
Demethylation of H3K9 and H3K27 Contributes to the Tubular Renal Damage Triggered by Endoplasmic Reticulum Stress



 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
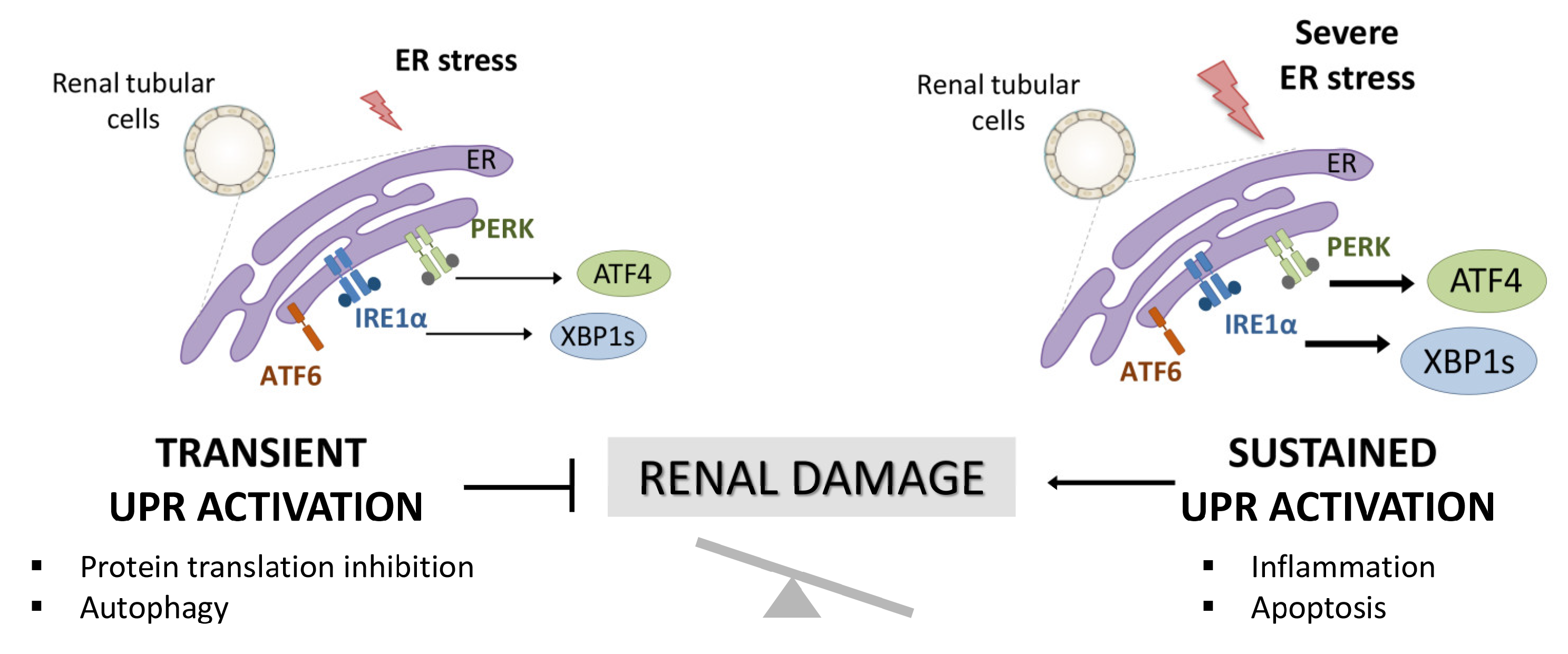
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatments
2.2. Cell Transfection and Gene Silencing
