

Actin Polymerization Defects Induce Mitochondrial Dysfunction in Cellular Models of Nemaline Myopathies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Patients and Cell Culture
2.3. Immunoblotting
2.4. Real-Time Quantitative PCR (qPCR)
2.5. Active RhoA Assay
2.6. Immunofluorescence Microscopy
2.7. Immunofluorescence Staining of Cytoskeletal F-Actin
2.8. Bioenergetics and Oxidative Stress Analysis
2.9. Mitotracker Staining: Analysis of Mitochondrial Network
2.10. Measurement of Intracellular Reactive Oxygen Species (ROS) Generation
2.11. Statistics
3. Results
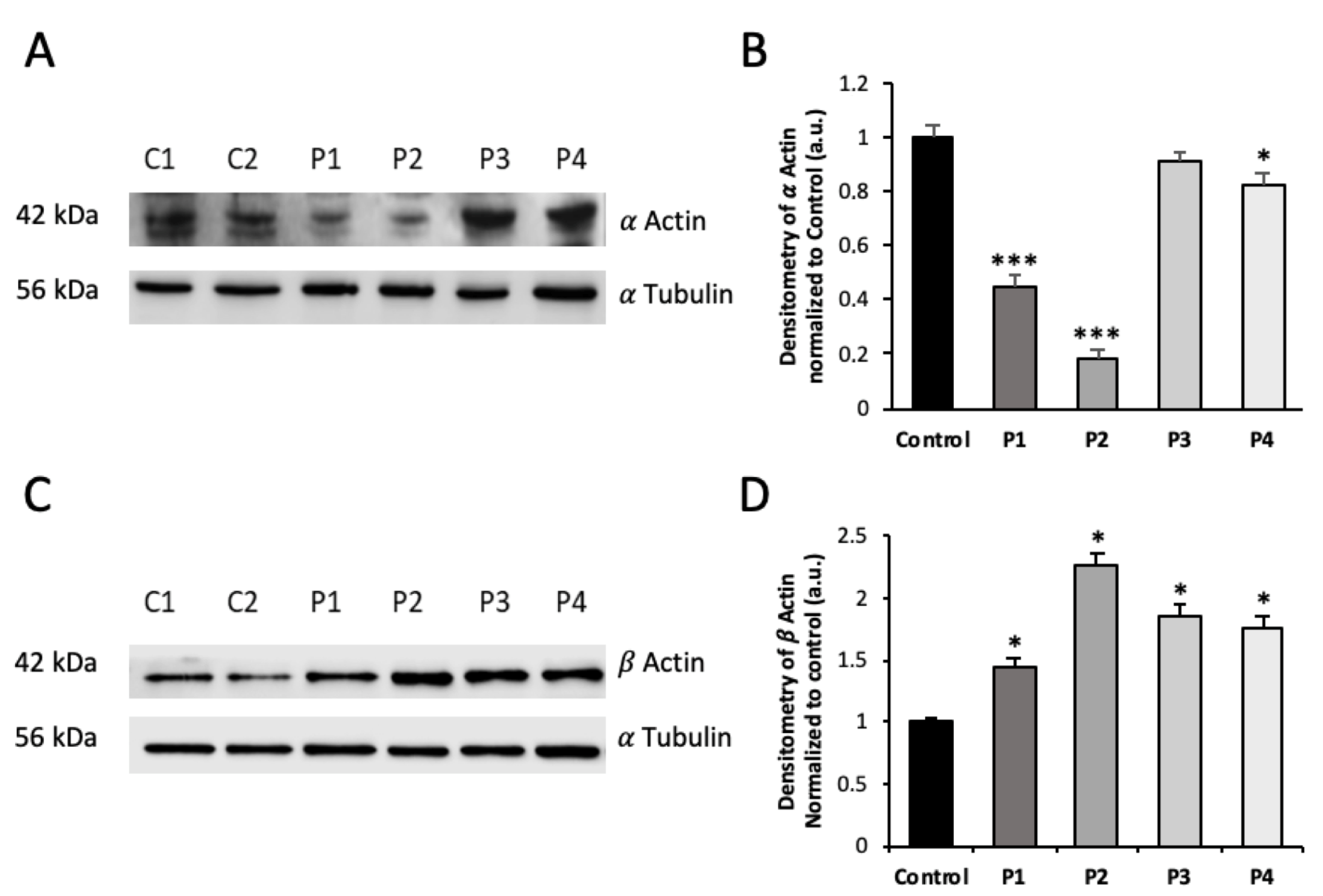
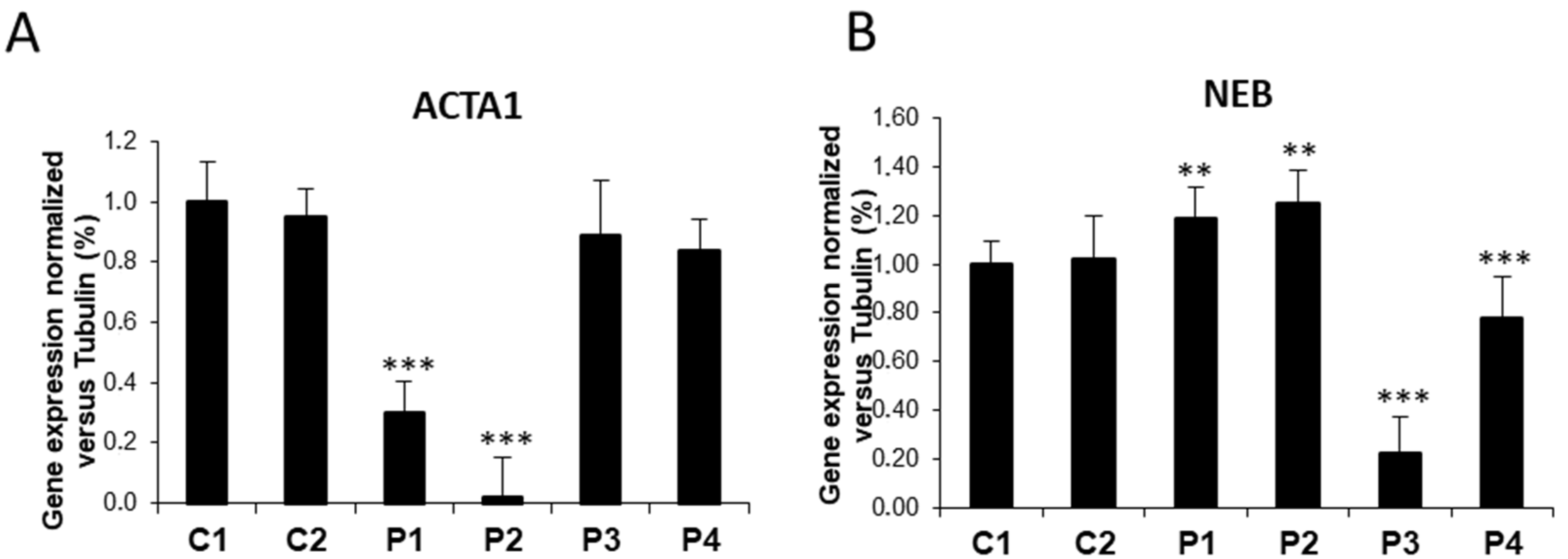
3.1. NM Fibroblasts Present Alterations in Actin Alpha 1 (ACTA1) and Nebulin (NEB) Expression Levels
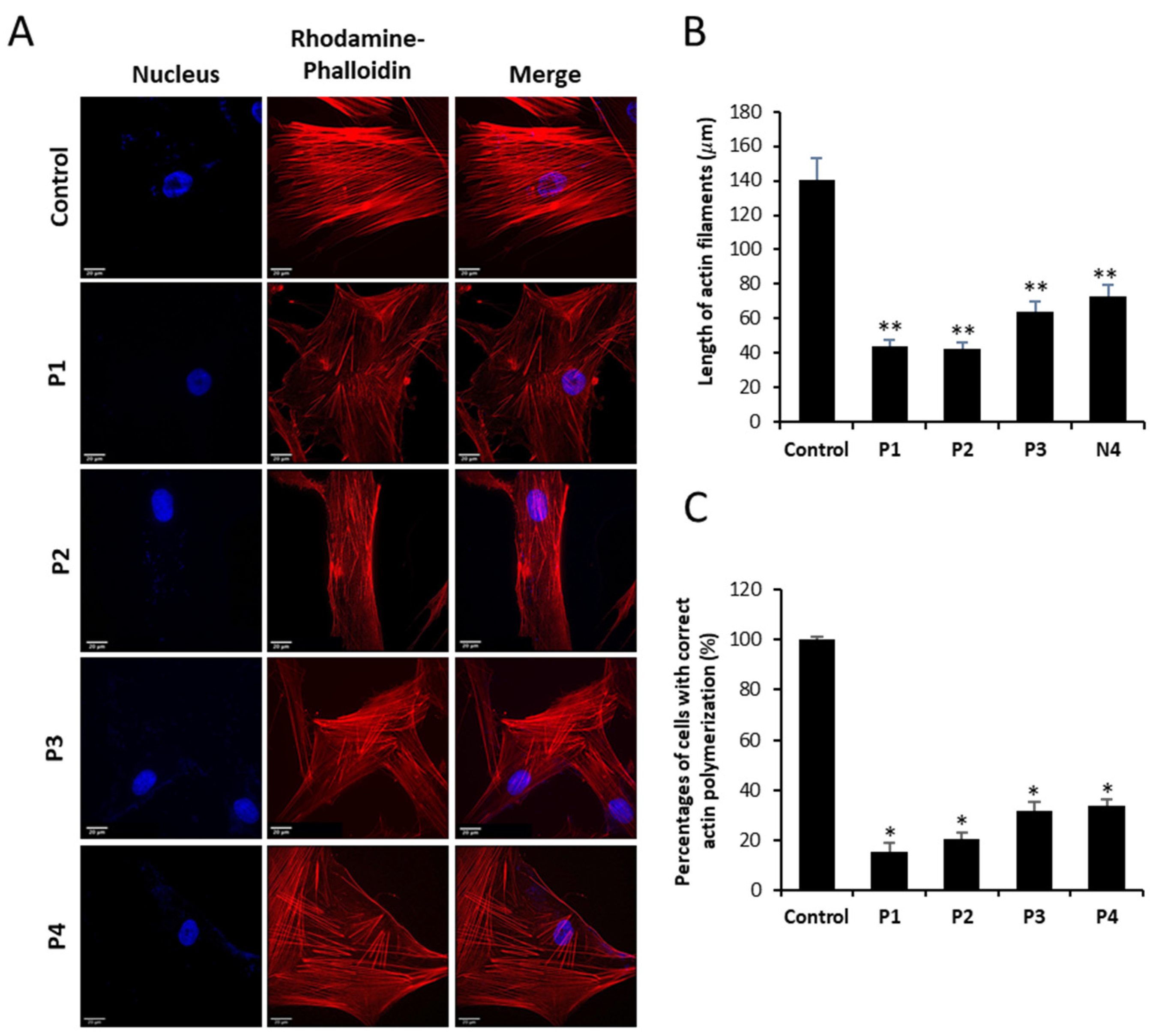
3.2. NM-Patient-Derived Fibroblasts Show Defects in Actin Filament Polymerization
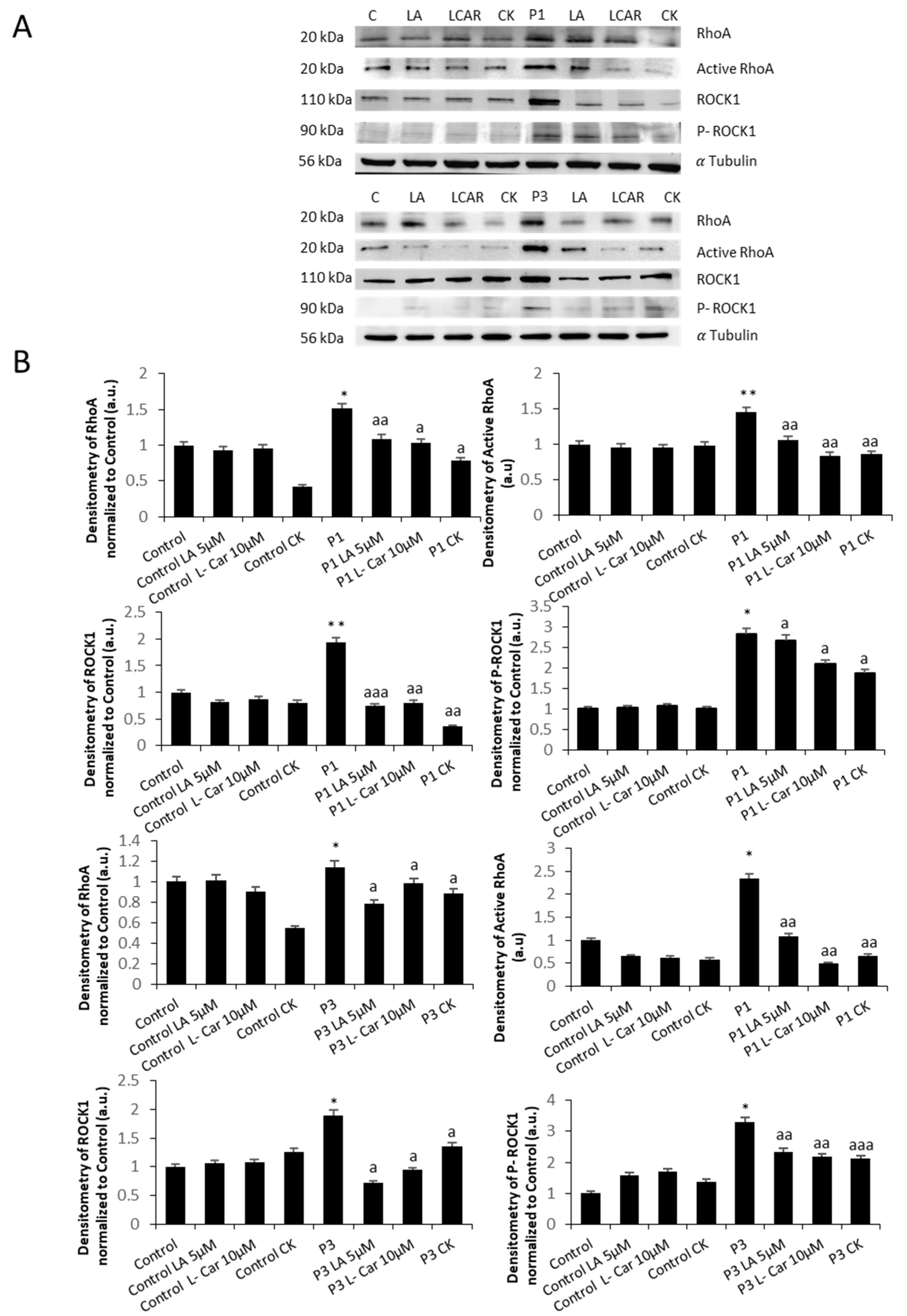
3.3. RhoA/ROCK Pathway Is Overactivated in NM Fibroblasts
3.4. NM Fibroblasts Display Alterations in Mitochondrial Bioenergetics and Network Morphology
3.5. NM-Patient-Derived Fibroblasts Present Increased ROS and Alterations in Antioxidant Enzyme Expression Levels
3.6. Supplementation with Linoleic Acid (LA) and L-Carnitine (LCAR) Restores Actin Polymerization
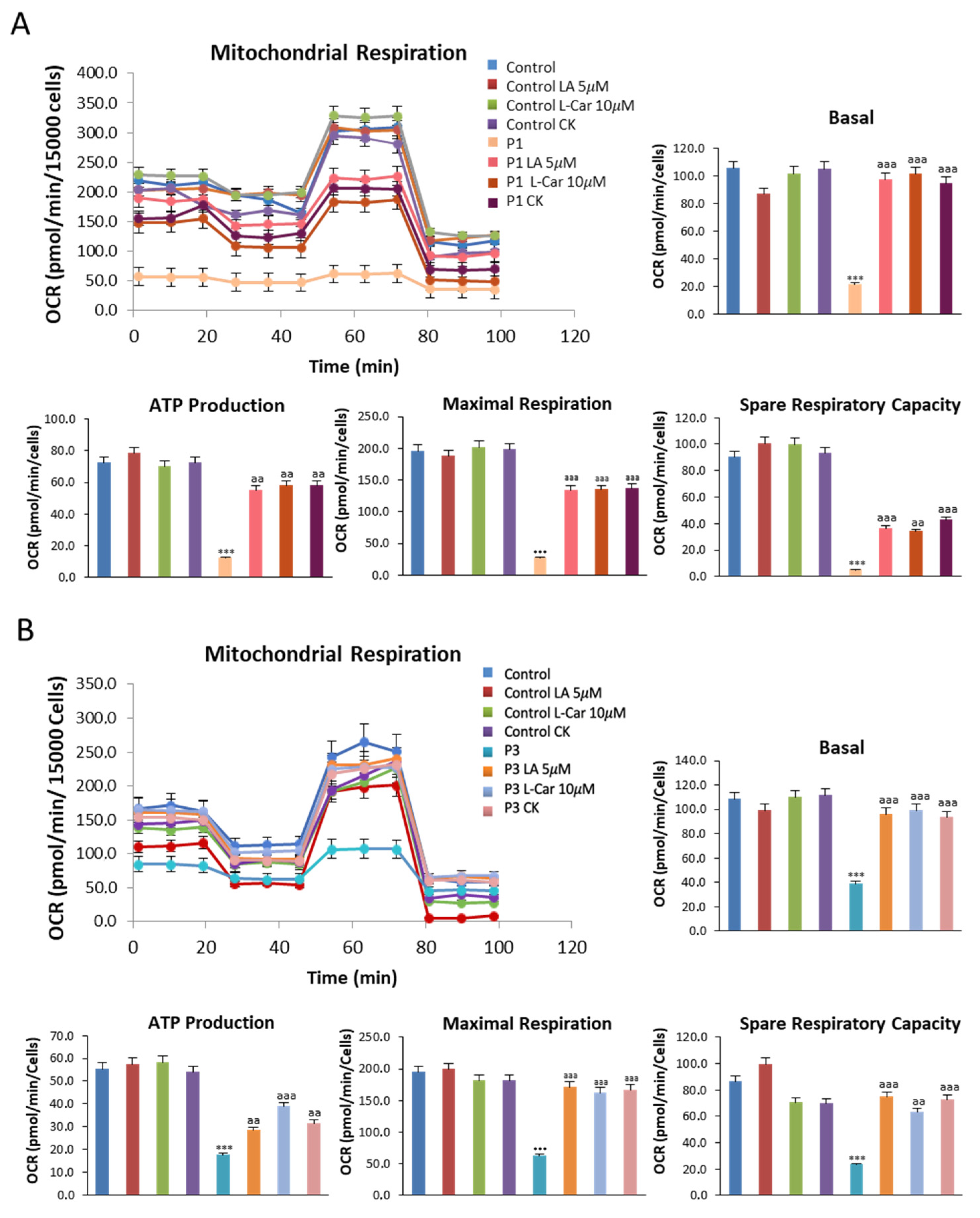
3.7. Supplementation with LA and LCAR Improves Mitochondrial Bioenergetics
3.8. Supplementation with LA and LCAR Corrects RhoA/ROCK Pathway Overactivation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- North, K.N.; Wang, C.H.; Clarke, N.; Jungbluth, H.; Vainzof, M.; Dowling, J.J.; Amburgey, K.; Quijano-Roy, S.; Beggs, A.H.; Sewry, C.; et al. Approach to the diagnosis of congenital myopathies. Neuromuscul. Disord. NMD 2014, 24, 97–116. [Google Scholar] [CrossRef]
- Shy, G.M.; Engel, W.K.; Somers, J.E.; Wanko, T. Nemaline Myopathy. A New Congenital Myopathy. Brain 1963, 86, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Sanoudou, D.; Beggs, A.H. Clinical and genetic heterogeneity in nemaline myopathy—A disease of skeletal muscle thin filaments. Trends Mol. Med. 2001, 7, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Wallgren-Pettersson, C.; Beggs, A.H.; Laing, N.G. 51st ENMC International Workshop: Nemaline Myopathy. 13-15 June 1997, Naarden, The Netherlands. Neuromuscul. Disord. NMD 1998, 8, 53–56. [Google Scholar] [PubMed]
- Ryan, M.M.; Schnell, C.; Strickland, C.D.; Shield, L.K.; Morgan, G.; Iannaccone, S.T.; Laing, N.G.; Beggs, A.H.; North, K.N. Nemaline myopathy: A clinical study of 143 cases. Ann. Neurol. 2001, 50, 312–320. [Google Scholar] [CrossRef]
- Gatayama, R.; Ueno, K.; Nakamura, H.; Yanagi, S.; Ueda, H.; Yamagishi, H.; Yasui, S. Nemaline myopathy with dilated cardiomyopathy in childhood. Pediatrics 2013, 131, e1986–e1990. [Google Scholar] [CrossRef]
- Ogasawara, M.; Nishino, I. A review of major causative genes in congenital myopathies. J. Hum. Genet. 2023, 68, 215–225. [Google Scholar] [CrossRef]
- Romero, N.B.; Sandaradura, S.A.; Clarke, N.F. Recent advances in nemaline myopathy. Curr. Opin. Neurol. 2013, 26, 519–526. [Google Scholar] [CrossRef]
- Pappas, C.T.; Bliss, K.T.; Zieseniss, A.; Gregorio, C.C. The Nebulin family: An actin support group. Trends Cell Biol. 2011, 21, 29–37. [Google Scholar] [CrossRef]
