
How Aging and Oxidative Stress Influence the Cytopathic and Inflammatory Effects of SARS-CoV-2 Infection: The Role of Cellular Glutathione and Cysteine Metabolism

 ,
,  ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Thiols in Maintaining the Redox Balance of Tissues
3. Age-Dependent Changes of Lung Thiols and the Role of GSH in Lung Diseases
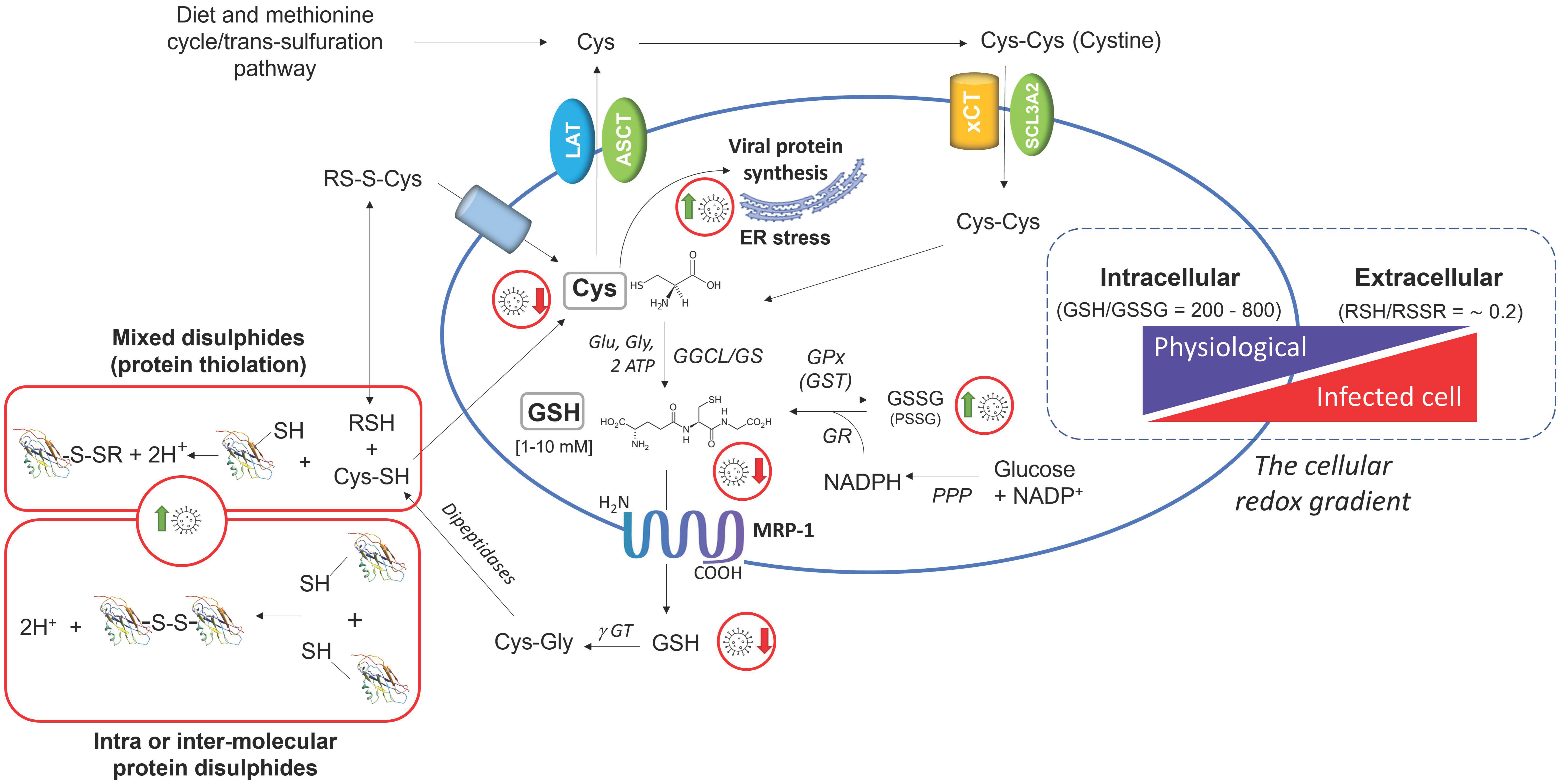
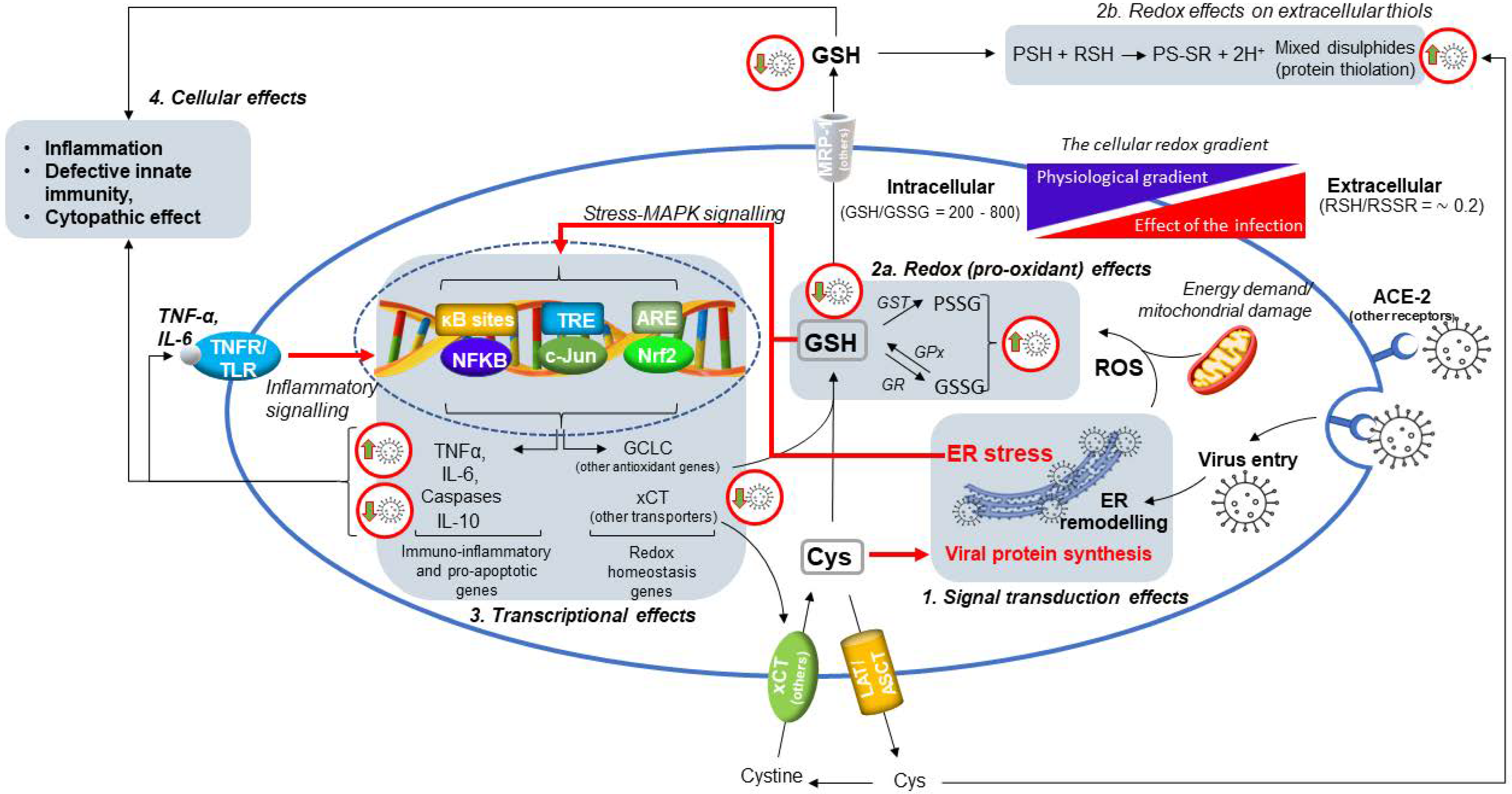
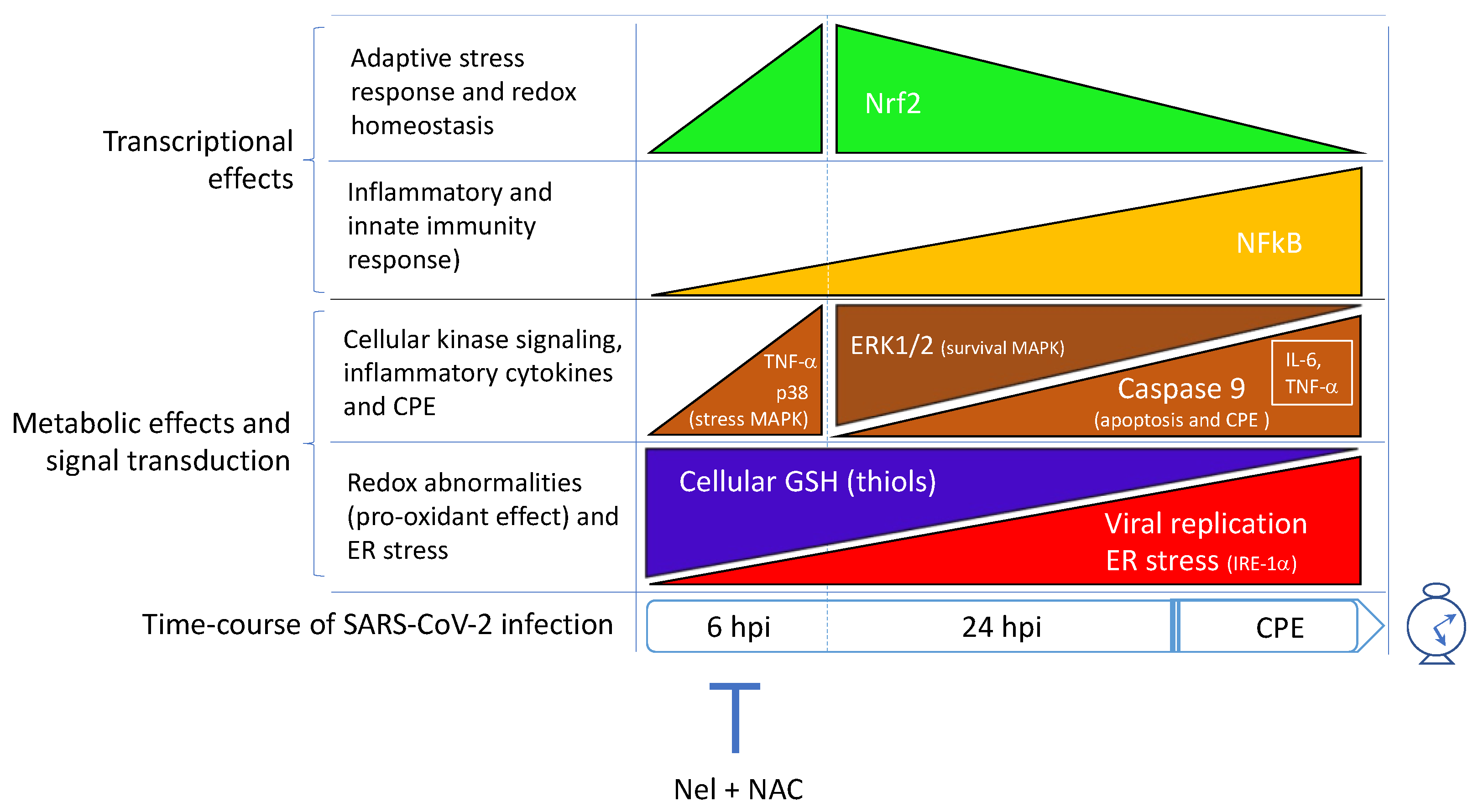
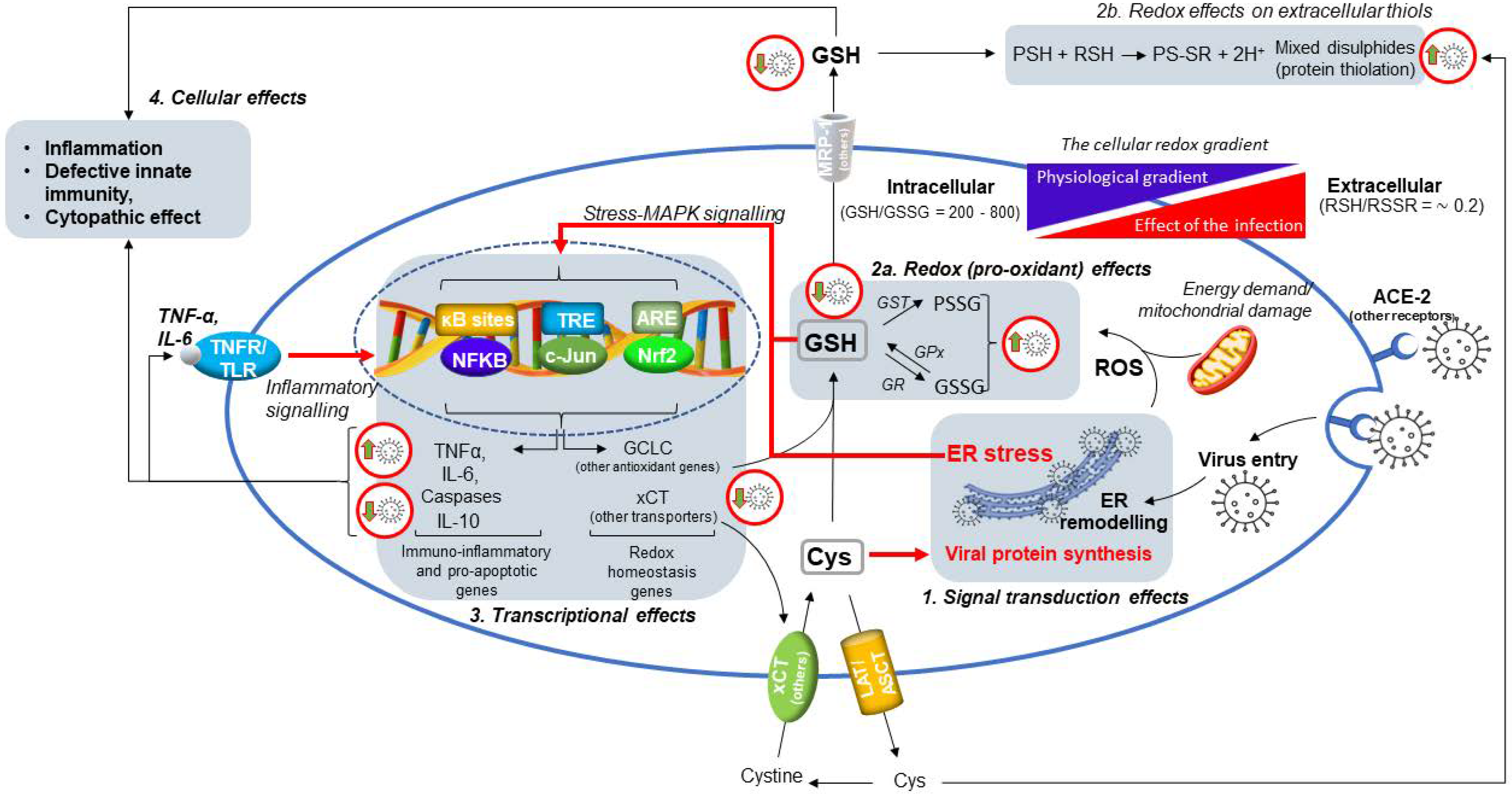
4. SARS-CoV-2 Infection Impairs the Metabolism and Redox Function of Cellular GSH
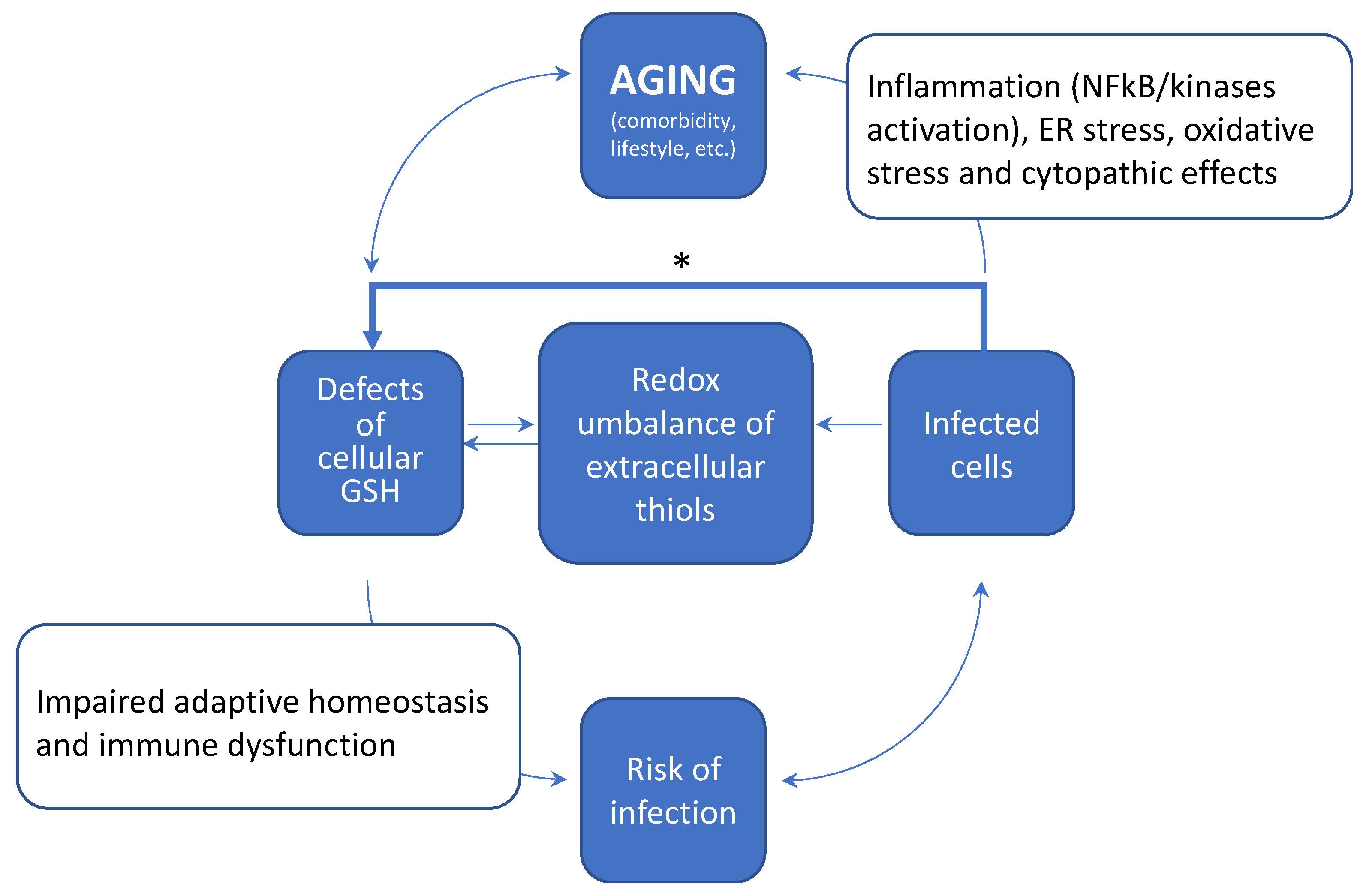
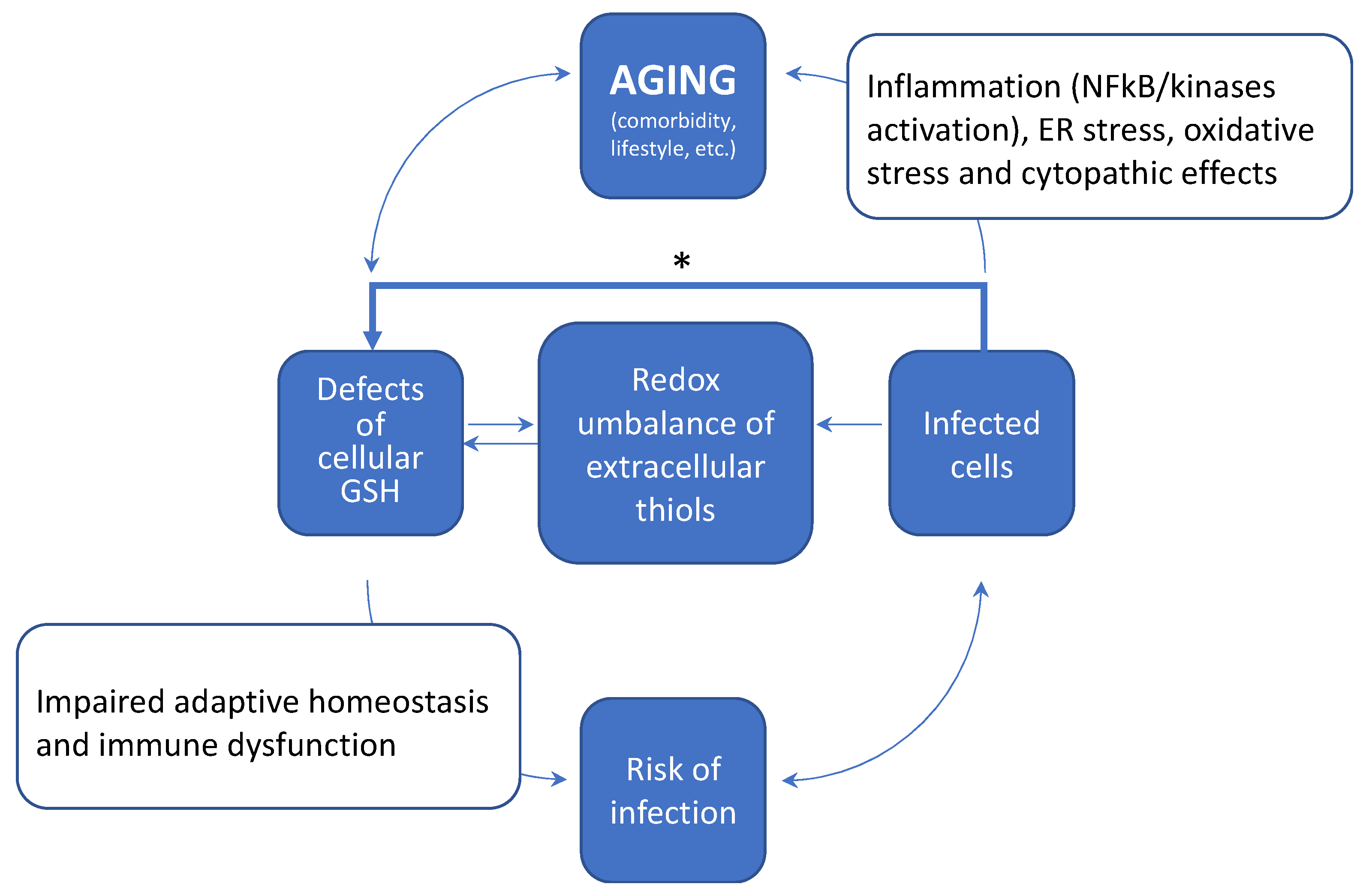
5. How Aging and a Defective Cellular Redox May Conspire to Increase the Risk of Infection and the Development of Severe COVID-19
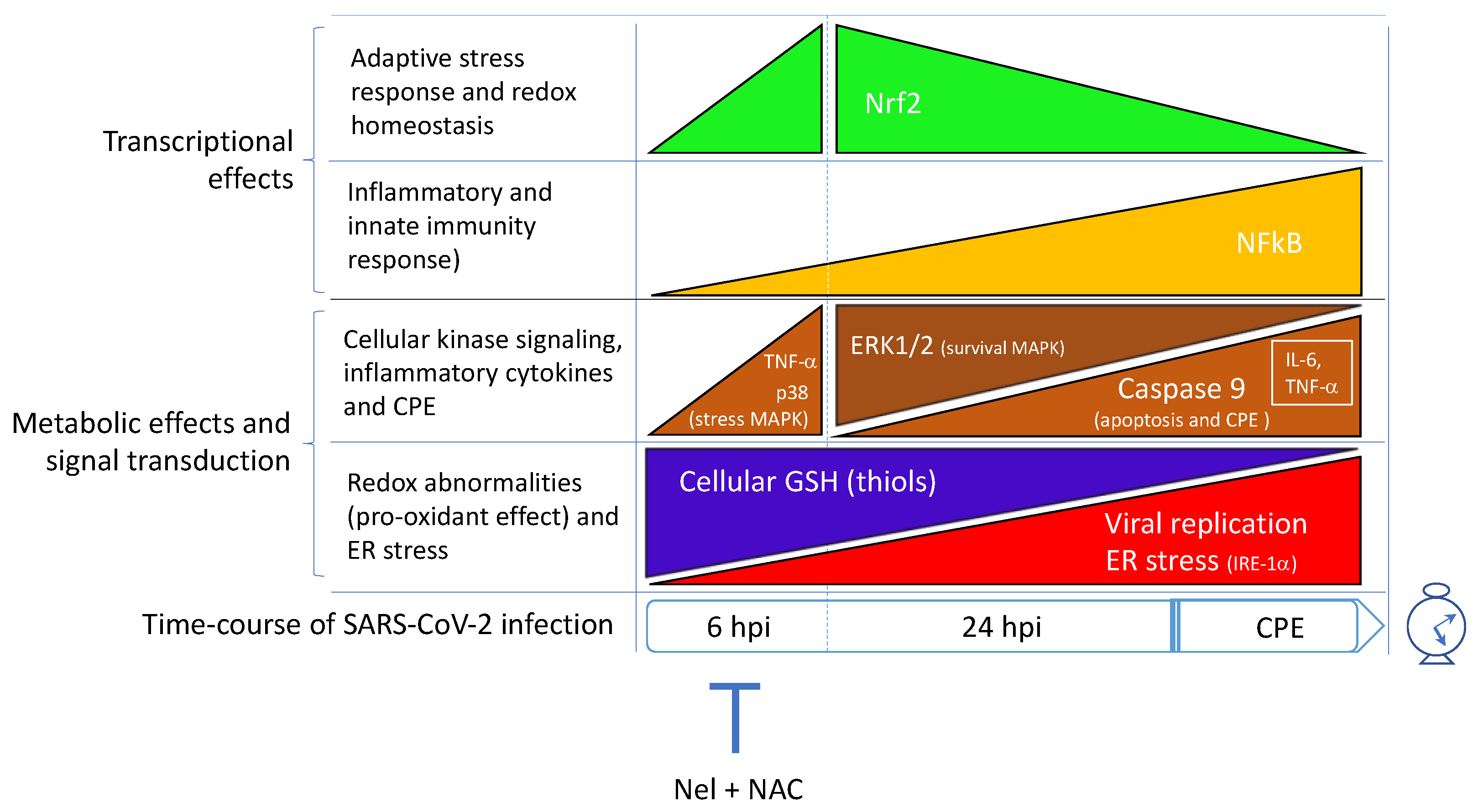
6. SARS-CoV-2 Infection Triggers NF-κB Activation and Inflammatory Cytokine Expression in the Host Cell
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mehra, M.R.; Desai, S.S.; Kuy, S.; Henry, T.D.; Patel, A.N. Cardiovascular Disease, Drug Therapy, and Mortality in COVID-19. N. Engl. J. Med. 2020, 382, e102. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Santucci, A.; Bartolini, D.; Galli, F.; Rossi, R. The Age-Dependent Decline of the Extracellular Thiol-Disulfide Balance and Its Role in SARS-CoV-2 Infection. Redox Biol. 2021, 41, 101902. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Dalle-Donne, I.; Lorenzini, S.; Selvi, E.; Colombo, G.; Milzani, A.; Fanti, P.; Rossi, R. Protein Thiolation Index (Pti) as a Biomarker of Oxidative Stress. Free Radic. Biol. Med. 2012, 53, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Dalle-Donne, I.; Lorenzini, S.; Milzani, A.; Rossi, R. Age-Related Influence on Thiol, Disulfide, and Protein-Mixed Disulfide Levels in Human Plasma. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Mody, V.C.; Carlson, J.L.; Lynn, M.J.; Sternberg, P. Redox Analysis of Human Plasma Allows Separation of Pro-Oxidant Events of Aging from Decline in Antioxidant Defenses. Free Radic. Biol. Med. 2002, 33, 1290–1300. [Google Scholar] [CrossRef]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef]
- Janssen-Heininger, Y.; Reynaert, N.L.; van der Vliet, A.; Anathy, V. Endoplasmic Reticulum Stress and Glutathione Therapeutics in Chronic Lung Diseases. Redox Biol. 2020, 33, 101516. [Google Scholar] [CrossRef]
- Bartolini, D.; Stabile, A.M.; Bastianelli, S.; Giustarini, D.; Pierucci, S.; Busti, C.; Vacca, C.; Gidari, A.; Francisci, D.; Castronari, R.; et al. SARS-CoV2 Infection Impairs the Metabolism and Redox Function of Cellular Glutathione. Redox Biol. 2021, 45, 102041. [Google Scholar] [CrossRef]
