City, CA, USA). The following sets of primers were used: Cathepsin K (5′-TGTATAACGCCACGGCAAA-3′ and 5′-GGTTCACATTATCACGGTCACA-3′), H+-atpase (5′-ACGGTGATGTCACAGCAGACGT-3′ and 5′-CCTCTGGATAGAGCCTGCCGCA-3′), Apt6v0d2 (5′-GAAGCTGTCAACATTGCAGA-3′ and 5′-TCACCGTGATCCTTGCAGAAT-3′), and Oscar (5′-CTGCTGGTAACGGATCAGCTCCCCAGA-3′ and 5′-CCAAGGAGCCAGAACCTTCGAAACT-3′), which encode for proteins that are key to extracellular matrix degradation and bone resorption. Nrf2 (5′-CGAGATATACGCAGGAGAGGTAAGA-3′ and 5′-GCTCGACAATGTTCTCCAGCTT-3′), Kelch-like ECH-associated protein-1, Keap1 (5′-GTGGCCGTCACCATGGA-3′ and 5′-GCTTCAGCAGGTACAGTTTTGTTG-3′), glutamate cysteine ligase catalytic subunit, Gclc (5′-TGGCCACTATCTGCCCAATT-3′ and 5′-GTCTGACACGTAGCCTCGGTAA-3′), glutamate cysteine ligase modifier subunit, Gclm (5′-ACATTGAAGCCCAGGATTGG-3′ and 5′-CCCCTGCTCTTCACGATGAC-3′), Heme oxygenase-1, Ho-1 (5′-CCTCACTGGCAGGAAATCATC-3′ and 5′-CCTCGTGGAGACGCTTTACATA-3′), NAD(P)H: quinine oxidoreductase-1, Nqo1 (5′-TATCCTTCCGAGTCATCTCTAGCA-3′ and 5′-TCTGCAGCTTCCAGCTTCTTG-3′) were also used. β-actin (5′-GTATGACTCCACTCACGGCAAA-3′ and 5′-GGTCTCGCTCCTGGAAGATG-3′) was used as the internal control in all cases.
2.6. Flow Cytometry
Intracellular ROS levels were measured using a DCFH-DA or MitoSOX fluorescent probe. RAW 264.7 cells were harvested and washed twice with PBS. The cells were then incubated for 30 min in PBS containing 5 μM DCFH-DA or 3 μM MitoSOX. After being washed with PBS, the cells were suspended in PBS and intracellular ROS production was measured by flow cytometry (Becton Dickinson FACSCanto II, Becton Dickinson, San Jose, CA, USA) using excitation/emission wavelengths of 488/525 nm for DCFH-DA and 396/610 nm for MitoSOX. FlowJO software (10.8.1) was used to analyze ROS levels.
2.7. MitoSOX Red and DCFH-DA Staining
RAW 264.7 cells were grown in 24-well plates at a density of 1 × 104/well overnight in the incubator until the cells reached 70–80%. The preparation of the DCFH-DA working solution occurred as follows: DCFH-DA and MitoSOX was diluted with serum-free medium for final concentrations of 5 μM or 4 μM, respectively. Serum-free medium, PBS, DCFH-DA, and MitoSOX working solution were pre-warmed in a 37 °C water bath before the experiment started; the 24-well plates containing cells were removed from the incubator and the medium was gently aspirated along the plate wall. The plate was washed twice with serum-free medium, gently, without touching the cells; next, 300 μL of DCFH-DA and/or MitoSOX working solution was added to each well. The incubation was continued at 37 °C for 30 min. The working solution was gently aspirated and washed twice with serum-free medium. Then, 300 μL of culture medium containing RANKL, NAC, DPI, or MitoQ was added to each well before being incubated again at 37 °C for 30 min. The treatment solution was discarded and washed once with PBS; 500 μL of PBS was added to each well again, and the cells were collected with a cell scraper and transferred into flow-through special glass tubes for the flow-through experiments.