- Laing, N.G.; Dye, D.E.; Wallgren-Pettersson, C.; Richard, G.; Monnier, N.; Lillis, S.; Winder, T.L.; Lochmuller, H.; Graziano, C.; Mitrani-Rosenbaum, S.; et al. Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1). Human. Mutat. 2009, 30, 1267–1277. [Google Scholar] [CrossRef]
- Bathe, F.S.; Rommelaere, H.; Machesky, L.M. Phenotypes of myopathy-related actin mutants in differentiated C2C12 myotubes. BMC Cell Biol. 2007, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.F.; Rommelaere, H.; Waterschoot, D.; Sethi, K.K.; Nowak, K.J.; Laing, N.G.; Ampe, C.; Machesky, L.M. Myopathy mutations in alpha-skeletal-muscle actin cause a range of molecular defects. J. Cell Sci. 2004, 117, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, D.; Lambert, E.; Waterschoot, D.; Tondeleir, D.; Vandekerckhove, J.; Machesky, L.M.; Constantin, B.; Rommelaere, H.; Ampe, C. Phenotypes induced by NM causing alpha-skeletal muscle actin mutants in fibroblasts, Sol 8 myoblasts and myotubes. BMC Res. Notes 2009, 2, 40. [Google Scholar] [CrossRef]
- Vandamme, D.; Rommelaere, H.; Lambert, E.; Waterschoot, D.; Vandekerckhove, J.; Constantin, B.; Ampe, C. alpha-Skeletal muscle actin mutants causing different congenital myopathies induce similar cytoskeletal defects in cell line cultures. Cell Motil. Cytoskelet. 2009, 66, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Levy, Y.; Ripolone, M.; Kolb, J.S.; Turmaine, M.; Holt, M.; Lindqvist, J.; Claeys, K.G.; Weis, J.; Monforte, M.; et al. Impairments in contractility and cytoskeletal organisation cause nuclear defects in nemaline myopathy. Acta Neuropathol. 2019, 138, 477–495. [Google Scholar] [CrossRef] [PubMed]
- Pula, S.; Urankar, K.; Norman, A.; Pierre, G.; Langton-Hewer, S.; Selby, V.; Mason, F.; Vijayakumar, K.; McFarland, R.; Taylor, R.W.; et al. A novel de novo ACTA1 variant in a patient with nemaline myopathy and mitochondrial Complex I deficiency. Neuromuscul. Disord. NMD 2020, 30, 159–164. [Google Scholar] [CrossRef]
- Lamont, P.J.; Thorburn, D.R.; Fabian, V.; Vajsar, J.; Hawkins, C.; Saada Reisch, A.; Durling, H.; Laing, N.G.; Nevo, Y. Nemaline rods and complex I deficiency in three infants with hypotonia, motor delay and failure to thrive. Neuropediatrics 2004, 35, 302–306. [Google Scholar] [CrossRef]
- Vandebrouck, A.; Domazetovska, A.; Mokbel, N.; Cooper, S.T.; Ilkovski, B.; North, K.N. In vitro analysis of rod composition and actin dynamics in inherited myopathies. J. Neuropathol. Exp. Neurol. 2010, 69, 429–441. [Google Scholar] [CrossRef]
- Illescas, M.; Penas, A.; Arenas, J.; Martin, M.A.; Ugalde, C. Regulation of Mitochondrial Function by the Actin Cytoskeleton. Front. Cell Dev. Biol. 2021, 9, 795838. [Google Scholar] [CrossRef]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Kast, D.J.; Dominguez, R. The Cytoskeleton-Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Zhang, Y.; Sampathkumar, A. Cytoskeleton Architecture Regulates Glycolysis Coupling Cellular Metabolism to Mechanical Cues. Trends Biochem. Sci. 2020, 45, 637–638. [Google Scholar] [CrossRef]
- Sewry, C.A.; Laitila, J.M.; Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 2019, 40, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.; Memo, M.; Messer, A.; Papadaki, M.; Nowak, K.; McNamara, E.; Ong, R.; El-Mezgueldi, M.; Li, X.; Lehman, W. Mutations in repeating structural motifs of tropomyosin cause gain of function in skeletal muscle myopathy patients. Human. Mol. Genet. 2013, 22, 4978–4987. [Google Scholar] [CrossRef] [PubMed]