- Checconi, P.; Limongi, D.; Baldelli, S.; Ciriolo, M.R.; Nencioni, L.; Palamara, A.T. Role of Glutathionylation in Infection and Inflammation. Nutrients 2019, 11, 1952. [Google Scholar] [CrossRef] [Green Version]
- Hati, S.; Bhattacharyya, S. Impact of Thiol–Disulfide Balance on the Binding of COVID-19 Spike Protein with Angiotensin-Converting Enzyme 2 Receptor. ACS Omega 2020, 5, 16292–16298. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516, Correction in Nat. Rev. Immunol. 2020, 20, 579. [Google Scholar] [CrossRef]
- Ciriolo, M.R.; Palamara, A.T.; Incerpi, S.; Lafavia, E.; Buè, M.C.; De Vito, P.; Garaci, E.; Rotilio, G. Loss of GSH, Oxidative Stress, and Decrease of Intracellular pH as Sequential Steps in Viral Infection. J. Biol. Chem. 1997, 272, 2700–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolini, D.; Stabile, A.M.; Vacca, C.; Pistilli, A.; Rende, M.; Gioiello, A.; Cruciani, G.; Galli, F. Endoplasmic Reticulum Stress and Nf-Kb Activation in SARS-CoV-2 Infected Cells and Their Response to Antiviral Therapy. IUBMB Life 2022, 74, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Michelson, A.P.; Foraker, R.; Zhan, M.; Payne, P.R.O. Computational Analysis to Repurpose Drugs for COVID-19 Based on Transcriptional Response of Host Cells to SARS-CoV-2. BMC Med. Inform. Decis. Mak. 2021, 21, 15. [Google Scholar] [CrossRef]
- Goel, S.; Sharif-Askari, F.S.; Askari, N.S.S.; Madkhana, B.; Alwaa, A.M.; Mahboub, B.; Zakeri, A.M.; Ratemi, E.; Hamoudi, R.; Hamid, Q.; et al. SARS-CoV-2 Switches ‘on’ Mapk and Nfκb Signaling Via the Reduction of Nuclear Dusp1 and Dusp5 Expression. Front. Pharmacol. 2021, 12, 631879. [Google Scholar] [CrossRef]
- Scire, A.; Cianfruglia, L.; Minnelli, C.; Bartolini, D.; Torquato, P.; Principato, G.; Galli, F.; Armeni, T. Glutathione Compartmentalization and Its Role in Glutathionylation and Other Regulatory Processes of Cellular Pathways. Biofactors 2019, 45, 152–168. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. Thiol/Disulfide Redox States in Signaling and Sensing. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechtel, T.J.; Weerapana, E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1600391. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, A. Antioxidant Function of Thioredoxin and Glutaredoxin Systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef]