2.3. Gene Expression Studies
2.4. Western Blot Analysis
2.5. Chromatin Immunoprecipitation Assay
2.6. Statistical Analysis
3. Results
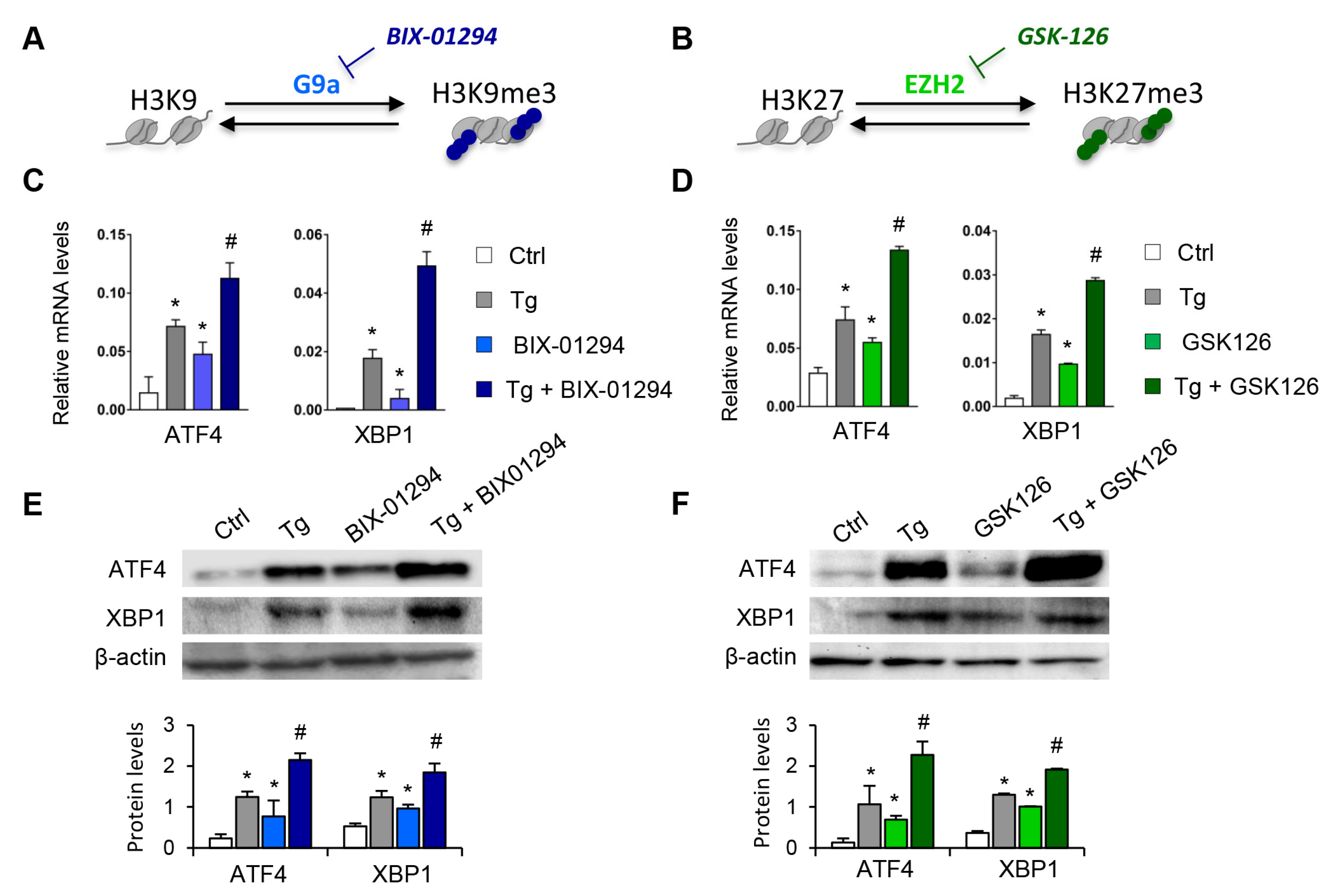
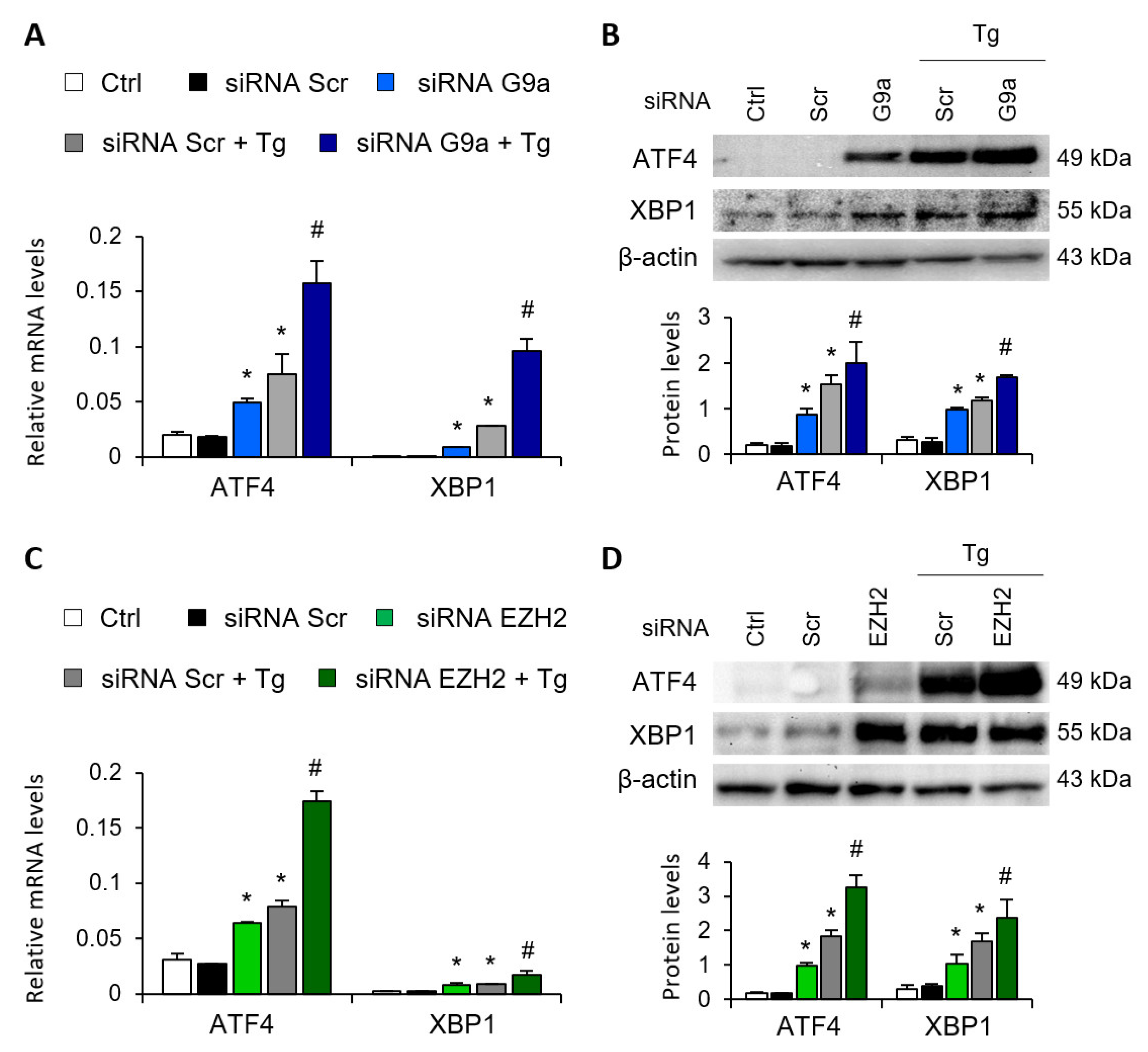
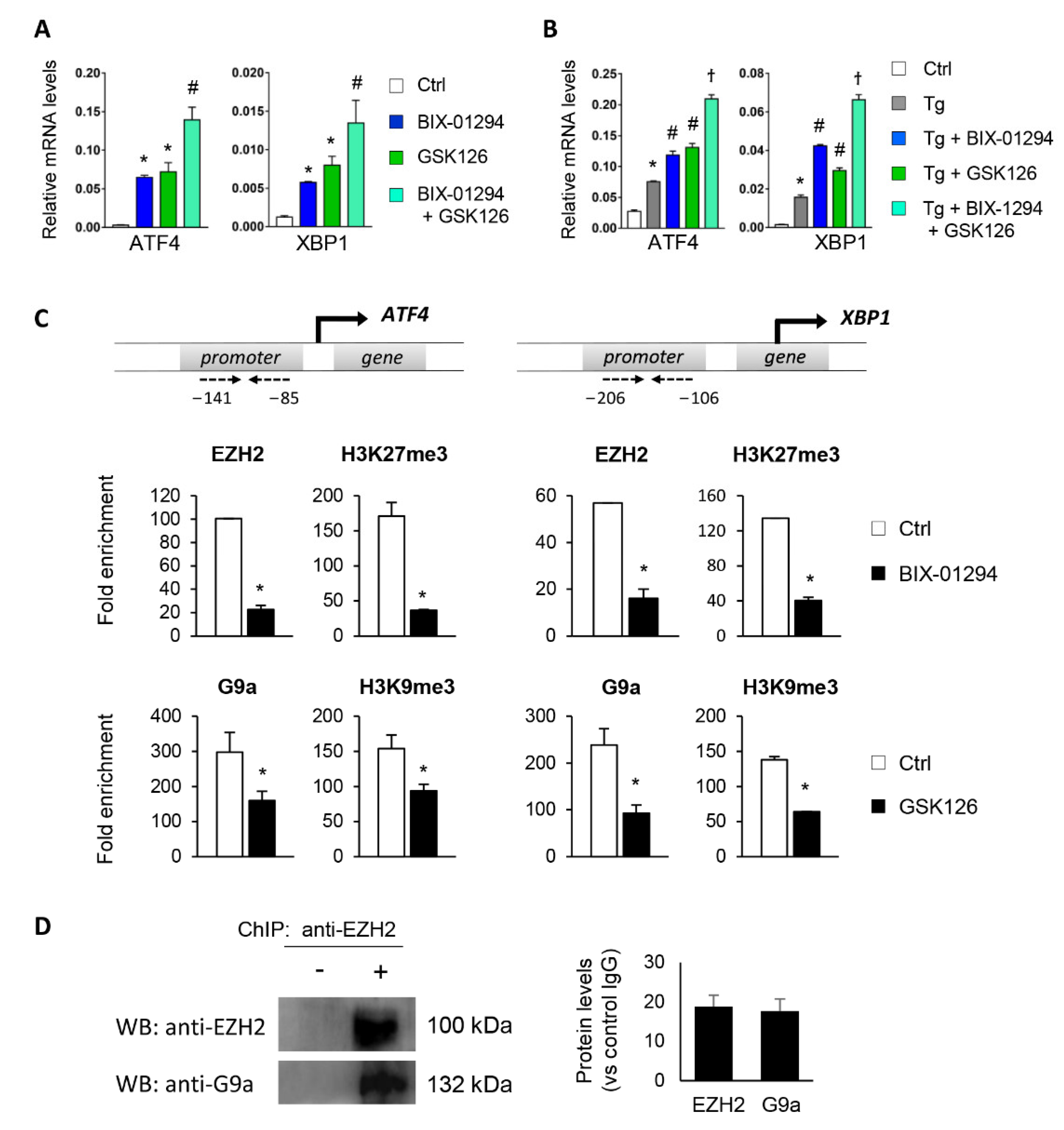
3.1. Blockage of the G9a and EZH2 HMTs