2.8. Western Blot Analysis
RAW 264.7 cells were rinsed with pre-cooled PBS and lysed by 1× lysis buffer that contained 1 mM phenylmethylsulfonyl fluoride (PMSF). The protein concentration was quantified using a BCA protein assay kit. Next, 50 μg of protein was separated by SDS–PAGE and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 3% BSA in Tris-buffered saline (TBS) containing 0.05% Tween-20 for 1 h. After blocking, the membranes were incubated with the corresponding primary antibodies overnight at 4 °C. NFATc1, GCLC, GCLM, HO-1, p-c-FOS, and β-ACTIN were used. Membranes were washed by 1× TBST, followed by incubation with anti-rabbit IgG-HRP for 1 h. Immunoreactive bands were visualized using Tanon 5500 (Tanon, Shanghai, China). The relative levels of the target proteins were normalized by β-ACTIN.
2.9. F-ACTIN Staining of Osteoclasts
RAW 264.7 cells were seeded in 6-well plates at 2 × 104/well, then put back into the incubator to continue to culture for 24 h. The culture medium containing 50 ng/mL RANKL and 30 ng/mL M-CSF was changed every other day for 5 days. Cells were washed using PBS and fixed with 4% paraformaldehyde for 15 min, permeabilized in 0.1% Triton X-100 for 5 min, and subsequently stained with rhodamine phalloidin (20 min) and DAPI (5 min). After washing with PBS, representative images of F-ACTIN belt formation were captured using a fluorescence microscope (DMi8, Lecia, Germany).
2.10. Statistics
All experiments were repeated at least three times. The normality of the data was evaluated using the Shapiro–Wilk test, while the similarity between variances was tested using the Brown–Forsythe test. In cases where the data exhibited normal distribution and homogeneity of variance, Student’s t-tests were conducted for comparisons between two groups. For comparisons among multiple groups, a one-way or two-way ANOVA with Bonferroni post hoc tests was employed. The Kruskal–Wallis test was used for data with a non-normal distribution and/or non-homogeneity of variance, followed by Dunn’s multiple comparisons test. The data are represented as the mean (m) ± standard deviation (SD) values of independent replicates. All statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA).
4. Discussion