- Hansen, R.E.; Roth, D.; Winther, J.R. Quantifying the global cellular thiol–disulfide status. Proc. Natl. Acad. Sci. USA 2009, 106, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, D.; Giustarini, D.; Pietrella, D.; Rossi, R.; Galli, F. Glutathione S-transferase P influences the Nrf2-dependent response of cellular thiols to seleno-compounds. Cell Biol. Toxicol. 2020, 36, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Pajares, M.; Benito, C.; Jimenez-Villegas, J.; Escoll, M.; Fernandez-Gines, R.; Yague, A.J.G.; Lastra, D.; Manda, G.; Rojo, A.I.; et al. Can Activation of Nrf2 Be a Strategy against COVID-19? Trends Pharmacol. Sci. 2020, 41, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolini, D.; Galli, F. The functional interactome of GSTP: A regulatory biomolecular network at the interface with the Nrf2 adaption response to oxidative stress. J. Chromatogr. B 2016, 1019, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Torquato, P.; Piroddi, M.; Galli, F. Targeting glutathione S-transferase P and its interactome with selenium compounds in cancer therapy. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1863, 130–143. [Google Scholar] [CrossRef]
- Strålin, P.; Karlsson, K.; Johansson, B.O.; Marklund, S.L. The Interstitium of the Human Arterial Wall Contains Very Large Amounts of Extracellular Superoxide Dismutase. Arter. Thromb. Vasc. Biol. 1995, 15, 2032–2036. [Google Scholar]
- Yi, M.C.; Khosla, C. Thiol–Disulfide Exchange Reactions in the Mammalian Extracellular Environment. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 197–222. [Google Scholar] [CrossRef] [Green Version]
- Pomatto, L.C.D.; Davies, K.J.A. The role of declining adaptive homeostasis in ageing. J. Physiol. 2017, 595, 7275–7309. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhang, H.; Davies, K.J.; Forman, H.J. Aging-related decline in the induction of Nrf2-regulated antioxidant genes in human bronchial epithelial cells. Redox Biol. 2017, 14, 35–40. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Colombo, G.; Milzani, A.; Santucci, A.; Rossi, R. Protein thiolation index in microvolumes of plasma. Anal. Biochem. 2021, 618, 114125. [Google Scholar] [CrossRef]
- Giustarini, D.; Galvagni, F.; Colombo, G.; Dalle-Donne, I.; Milzani, A.; Aloisi, A.M.; Rossi, R. Determination of protein thiolation index (PTI) as a biomarker of oxidative stress in human serum. Anal. Biochem. 2017, 538, 38–41. [Google Scholar] [CrossRef]
- Gould, N.S.; Min, E.; Gauthier, S.; Chu, H.W.; Martin, R.; Day, B.J. Aging Adversely Affects the Cigarette Smoke–induced Glutathione Adaptive Response in the Lung. Am. J. Respir. Crit. Care Med. 2010, 182, 1114–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, I.; MacNee, W. Lung glutathione and oxidative stress: Implications in cigarette smoke-induced airway disease. Am. J. Physiol. Cell. Mol. Physiol. 1999, 277, L1067–L1088. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Hollinger, M.; Lachowicz-Scroggins, M.E.; Kerr, S.C.; Dunican, E.M.; Daniel, B.M.; Ghosh, S.; Erzurum, S.C.; Willard, B.; Hazen, S.L.; et al. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci. Transl. Med. 2015, 7, 276ra27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, F.; Battistoni, A.; Gambari, R.; Pompella, A.; Bragonzi, A.; Pilolli, F.; Iuliano, L.; Piroddi, M.; Dechecchi, M.C.; Cabrini, G. Oxidative Stress and Antioxidant Therapy in Cystic Fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 690–713. [Google Scholar] [CrossRef] [Green Version]
- Giustarini, D.; Dalle-Donne, I.; Milzani, A.D.G.; Rossi, R. Low molecular mass thiols, disulfides and protein mixed disulfides in rat tissues: Influence of sample manipulation, oxidative stress and ageing. Mech. Ageing Dev. 2011, 132, 141–148. [Google Scholar] [CrossRef]
- Iyer, S.S.; Ramirez, A.M.; Ritzenthaler, J.D.; Torres-Gonzalez, E.; Roser-Page, S.; Mora, A.L.; Brigham, K.L.; Jones, D.P.; Roman, J.; Rojas, M. Oxidation of extracellular cysteine/cystine redox state in bleomycin-induced lung fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L37–L45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elko, E.A.; Mahoney, J.M.; Vacek, P.; van der Vliet, A.; Anathy, V.; van der Velden, J.L.; Janssen-Heininger, Y.M.; Seward, D.J. Age-dependent dysregulation of redox genes may contribute to fibrotic pulmonary disease susceptibility. Free Radic. Biol. Med. 2019, 141, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Gould, N.S.; Min, E.; Huang, J.; Chu, H.W.; Good, J.; Martin, R.J.; Day, B.J. Glutathione Depletion Accelerates Cigarette Smoke-Induced Inflammation and Airspace Enlargement. Toxicol. Sci. 2015, 147, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Fanti, P.; Giustarini, D.; Rossi, R.; Cunningham, S.E.; Folli, F.; Khazim, K.; Cornell, J.; Matteucci, E.; Bansal, S. Dietary Intake of Proteins and Calories Is Inversely Associated With The Oxidation State of Plasma Thiols in End-Stage Renal Disease Patients. J. Ren. Nutr. 2015, 25, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Park, Y.; Gletsu-Miller, N.; Liang, Y.; Yu, T.; Accardi, C.J.; Ziegler, T.R. Dietary sulfur amino acid effects on fasting plasma cysteine/cystine redox potential in humans. Nutrition 2011, 27, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Ziegler, T.R.; Gletsu-Miller, N.; Liang, Y.; Yu, T.; Accardi, C.J.; Jones, D.P. Postprandial Cysteine/Cystine Redox Potential in Human Plasma Varies with Meal Content of Sulfur Amino Acids. J. Nutr. 2010, 140, 760–765. [Google Scholar] [CrossRef] [Green Version]