- Ilkovski, B.; Nowak, K.J.; Domazetovska, A.; Maxwell, A.L.; Clement, S.; Davies, K.E.; Laing, N.G.; North, K.N.; Cooper, S.T. Evidence for a dominant-negative effect in ACTA1 nemaline myopathy caused by abnormal folding, aggregation and altered polymerization of mutant actin isoforms. Human. Mol. Genet. 2004, 13, 1727–1743. [Google Scholar] [CrossRef][Green Version]
- Clayton, J.S.; Scriba, C.K.; Romero, N.B.; Malfatti, E.; Saker, S.; Larmonier, T.; Nowak, K.J.; Ravenscroft, G.; Laing, N.G.; Taylor, R.L. Generation of two isogenic induced pluripotent stem cell lines from a 4-month-old severe nemaline myopathy patient with a heterozygous dominant c.553C > A (p.Arg183Ser) variant in the ACTA1 gene. Stem Cell Res. 2021, 53, 102273. [Google Scholar] [CrossRef]
- Villalon-Garcia, I.; Alvarez-Cordoba, M.; Suarez-Rivero, J.M.; Povea-Cabello, S.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Sanchez-Alcazar, J.A. Precision Medicine in Rare Diseases. Diseases 2020, 8, 42. [Google Scholar] [CrossRef]
- Vangipuram, M.; Ting, D.; Kim, S.; Diaz, R.; Schule, B. Skin punch biopsy explant culture for derivation of primary human fibroblasts. J. Vis. Exp. 2013, e3779. [Google Scholar] [CrossRef]
- Alvarez-Cordoba, M.; Fernandez Khoury, A.; Villanueva-Paz, M.; Gomez-Navarro, C.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotan, D.; Talaveron-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2019, 56, 3638–3656. [Google Scholar] [CrossRef]
- Villalon-Garcia, I.; Alvarez-Cordoba, M.; Povea-Cabello, S.; Talaveron-Rey, M.; Villanueva-Paz, M.; Luzon-Hidalgo, R.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; et al. Vitamin E prevents lipid peroxidation and iron accumulation in PLA2G6-Associated Neurodegeneration. Neurobiol. Dis. 2022, 165, 105649. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kano, Y.; Amano, M.; Kaibuchi, K.; Fujiwara, K. Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am. J. Physiology. Cell Physiol. 2001, 280, C1669–C1679. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.D.; Gerlach, B.D. The roles and regulation of the actin cytoskeleton, intermediate filaments and microtubules in smooth muscle cell migration. Respir. Res. 2017, 18, 54. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, K.; Mathew, A.; Koushika, S.P. Role of actin in organelle trafficking in neurons. Cytoskeleton 2020, 77, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Szent-Gyorgyi, A.G. The early history of the biochemistry of muscle contraction. J. Gen. Physiol. 2004, 123, 631–641. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef]
- Vandekerckhove, J.; Weber, K. At least six different actins are expressed in a higher mammal: An analysis based on the amino acid sequence of the amino-terminal tryptic peptide. J. Mol. Biol. 1978, 126, 783–802. [Google Scholar] [CrossRef]
- Hightower, R.C.; Meagher, R.B. The molecular evolution of actin. Genetics 1986, 114, 315–332. [Google Scholar] [CrossRef]
- Gunning, P.; Ponte, P.; Kedes, L.; Eddy, R.; Shows, T. Chromosomal location of the co-expressed human skeletal and cardiac actin genes. Proc. Natl. Acad. Sci. USA 1984, 81, 1813–1817. [Google Scholar] [CrossRef]
- Malek, N.; Michrowska, A.; Mazurkiewicz, E.; Mrowczynska, E.; Mackiewicz, P.; Mazur, A.J. The origin of the expressed retrotransposed gene ACTBL2 and its influence on human melanoma cells’ motility and focal adhesion formation. Sci. Rep. 2021, 11, 3329. [Google Scholar] [CrossRef] [PubMed]