Numerous reports have demonstrated a significant association between oxidative stress and osteoporosis in the general population. However, the precise mechanism underlying this relationship, particularly the impact of advanced age and estrogen deficiency on osteoclast differentiation, remains elusive. While a substantial body of literature suggests that ROS may serve as a secondary messenger in osteoclast differentiation, further investigation is required in order to elucidate the role of abnormally elevated ROS in individuals with osteoporosis. Consequently, this study aims to examine the influence of ROS on osteoclast differentiation. In this study, we found that a deficiency of
Nrf2 in BMMs and RAW 264.7 cells resulted in elevated osteoclast differentiation, where overexpression of
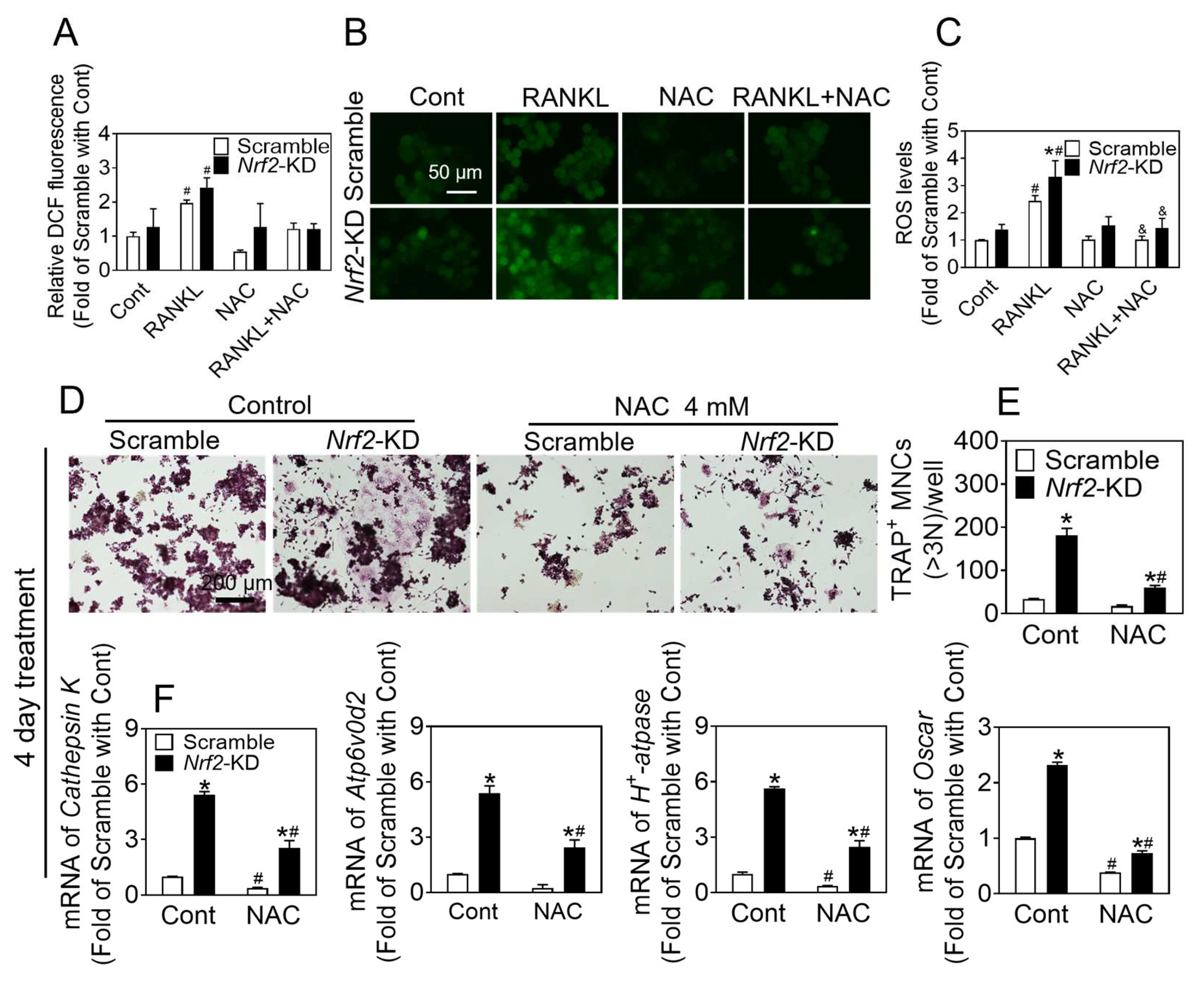
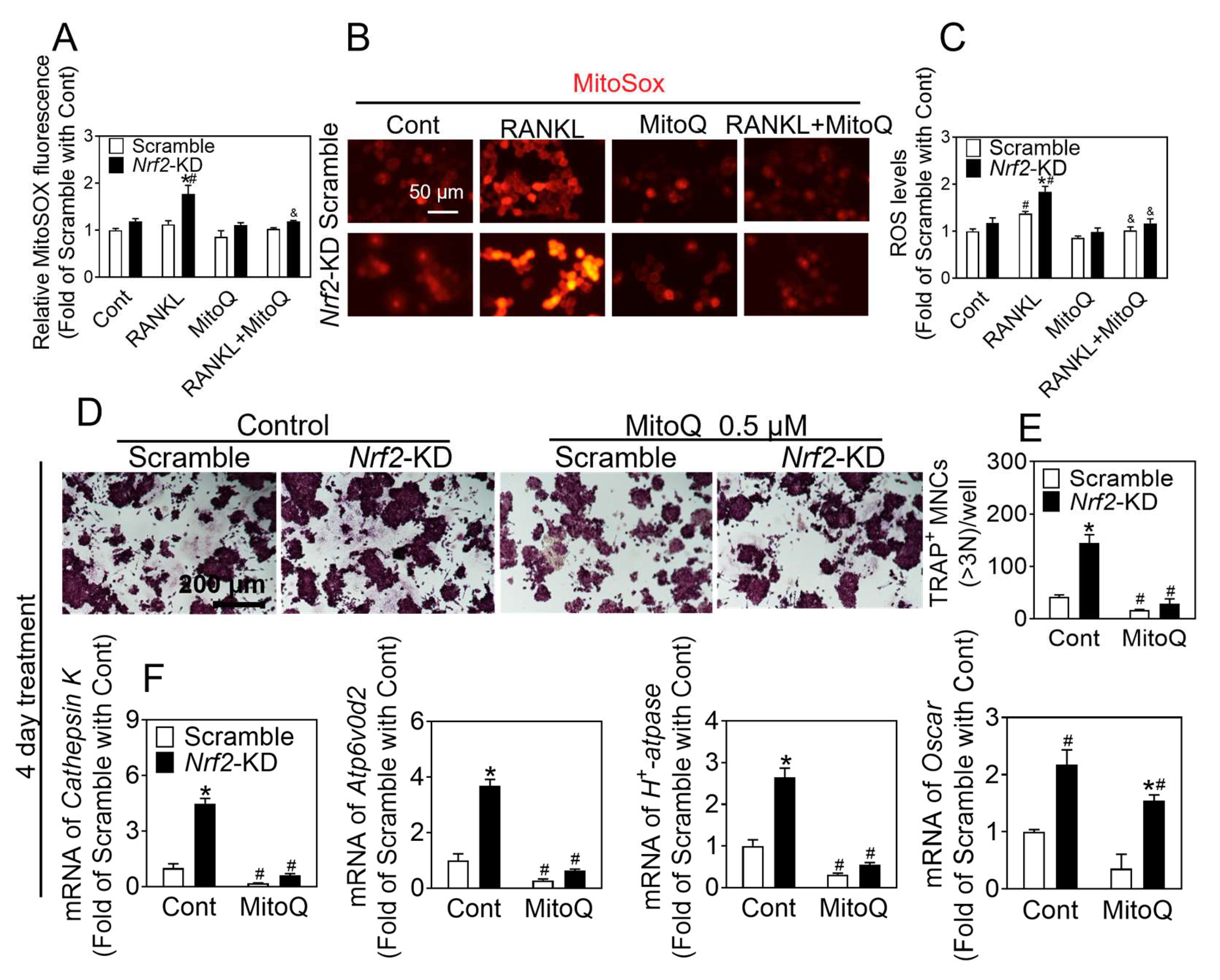
Nrf2 in RAW 264.7 cells inhibited osteoclast differentiation. Analysis of the intracellular ROS level in response to RANKL treatment revealed that the administration of RANKL quickly resulted in a peak of ROS levels within the cytoplasm at the 15 min mark. The treatment of RANKL for 24 and 48 h led to an elevation in both cytoplasmic and mitochondrial ROS levels.