- Moriarty-Craige, S.E.; Jones, D.P. Extracellular thiols and thiol/disulfide redox in metabolism. Annu. Rev. Nutr. 2004, 24, 481–509. [Google Scholar] [CrossRef]
- Canestrari, F.; Buoncristiani, U.; Galli, F.; Giorgini, A.; Albertini, M.; Carobi, C.; Pascucci, M.; Bossù, M. Redox state, antioxidative activity and lipid peroxidation in erythrocytes and plasma of chronic ambulatory peritoneal dialysis patients. Clin. Chim. Acta 1995, 234, 127–136. [Google Scholar] [CrossRef]
- Canestrari, F.; Galli, F.; Giorgini, A.; Albertini, M.C.; Galiotta, P.; Pascucci, M.; Bossù, M. Erythrocyte Redox State in Uremic Anemia: Effects of Hemodialysis and Relevance of Glutathione Metabolism. Acta Haematol. 1994, 91, 187–193. [Google Scholar] [CrossRef]
- Garavaglia, M.L.; Giustarini, D.; Colombo, G.; Reggiani, F.; Finazzi, S.; Calatroni, M.; Landoni, L.; Portinaro, N.M.; Milzani, A.; Badalamenti, S.; et al. Blood Thiol Redox State in Chronic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 2853. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Piroddi, M.; Bartolini, D.; Ciffolilli, S.; Buoncristiani, E.; Ricci, G.; Buoncristianí, U. Blood thiol status and erythrocyte glutathione-S-transferase in chronic kidney disease patients on treatment with frequent (daily) hemodialysis. Free Radic. Res. 2013, 48, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J. Redox Biology and Therapeutics in Chronic Lung Disease (Lung Redox Therapeutics—Special Issue). Redox Biol. 2020, 33, 101579. [Google Scholar] [CrossRef]
- Derouiche, S. Oxidative Stress Associated with SARS-CoV-2 (COVID-19) Increases the Severity of the Lung Disease—A Systematic Review. J. Infect. Dis. Epidemiol. 2020, 6, 121. [Google Scholar]
- D’Angelo, J.A.; Dehlink, E.; Platzer, B.; Dwyer, P.; Circu, M.L.; Garay, J.; Aw, T.Y.; Fiebiger, E.; Dickinson, B.L. The Cystine/Glutamate Antiporter Regulates Dendritic Cell Differentiation and Antigen Presentation. J. Immunol. 2010, 185, 3217–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Los, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S. Endoplasmic Reticulum as a Potential Therapeutic Target for COVID-19 Infection Management? Eur. J. Pharmacol. 2020, 882, 173288. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Balansky, R.; la Maestra, S. Rationale for the Use of N-Acetylcysteine in Both Prevention and Adjuvant Therapy of COVID-19. FASEB J. 2020, 34, 13185–13193. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef]