- Ampe, C.; Van Troys, M. Mammalian Actins: Isoform-Specific Functions and Diseases. Handb. Exp. Pharmacol. 2017, 235, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Moradi, M.; Sivadasan, R.; Saal, L.; Luningschror, P.; Dombert, B.; Rathod, R.J.; Dieterich, D.C.; Blum, R.; Sendtner, M. Differential roles of alpha-, beta-, and gamma-actin in axon growth and collateral branch formation in motoneurons. J. Cell Biol. 2017, 216, 793–814. [Google Scholar] [CrossRef] [PubMed]
- Sandbo, N.; Dulin, N. Actin cytoskeleton in myofibroblast differentiation: Ultrastructure defining form and driving function. Transl. Res. 2011, 158, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Tondeleir, D.; Lambrechts, A.; Muller, M.; Jonckheere, V.; Doll, T.; Vandamme, D.; Bakkali, K.; Waterschoot, D.; Lemaistre, M.; Debeir, O.; et al. Cells lacking beta-actin are genetically reprogrammed and maintain conditional migratory capacity. Mol. Cell Proteom. 2012, 11, 255–271. [Google Scholar] [CrossRef]
- Labeit, S.; Ottenheijm, C.A.; Granzier, H. Nebulin, a major player in muscle health and disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 822–829. [Google Scholar] [CrossRef]
- Laflamme, N.; Lace, B.; Thonta Setty, S.; Rioux, N.; Labrie, Y.; Droit, A.; Chrestian, N.; Rivest, S. A Homozygous Deep Intronic Mutation Alters the Splicing of Nebulin Gene in a Patient With Nemaline Myopathy. Front. Neurol. 2021, 12, 660113. [Google Scholar] [CrossRef]
- Marttila, M.; Hanif, M.; Lemola, E.; Nowak, K.J.; Laitila, J.; Gronholm, M.; Wallgren-Pettersson, C.; Pelin, K. Nebulin interactions with actin and tropomyosin are altered by disease-causing mutations. Skelet. Muscle 2014, 4, 15. [Google Scholar] [CrossRef]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef]
- van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef]
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria-hubs for regulating cellular biochemistry: Emerging concepts and networks. Open Biol. 2019, 9, 190126. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Okamoto, K. Mitochondrial clearance: Mechanisms and roles in cellular fitness. FEBS Lett. 2021, 595, 1239–1263. [Google Scholar] [CrossRef]
- Choubey, V.; Zeb, A.; Kaasik, A. Molecular Mechanisms and Regulation of Mammalian Mitophagy. Cells 2021, 11, 38. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Sandri, M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2015, 6, 422. [Google Scholar] [CrossRef]
- Baechler, B.L.; Bloemberg, D.; Quadrilatero, J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy 2019, 15, 1606–1619. [Google Scholar] [CrossRef]
- McMillan, E.M.; Quadrilatero, J. Autophagy is required and protects against apoptosis during myoblast differentiation. Biochem. J. 2014, 462, 267–277. [Google Scholar] [CrossRef]
- Sin, J.; Andres, A.M.; Taylor, D.J.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Chakrabarti, R.; Higgs, H.N. The multiple links between actin and mitochondria. Nat. Rev. Mol. Cell Biol. 2023, 24, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Holzbaur, E.L.F. Mitochondrial-cytoskeletal interactions: Dynamic associations that facilitate network function and remodeling. Curr. Opin. Physiol. 2018, 3, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.R.; Zenezini Chiozzi, R.; Roelofs, M.C.; Hevler, J.F.; Ravi, R.T.; Maitan, P.; Zhang, M.; Henning, H.; Bromfield, E.G.; Howes, S.C.; et al. In-cell structures of conserved supramolecular protein arrays at the mitochondria-cytoskeleton interface in mammalian sperm. Proc. Natl. Acad. Sci. USA 2021, 118, e2110996118. [Google Scholar] [CrossRef]