Induces Expression of ATF4 and XBP1
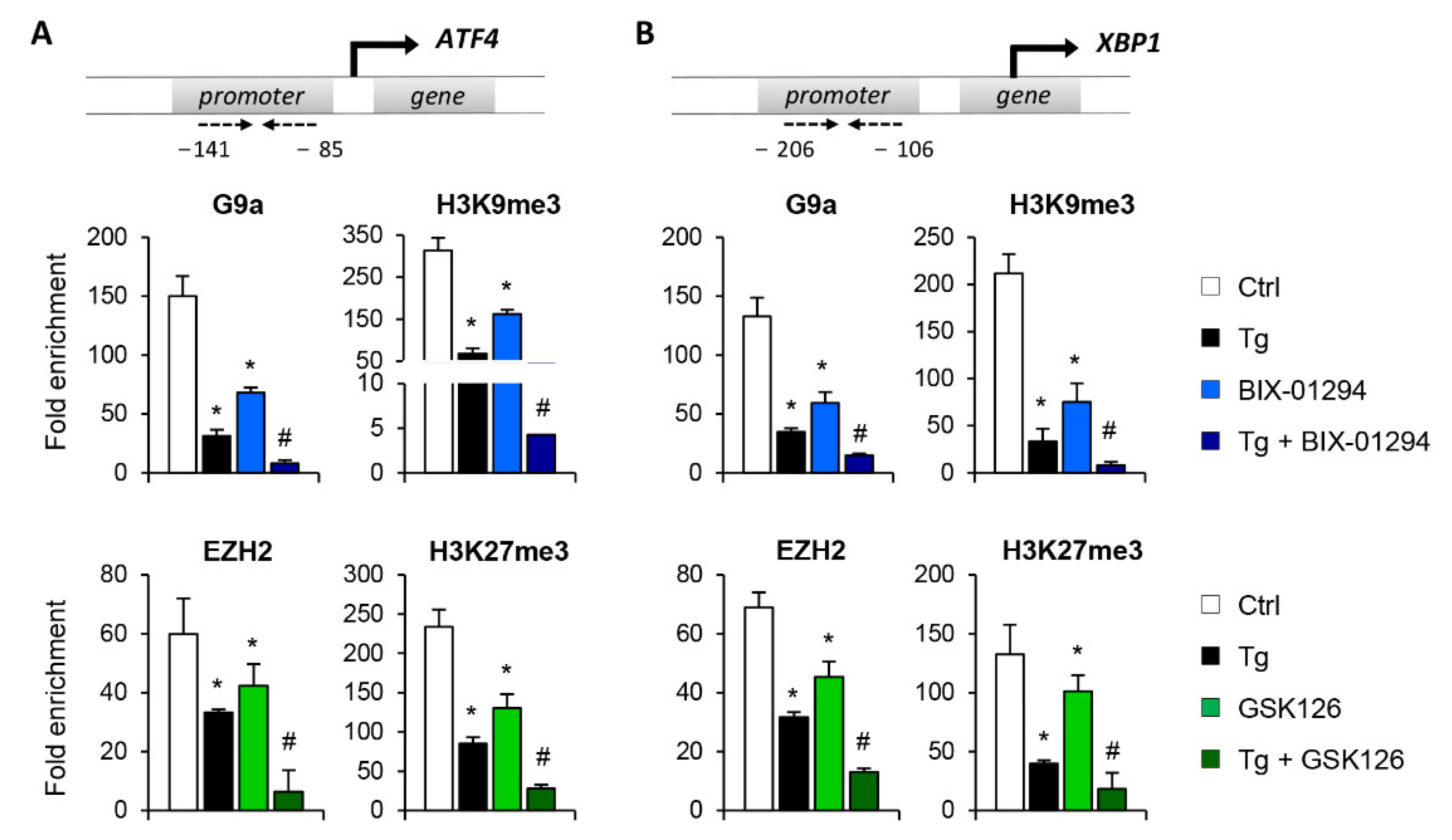
3.2. G9a and EZH2 HMTs Bind to ATF4 and XBP1 Promoter, Allowing the Formation