Nrf2 deficiency increased the sensitivity of BMMs and RAW 264.7 to RANKL-induced ROS production. Treatment with ROS inhibitors such as NAC, DPI, and MitoQ significantly blocked osteoclast differentiation, which was elevated by
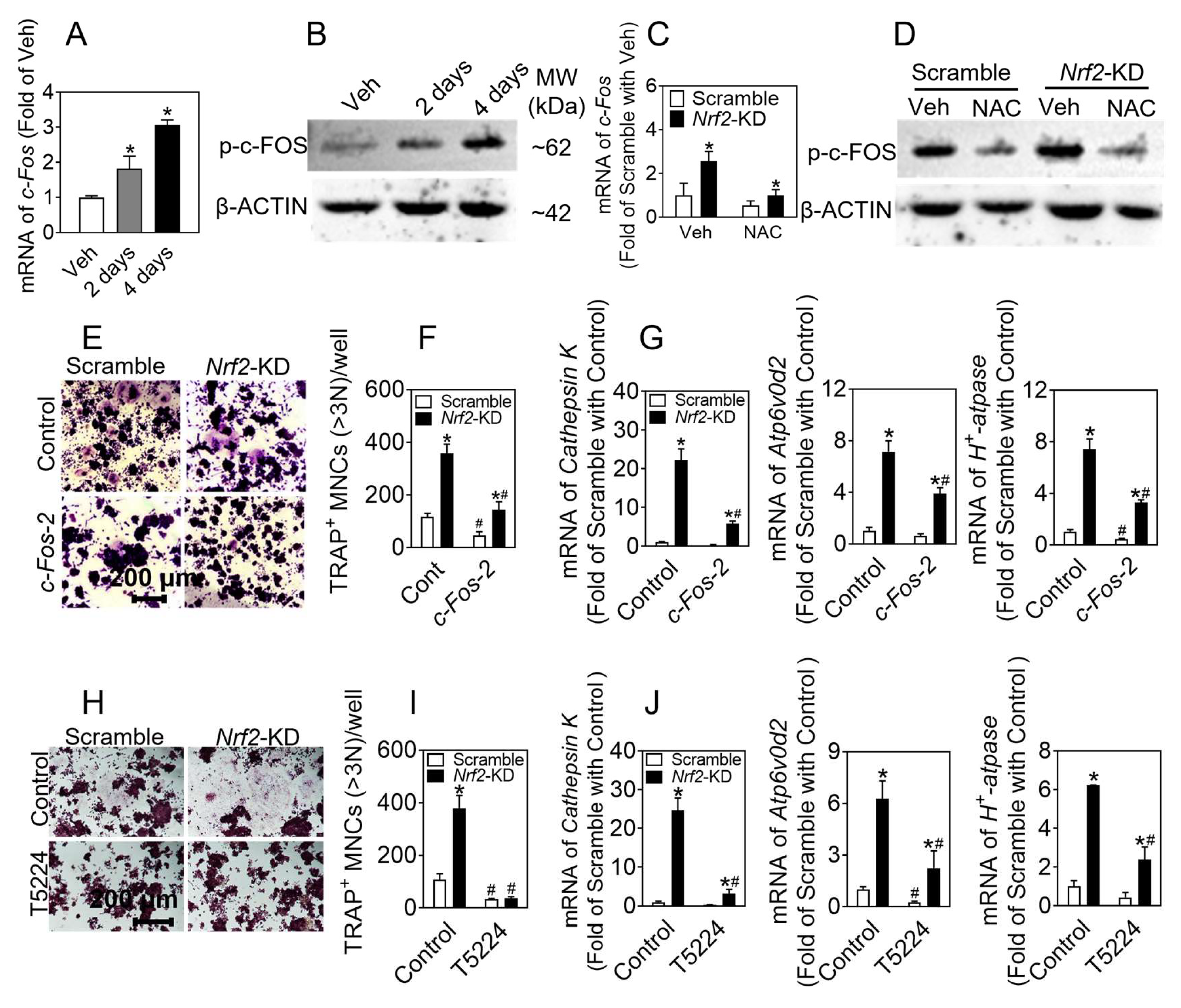
Nrf2 deficiency. During the process of osteoclast differentiation, an upregulation of c-FOS expression was noted, with a more pronounced effect observed in cells lacking
Nrf2. Additionally, the reduction in c-FOS levels hindered the process of osteoclastic differentiation. Moreover, it was discovered that antioxidants effectively suppressed the abnormal elevation of c-FOS levels induced by
Nrf2 deficiency, subsequently impeding osteoclast differentiation (
Figure 7). The data presented in this study offer compelling evidence that Nrf2 plays a crucial role in osteoclast differentiation through the modulation of ROS and c-FOS.