- Solomon, T.; Rajendran, M.; Rostovtseva, T.; Hool, L. How cytoskeletal proteins regulate mitochondrial energetics in cell physiology and diseases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2022, 377, 20210324. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 2015, 4, e8828. [Google Scholar] [CrossRef]
- Ji, W.K.; Hatch, A.L.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. eLife 2015, 4, e11553. [Google Scholar] [CrossRef]
- Simon, V.R.; Swayne, T.C.; Pon, L.A. Actin-dependent mitochondrial motility in mitotic yeast and cell-free systems: Identification of a motor activity on the mitochondrial surface. J. Cell Biol. 1995, 130, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.R.; Yang, H.C.; Nowakowski, W.D.; Karmon, S.L.; Hays, L.G.; Yates, J.R., 3rd; Pon, L.A. Arp2/3 complex and actin dynamics are required for actin-based mitochondrial motility in yeast. Proc. Natl. Acad. Sci. USA 2001, 98, 3162–3167. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; He, J.; Mao, C.C.; Bailey, L.J.; Di Re, M.; Sembongi, H.; Kazak, L.; Dzionek, K.; Holmes, J.B.; Cluett, T.J.; et al. Actin and myosin contribute to mammalian mitochondrial DNA maintenance. Nucleic Acids Res. 2011, 39, 5098–5108. [Google Scholar] [CrossRef]
- Clayton, D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef]
- Spelbrink, J.N. Functional organization of mammalian mitochondrial DNA in nucleoids: History, recent developments, and future challenges. IUBMB Life 2010, 62, 19–32. [Google Scholar] [CrossRef]
- Rampelt, H.; Pfanner, N. Coordination of Two Genomes by Mitochondrial Translational Plasticity. Cell 2016, 167, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.R.; Pon, L.A. Interactions of mitochondria with the actin cytoskeleton. Biochim. Et Biophys. Acta 2006, 1763, 450–462. [Google Scholar] [CrossRef]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef]
- Dadsena, S.; King, L.E.; Garcia-Saez, A.J. Apoptosis regulation at the mitochondria membrane level. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183716. [Google Scholar] [CrossRef]
- Xie, X.; Venit, T.; Drou, N.; Percipalle, P. In Mitochondria ?-Actin Regulates mtDNA Transcription and Is Required for Mitochondrial Quality Control. iScience 2018, 3, 226–237. [Google Scholar] [CrossRef]
- Arnold, H.; Pette, D. Binding of glycolytic enzymes to structure proteins of the muscle. Eur. J. Biochem. 1968, 6, 163–171. [Google Scholar] [CrossRef]
- Takahashi, K.; Miura, Y.; Ohsawa, I.; Shirasawa, T.; Takahashi, M. In vitro rejuvenation of brain mitochondria by the inhibition of actin polymerization. Sci. Rep. 2018, 8, 15585. [Google Scholar] [CrossRef]
- Slick, R.A.; Tinklenberg, J.A.; Sutton, J.; Zhang, L.; Meng, H.; Beatka, M.; Avond, M.V.; Prom, M.J.; Ott, E.; Montanaro, F.; et al. Aberrations in Energetic Metabolism and Stress-Related Pathways Contribute to Pathophysiology in the Neb Conditional Knockout Mouse Model of Nemaline Myopathy. Am. J. Pathol. 2023, 193, 1528–1547. [Google Scholar] [CrossRef]
- Tinklenberg, J.A.; Slick, R.A.; Sutton, J.; Zhang, L.; Meng, H.; Beatka, M.J.; Avond, M.V.; Prom, M.J.; Ott, E.; Montanaro, F.; et al. Different Mouse Models of Nemaline Myopathy Harboring Acta1 Mutations Display Differing Abnormalities Related to Mitochondrial Biology. Am. J. Pathol. 2023, 193, 1548–1567. [Google Scholar] [CrossRef] [PubMed]