- Liu, R.M.; Vayalil, P.K.; Ballinger, C.; Dickinson, D.A.; Huang, W.T.; Wang, S.; Kavanagh, T.J.; Matthews, Q.L.; Postlethwait, E.M. Transforming Growth Factor Β Suppresses Glutamate-Cysteine Ligase Gene Expression and Induces Oxidative Stress in a Lung Fibrosis Model. Free Radic. Biol. Med. 2012, 53, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, D.; Arato, I.; Mancuso, F.; Giustarini, D.; Bellucci, C.; Vacca, C.; Aglietti, M.C.; Stabile, A.M.; Rossi, R.; Cruciani, G.; et al. Melatonin Modulates Nrf2 Activity to Protect Porcine Pre-Pubertal Sertoli Cells from the Abnormal HH2O2 generation and reductive stress effects of cadmium. J. Pineal Res. 2022, e12806, online ahead of print. [Google Scholar]
- El-Fattah, E.E.A.; Saber, S.; Mourad, A.A.; El-Ahwany, E.; Amin, N.A.; Cavalu, S.; Yahya, G.; Saad, A.S.; Alsharidah, M.; Shata, A.; et al. The dynamic interplay between AMPK/NFκB signaling and NLRP3 is a new therapeutic target in inflammation: Emerging role of dapagliflozin in overcoming lipopolysaccharide-mediated lung injury. Biomed. Pharmacother. 2022, 147, 112628. [Google Scholar] [CrossRef]
- Hemmat, N.; Asadzadeh, Z.; Ahangar, N.K.; Alemohammad, H.; Najafzadeh, B.; Derakhshani, A.; Baghbanzadeh, A.; Baghi, H.B.; Javadrashid, D.; Najafi, S.; et al. The Roles of Signaling Pathways in SARS-CoV-2 Infection; Lessons Learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021, 166, 675–696. [Google Scholar] [CrossRef]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Checconi, P.; De Angelis, M.; Marcocci, M.E.; Fraternale, A.; Magnani, M.; Palamara, A.T.; Nencioni, L. Redox-Modulating Agents in the Treatment of Viral Infections. Int. J. Mol. Sci. 2020, 21, 4084. [Google Scholar] [CrossRef]
- Herzenberg, L.A.; De Rosa, S.C.; Dubs, J.G.; Roederer, M.; Anderson, M.T.; Ela, S.W.; Deresinski, S.C. Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. USA 1997, 94, 1967–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogberg, J.; Kristoferson, A. A Correlation between Glutathione Levels and Cellular Damage in Isolated Hepatocytes. JBIC J. Biol. Inorg. Chem. 1977, 74, 77–82. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Yan, H.; Xiao, G.; Zhang, J.; Hu, Y.; Yuan, F.; Cole, D.K.; Zheng, C.; Gao, G.F. SARS coronavirus induces apoptosis in Vero E6 Cells. J. Med. Virol. 2004, 73, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef] [Green Version]
- De Nicola, M.; Ghibelli, L. Glutathione depletion in survival and apoptotic pathways. Front. Pharmacol. 2014, 5, 267. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Cidlowski, J.A. Glutathione Efflux and Cell Death. Antioxid. Redox Signal. 2012, 17, 1694–1713. [Google Scholar] [CrossRef] [Green Version]