Nrf2, a redox-sensitive transcription factor, plays a crucial role in providing adaptive protection against oxidative stress through the activation of various cytoprotective genes. Nrf2 has been found to be linked to age-related bone loss, specifically senile osteoporosis, as well as postmenopausal osteoporosis. The administration of the Nrf2 agonist dimethyl fumarate has been observed to impede the progression of osteoporosis in ovariectomized mice [
29]. Consequently, an imbalance in this mechanism leads to an elevation in oxidative stress and a decrease in Nrf2 levels. Previous studies have demonstrated that the absence of
Nrf2 contributes to the differentiation of osteoclasts induced by oxidative stress. Therefore, in order to mitigate the development of osteoporosis by reducing the levels of ROS, the Nrf2-mediated regulation of antioxidant enzymes such as
Ho-1,
Nqo1,
Gclc, and
Gclm should be taken into account [
30]. During osteoclast differentiation, our study revealed that treatment with RANKL and M-CSF resulted in a notable decrease in Nrf2 expression. Furthermore, the absence of
Nrf2 in both RAW 264.7 cells and BMMs augmented the osteoclast differentiation induced by RANKL and M-CSF. Conversely, the overexpression of
Nrf2 in RAW 264.7 cells hindered the osteoclast differentiation. Additionally, our findings demonstrated that the deficiency of
Nrf2 during osteoclast differentiation led to a reduction in the expression levels of antioxidant enzymes. Moreover, the levels of ROS in the cytoplasm and mitochondria of osteoclasts increased, with a more pronounced increase observed in osteoclasts lacking
Nrf2. The above results suggest that osteoclast differentiation can be promoted by decreasing intracellular Nrf2 levels, and when
Nrf2 is absent, the level of antioxidant enzymes mediated by
Nrf2 decreases, leading to enhanced osteoclast differentiation.
Oxidative stress frequently occurs as a pathological condition resulting from estrogen deficiency, aging, hyperglycemia, and hyperlipemia [
31]. The redox state changes are also related to the bone remodeling process, which allows for continuous bone regeneration [
32,
33]. In osteoclast differentiation, redundant ROS in cells can trigger many disorders, including inflammation, aging, metabolic disturbance, and osteoporosis. Oxidative stress can activate the differentiation of pre-osteoclasts to osteoclasts and increase bone resorption [
34,
35]. The regulation of redox status during osteoclast differentiation plays an important role in maintaining bone homeostasis [
36]. A plethora of natural plant active compounds have been found to be capable of inhibiting osteoclast-specific marker genes, including transcription factors such as c-FOS, NFATc1, and c-Src. These compounds also counteract the effects of local factors, such as ROS and NO, while suppressing the activation of diverse signaling pathways, such as MAPK and NF-κB, thereby impeding osteoclast differentiation [
31]. Mitochondrial ROS are essential for the hypoxic enhancement of osteoclast differentiation. In osteoclast differentiation, TRAF6 plays a key linkage role in ROS production by RANKL. Rac1 and NOX form the sequential order of the signaling cascade of ROS production. In this study, it was observed that RANKL stimulation led to a dose-dependent increase in ROS production. The levels of ROS rapidly reached their peak at approximately 15 min and subsequently declined after 30 min in the cytoplasm. Additionally, the levels of ROS in both the cytoplasm and the mitochondria increased after 24 and 48 h of RANKL treatment. Notably, the
Nrf2-silenced group exhibited higher levels of ROS production compared to the scramble group in response to RANKL treatment. These findings provide clear evidence that
Nrf2 deficiency enhances RANKL-induced ROS production, highlighting the crucial role of Nrf2 in osteoclastogenesis.