- Sandri, J.; Viala, J. Direct Preparation of (Z,Z)-1,4-Dienic Units with a New C6 Homologating Agent: Synthesis of α-Linolenic Acid. Synthesis 1995, 1995, 271–275. [Google Scholar] [CrossRef]
- Yuan, Q.; Xie, F.; Huang, W.; Hu, M.; Yan, Q.; Chen, Z.; Zheng, Y.; Liu, L. The review of alpha-linolenic acid: Sources, metabolism, and pharmacology. Phytother. Res. 2022, 36, 164–188. [Google Scholar] [CrossRef] [PubMed]
- Egert, S.; Baxheinrich, A.; Lee-Barkey, Y.H.; Tschoepe, D.; Stehle, P.; Stratmann, B.; Wahrburg, U. Effects of a hypoenergetic diet rich in alpha-linolenic acid on fatty acid composition of serum phospholipids in overweight and obese patients with metabolic syndrome. Nutrition 2018, 49, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Lee, M.H.; Lee, S.; Cho, E.J. Alpha-Linolenic Acid from Perilla frutescens var. japonica Oil Protects Abeta-Induced Cognitive Impairment through Regulation of APP Processing and Abeta Degradation. J. Agric. Food Chem. 2017, 65, 10719–10729. [Google Scholar] [CrossRef]
- Litwiniuk, A.; Domanska, A.; Chmielowska, M.; Martynska, L.; Bik, W.; Kalisz, M. The Effects of Alpha-Linolenic Acid on the Secretory Activity of Astrocytes and beta Amyloid-Associated Neurodegeneration in Differentiated SH-SY5Y Cells: Alpha-Linolenic Acid Protects the SH-SY5Y cells against beta Amyloid Toxicity. Oxid. Med. Cell Longev. 2020, 2020, 8908901. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Kagan, V.E.; Chu, C.T.; Tyurina, Y.Y.; Cheikhi, A.; Bayir, H. Cardiolipin asymmetry, oxidation and signaling. Chem. Phys. Lipids 2014, 179, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurina, Y.Y.; Tyurin, V.A.; Mohammadyani, D.; Angeli, J.P.; Baranov, S.V.; Klein-Seetharaman, J.; Friedlander, R.M.; Mallampalli, R.K.; Conrad, M.; et al. Cardiolipin signaling mechanisms: Collapse of asymmetry and oxidation. Antioxid. Redox Signal 2015, 22, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Cardiolipin and mitochondrial function in health and disease. Antioxid. Redox Signal 2014, 20, 1925–1953. [Google Scholar] [CrossRef] [PubMed]
- Jalmar, O.; Francois-Moutal, L.; Garcia-Saez, A.J.; Perry, M.; Granjon, T.; Gonzalvez, F.; Gottlieb, E.; Ayala-Sanmartin, J.; Klosgen, B.; Schwille, P.; et al. Caspase-8 binding to cardiolipin in giant unilamellar vesicles provides a functional docking platform for bid. PLoS ONE 2013, 8, e55250. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Paradies, G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: Induction of permeability transition and cytochrome c release. FEBS Lett. 2006, 580, 6311–6316. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; McMeekin, L.; Saint, C.; LeBlanc, P.J. Cardiolipin linoleic acid content and mitochondrial cytochrome c oxidase activity are associated in rat skeletal muscle. Chem. Phys. Lipids 2015, 187, 50–55. [Google Scholar] [CrossRef]
- Bazan, S.; Mileykovskaya, E.; Mallampalli, V.K.; Heacock, P.; Sparagna, G.C.; Dowhan, W. Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. J. Biol. Chem. 2013, 288, 401–411. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Letts, J.A.; Degliesposti, G.; Kaszuba, K.; Skehel, M.; Sazanov, L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nature 2016, 538, 406–410. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids 2014, 179, 42–48. [Google Scholar] [CrossRef]