- Sgarbanti, R.; Nencioni, L.; Amatore, D.; Coluccio, P.; Fraternale, A.; Sale, P.; Mammola, C.L.; Carpino, G.; Gaudio, E.; Magnani, M.; et al. Redox Regulation of the Influenza Hemagglutinin Maturation Process: A New Cell-Mediated Strategy for Anti-Influenza Therapy. Antioxid. Redox Signal. 2011, 15, 593–606. [Google Scholar] [CrossRef]
- Amatore, D.; Celestino, I.; Brundu, S.; Galluzzi, L.; Coluccio, P.; Checconi, P.; Magnani, M.; Palamara, A.T.; Fraternale, A.; Nencioni, L. Glutathione increase by the n-butanoyl glutathione derivative (GSH-C4) inhibits viral replication and induces a predominant Th1 immune profile in old mice infected with influenza virus. FASEB BioAdv. 2019, 1, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Zhitkovich, A. Ascorbate: Antioxidant and biochemical activities and their importance for in vitro models. Arch. Toxicol. 2021, 95, 3623–3631. [Google Scholar] [CrossRef]
- Bartolini, D.; Wang, Y.; Zhang, J.; Giustarini, D.; Rossi, R.; Wang, G.Y.; Torquato, P.; Townsend, D.M.; Tew, K.D.; Galli, F. A seleno-hormetine protects bone marrow hematopoietic cells against ionizing radiation-induced toxicities. PLoS ONE 2019, 14, e0205626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Tsutsuki, H.; Islam, W.; Ono, K.; Takeda, K.; Akaike, T.; Sawa, T. ATP exposure stimulates glutathione efflux as a necessary switch for NLRP3 inflammasome activation. Redox Biol. 2021, 41, 101930. [Google Scholar] [CrossRef] [PubMed]
- Fenouillet, E.; Barbouche, R.; Jones, I.M. Cell Entry by Enveloped Viruses: Redox Considerations for HIV and SARS-Coronavirus. Antioxid. Redox Signal. 2007, 9, 1009–1034. [Google Scholar] [CrossRef] [PubMed]
- Lavillette, D.; Barbouche, R.; Yao, Y.; Boson, B.; Cosset, F.L.; Jones, I.M.; Fenouillet, E. Significant Redox Insensitivity of the Functions of the SARS-CoV Spike Glycoprotein: Comparison with Hiv Envelope. J. Biol. Chem. 2006, 281, 9200–9204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on Ace2 and Tmprss2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, I.; Tragni, V.; Busto, F.; de Grassi, A.; Pierri, C.L. Protein Structure Analysis of the Interactions between SARS-CoV-2 Spike Protein and the Human Ace2 Receptor: From Conformational Changes to Novel Neutralizing Antibodies. Cell Mol. Life Sci. 2021, 78, 1501–1522. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human Ace2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Nawijn, M.C.; Timens, W. Can Ace2 Expression Explain SARS-CoV-2 Infection of the Respiratory Epithelia in COVID-19? Mol. Syst. Biol. 2020, 16, e9841. [Google Scholar] [CrossRef]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of Ace2, Cd147, Cd26, and Other SARS-CoV-2 Associated Molecules in Tissues and Immune Cells in Health and in Asthma, Copd, Obesity, Hypertension, and COVID-19 Risk Factors. Allergy 2020, 75, 2829–2845. [Google Scholar] [CrossRef]