NAC has been shown to significantly reduce ROS concentrations. DPI, a non-reversible inhibitor of flavoprotein, NOX, and xanthine oxidase, has also been found to decrease ROS levels. Additionally, MitoQ, a mitochondria-targeted antioxidant, has been demonstrated to inhibit the production of ROS within the mitochondria. The levels of intracellular ROS directly impact RANKL-induced osteoclast differentiation and bone density. Antioxidants such as NAC, DPI, and MitoQ effectively suppress osteoclastogenesis by scavenging ROS activity. In this study, it was observed that the rapid augmentation in ROS production induced by RANKL reached its maximum level at approximately 15 min. Pretreatment with NAC and DPI demonstrated significant inhibition of the transient rise in ROS levels caused by Nrf2 deficiency in response to RANKL treatment. However, these pretreatments did not exhibit a significant inhibitory effect on the promotion of osteoclast differentiation caused by Nrf2 deficiency. These findings suggest that the transient elevation of ROS resulting from RANKL treatment does not exert any influence on the process of osteoclast differentiation. The detection of ROS levels was conducted using DCFH-DA and MitoSOX. The results revealed increased ROS levels in both the cytoplasm and mitochondria following 48 h of RANKL treatment. To investigate the impact of antioxidants (NAC, DPI, and MitoQ) on osteoclast differentiation, the cells were treated with these compounds for the first two days and throughout the differentiation process. It was observed that NAC, DPI, and MitoQ effectively inhibited osteoclast differentiation. These findings suggest that Nrf2 deficiency enhances the accumulation of RANKL-induced oxidative stress during osteoclastogenesis, thereby promoting osteoclast differentiation. Hence, the utilization of Nrf2 as a therapeutic target to impede osteoclast differentiation can be achieved through the activation of Nrf2 and the augmentation of intracellular antioxidant enzymes, while concurrently suppressing intracellular ROS levels.
c-FOS belongs to the FOS gene family, including c-FOS, FosB, Fra-1, and Fra-2, which is a component of the AP-1 family of transcriptional activators. RANKL binds to RANK, resulting in the recruitment of TRAF6 in the cytoplasm, then triggers the downstream NF-κB and MAPK signaling, which subsequently activates transcription factors c-FOS and NFATc1. This enhances the expression of osteoclastogenesis-related markers, eventually leading to the differentiation and maturation of osteoclasts [
37]. The RANKL signaling pathway has the potential to induce the activation of the c-FOS signaling pathway through the elevation of intracellular ROS levels, thereby facilitating the process of osteoclast differentiation. Further investigation is warranted to elucidate the precise influence of intracellular ROS levels on c-FOS expression during osteoclast differentiation, as well as the role of Nrf2 in this process. In the preceding findings of this investigation, it was observed that the deficiency of
Nrf2 had a more pronounced impact on the elevation of ROS levels induced by RNAKL treatment. Additionally, the protein levels of c-FOS generated during differentiation were significantly inhibited by NAC treatment in
Nrf2-deficient cells. The results of this investigation indicate that the lack of
Nrf2 may exert an influence on osteoclast differentiation by increasing the levels of ROS in both the cytoplasm and mitochondria, thereby affecting the expression of c-FOS within the cells. To further investigate the role of c-FOS in osteoclast differentiation, we conducted experiments involving the silencing of
c-Fos and
Nrf2 to induce this process. Our findings indicate that the absence of
c-Fos hinders the promotion of osteoclast differentiation caused by
Nrf2 deletion. Additionally, the compound T5224, known for its specific inhibition of c-FOS, effectively impedes the increase in osteoclast differentiation that results from
Nrf2 deletion. The presented data indicate that c-FOS significantly contributes to the promotion of osteoclast differentiation in cells lacking
Nrf2, thereby suggesting that c-FOS serves as a prominent downstream target of Nrf2 in osteoclasts.
Our current study has several limitations. Firstly, osteoclast differentiation involves multiple signaling pathways, including NF-κB and MAPK. Therefore, it is necessary to conduct further analysis on the impact of increased ROS levels resulting from Nrf2 deficiency on these signaling pathways at different stages of differentiation. Secondly, our study primarily relied on in vitro experiments, and additional evidence from in vivo experiments is required in order to gain a deeper understanding of the role of Nrf2 in osteoclast differentiation. Furthermore, it is imperative to identify specific activators of Nrf2 in forthcoming research endeavors, as this will establish a theoretical framework for the prevention and treatment of osteoporosis.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}