- Schwall, C.T.; Greenwood, V.L.; Alder, N.N. The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim. Biophys. Acta 2012, 1817, 1588–1596. [Google Scholar] [CrossRef]
- Maekawa, S.; Takada, S.; Nambu, H.; Furihata, T.; Kakutani, N.; Setoyama, D.; Ueyanagi, Y.; Kang, D.; Sabe, H.; Kinugawa, S. Linoleic acid improves assembly of the CII subunit and CIII2/CIV complex of the mitochondrial oxidative phosphorylation system in heart failure. Cell Commun. Signal 2019, 17, 128. [Google Scholar] [CrossRef] [PubMed]
- Snoke, D.B.; Mahler, C.A.; Angelotti, A.; Cole, R.M.; Sparagna, G.C.; Baskin, K.K.; Belury, M.A. Linoleic Acid-Enriched Diet Increases Mitochondrial Tetralinoleoyl Cardiolipin, OXPHOS Protein Levels, and Uncoupling in Interscapular Brown Adipose Tissue during Diet-Induced Weight Gain. Biology 2022, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 2002, 277, 43553–43556. [Google Scholar] [CrossRef]
- Valianpour, F.; Wanders, R.J.; Overmars, H.; Vaz, F.M.; Barth, P.G.; van Gennip, A.H. Linoleic acid supplementation of Barth syndrome fibroblasts restores cardiolipin levels: Implications for treatment. J. Lipid Res. 2003, 44, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, Y. Conjugated linoleic acid (CLA) stimulates mitochondrial biogenesis signaling by the upregulation of PPARgamma coactivator 1alpha (PGC-1alpha) in C2C12 cells. Lipids 2015, 50, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reyes, C.; Marcial-Medina, C.; Cervantes-Anaya, N.; Cortes-Reynosa, P.; Salazar, E.P. Migration and invasion induced by linoleic acid are mediated through fascin in MDA-MB-231 breast cancer cells. Mol. Cell Biochem. 2018, 443, 1–10. [Google Scholar] [CrossRef]
- Desale, S.E.; Chinnathambi, S. alpha-Linolenic acid induces clearance of Tau seeds via Actin-remodeling in Microglia. Mol. Biomed. 2021, 2, 4. [Google Scholar] [CrossRef]
- Masner, M.; Lujea, N.; Bisbal, M.; Acosta, C.; Kunda, P. Linoleic and oleic acids enhance cell migration by altering the dynamics of microtubules and the remodeling of the actin cytoskeleton at the leading edge. Sci. Rep. 2021, 11, 14984. [Google Scholar] [CrossRef]
- Jung, C.H.; Lee, W.J.; Hwang, J.Y.; Seol, S.M.; Kim, Y.M.; Lee, Y.L.; Ahn, J.H.; Park, J.Y. The role of Rho/Rho-kinase pathway in the expression of ICAM-1 by linoleic acid in human aortic endothelial cells. Inflammation 2012, 35, 1041–1048. [Google Scholar] [CrossRef]
- Bremer, J. Carnitine--metabolism and functions. Physiol. Rev. 1983, 63, 1420–1480. [Google Scholar] [CrossRef]
- Tragni, V.; Primiano, G.; Tummolo, A.; Cafferati Beltrame, L.; La Piana, G.; Sgobba, M.N.; Cavalluzzi, M.M.; Paterno, G.; Gorgoglione, R.; Volpicella, M.; et al. Personalized Medicine in Mitochondrial Health and Disease: Molecular Basis of Therapeutic Approaches Based on Nutritional Supplements and Their Analogs. Molecules 2022, 27, 3494. [Google Scholar] [CrossRef] [PubMed]
- Liufu, T.; Wang, Z. Treatment for mitochondrial diseases. Rev. Neurosci. 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Modanloo, M.; Shokrzadeh, M. Analyzing Mitochondrial Dysfunction, Oxidative Stress, and Apoptosis: Potential Role of L-carnitine. Iran. J. Kidney Dis. 2019, 13, 74–86. [Google Scholar] [PubMed]
- Cabello, A.; Martinez-Martin, P.; Gutierrez-Rivas, E.; Madero, S. Myopathy with nemaline structures associated with HIV infection. J. Neurol. 1990, 237, 64–65. [Google Scholar] [CrossRef]