- AlGhatrif, M.; Cingolani, O.; Lakatta, E.G. The Dilemma of Coronavirus Disease 2019, Aging, and Cardiovascular Disease: Insights from Cardiovascular Aging Science. JAMA Cardiol. 2020, 5, 747–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debnath, U.; Dewaker, V.; Prabhakar, Y.S.; Bhattacharyya, B.; Mandal, A. Conformational Perturbation of SARS-CoV-2 Spike Protein Using N-Acetyl Cysteine, a Molecular Scissor: A Probable Strategy to Combat COVID-19. ChemRxiv 2020, 29, 1657. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared Principles in Nf-Kappab Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. Er Stress Activates Nf-Kappab by Integrating Functions of Basal Ikk Activity, Ire1 and Perk. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef] [Green Version]
- Aydemir, M.N.; Aydemir, H.B.; Korkmaz, E.M.; Budak, M.; Cekin, N.; Pinarbasi, E. Computationally predicted SARS-CoV-2 encoded microRNAs target NFKB, JAK/STAT and TGFB signaling pathways. Gene Rep. 2020, 22, 101012. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galli, F.; Marcantonini, G.; Giustarini, D.; Albertini, M.C.; Migni, A.; Zatini, L.; Gioiello, A.; Rossi, R.; Bartolini, D. How Aging and Oxidative Stress Influence the Cytopathic and Inflammatory Effects of SARS-CoV-2 Infection: The Role of Cellular Glutathione and Cysteine Metabolism. Antioxidants 2022, 11, 1366. https://doi.org/10.3390/antiox11071366
Galli F, Marcantonini G, Giustarini D, Albertini MC, Migni A, Zatini L, Gioiello A, Rossi R, Bartolini D. How Aging and Oxidative Stress Influence the Cytopathic and Inflammatory Effects of SARS-CoV-2 Infection: The Role of Cellular Glutathione and Cysteine Metabolism. Antioxidants. 2022; 11(7):1366. https://doi.org/10.3390/antiox11071366
Chicago/Turabian StyleGalli, Francesco, Giada Marcantonini, Daniela Giustarini, Maria Cristina Albertini, Anna Migni, Linda Zatini, Antimo Gioiello, Ranieri Rossi, and Desirée Bartolini. 2022. "How Aging and Oxidative Stress Influence the Cytopathic and Inflammatory Effects of SARS-CoV-2 Infection: The Role of Cellular Glutathione and Cysteine Metabolism" Antioxidants 11, no. 7: 1366. https://doi.org/10.3390/antiox11071366
APA StyleGalli, F., Marcantonini, G., Giustarini, D., Albertini, M. C., Migni, A., Zatini, L., Gioiello, A., Rossi, R., & Bartolini, D. (2022). How Aging and Oxidative Stress Influence the Cytopathic and Inflammatory Effects of SARS-CoV-2 Infection: The Role of Cellular Glutathione and Cysteine Metabolism. Antioxidants, 11(7), 1366. https://doi.org/10.3390/antiox11071366