- Hsu, P.J.; Wang, H.D.; Tseng, Y.C.; Pan, S.W.; Sampurna, B.P.; Jong, Y.J.; Yuh, C.H. L-Carnitine ameliorates congenital myopathy in a tropomyosin 3 de novo mutation transgenic zebrafish. J. Biomed. Sci. 2021, 28, 8. [Google Scholar] [CrossRef]
- Niedbalska-Tarnowska, J.; Ochenkowska, K.; Migocka-Patrzalek, M.; Dubinska-Magiera, M. Assessment of the Preventive Effect of L-carnitine on Post-statin Muscle Damage in a Zebrafish Model. Cells 2022, 11, 1297. [Google Scholar] [CrossRef]
- Jang, J.; Park, J.; Chang, H.; Lim, K. l-Carnitine supplement reduces skeletal muscle atrophy induced by prolonged hindlimb suspension in rats. Appl. Physiol. Nutr. Metab. 2016, 41, 1240–1247. [Google Scholar] [CrossRef]
- Gimenes, A.C.; Bravo, D.M.; Napolis, L.M.; Mello, M.T.; Oliveira, A.S.; Neder, J.A.; Nery, L.E. Effect of L-carnitine on exercise performance in patients with mitochondrial myopathy. Braz. J. Med. Biol. Res. 2015, 48, 354–362. [Google Scholar] [CrossRef]
- Ochi, H.; Westerfield, M. Signaling networks that regulate muscle development: Lessons from zebrafish. Dev. Growth Differ. 2007, 49, 1–11. [Google Scholar] [CrossRef]
- Fujiwara, I.; Vavylonis, D.; Pollard, T.D. Polymerization kinetics of ADP- and ADP-Pi-actin determined by fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2007, 104, 8827–8832. [Google Scholar] [CrossRef]
- Pollard, T.D. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J. Cell Biol. 1986, 103, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Hallett, M.A.; Dagher, P.C.; Atkinson, S.J. Rho GTPases show differential sensitivity to nucleotide triphosphate depletion in a model of ischemic cell injury. Am. J. Physiol. Cell Physiol. 2003, 285, C129–C138. [Google Scholar] [CrossRef] [PubMed][Green Version]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piñero-Pérez, R.; López-Cabrera, A.; Álvarez-Córdoba, M.; Cilleros-Holgado, P.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Gómez-Fernández, D.; Reche-López, D.; Romero-González, A.; et al. Actin Polymerization Defects Induce Mitochondrial Dysfunction in Cellular Models of Nemaline Myopathies. Antioxidants 2023, 12, 2023. https://doi.org/10.3390/antiox12122023
Piñero-Pérez R, López-Cabrera A, Álvarez-Córdoba M, Cilleros-Holgado P, Talaverón-Rey M, Suárez-Carrillo A, Munuera-Cabeza M, Gómez-Fernández D, Reche-López D, Romero-González A, et al. Actin Polymerization Defects Induce Mitochondrial Dysfunction in Cellular Models of Nemaline Myopathies. Antioxidants. 2023; 12(12):2023. https://doi.org/10.3390/antiox12122023
Chicago/Turabian StylePiñero-Pérez, Rocío, Alejandra López-Cabrera, Mónica Álvarez-Córdoba, Paula Cilleros-Holgado, Marta Talaverón-Rey, Alejandra Suárez-Carrillo, Manuel Munuera-Cabeza, David Gómez-Fernández, Diana Reche-López, Ana Romero-González, and et al. 2023. "Actin Polymerization Defects Induce Mitochondrial Dysfunction in Cellular Models of Nemaline Myopathies" Antioxidants 12, no. 12: 2023. https://doi.org/10.3390/antiox12122023
APA StylePiñero-Pérez, R., López-Cabrera, A., Álvarez-Córdoba, M., Cilleros-Holgado, P., Talaverón-Rey, M., Suárez-Carrillo, A., Munuera-Cabeza, M., Gómez-Fernández, D., Reche-López, D., Romero-González, A., Romero-Domínguez, J. M., de Pablos, R. M., & Sánchez-Alcázar, J. A. (2023). Actin Polymerization Defects Induce Mitochondrial Dysfunction in Cellular Models of Nemaline Myopathies. Antioxidants, 12(12), 2023. https://doi.org/10.3390/antiox12122023