of a Repressive Chromatin Structure
3.3. Crosstalk between G9a and EZH2 Represses Expression of ATF4 and XBP1
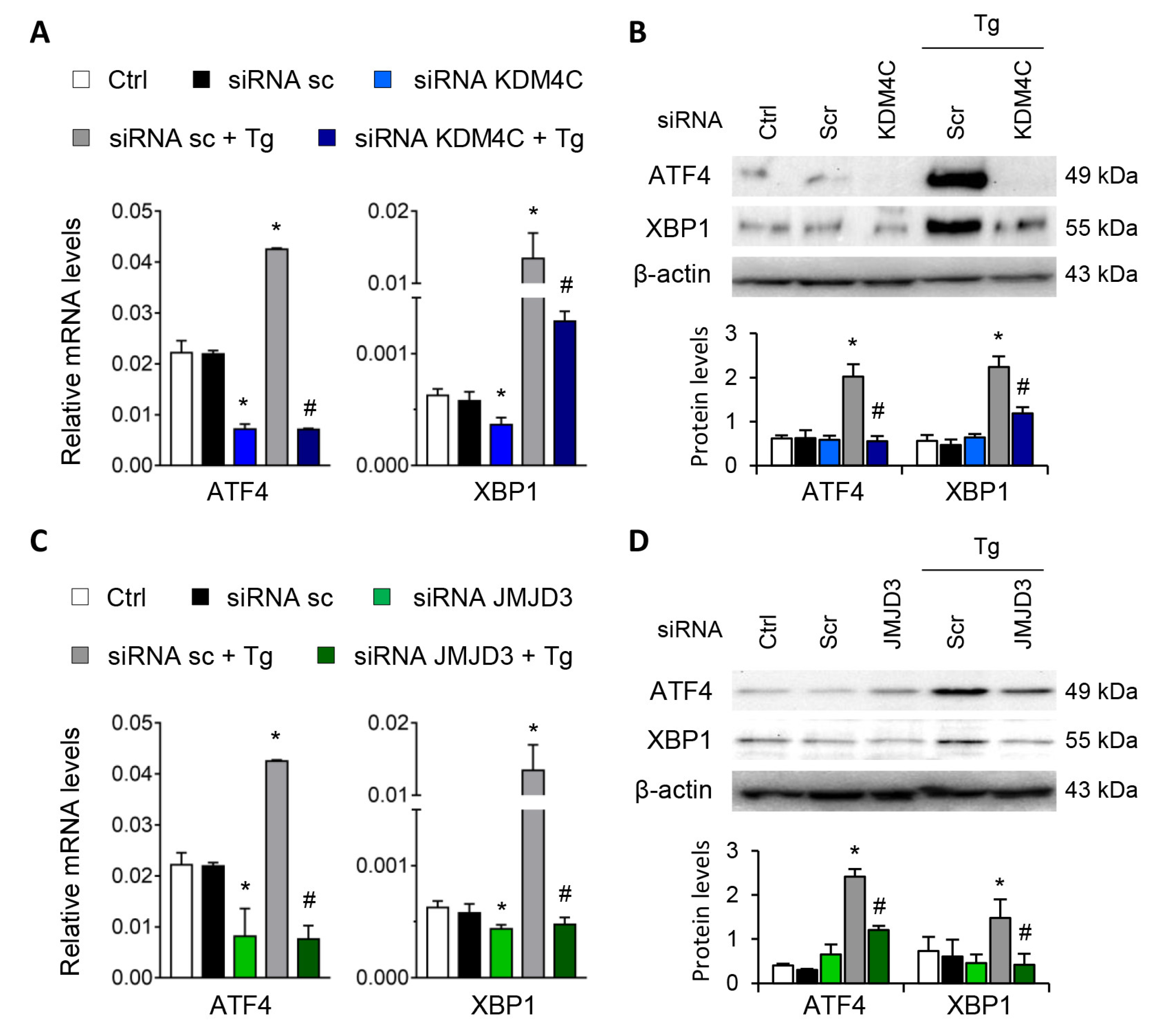
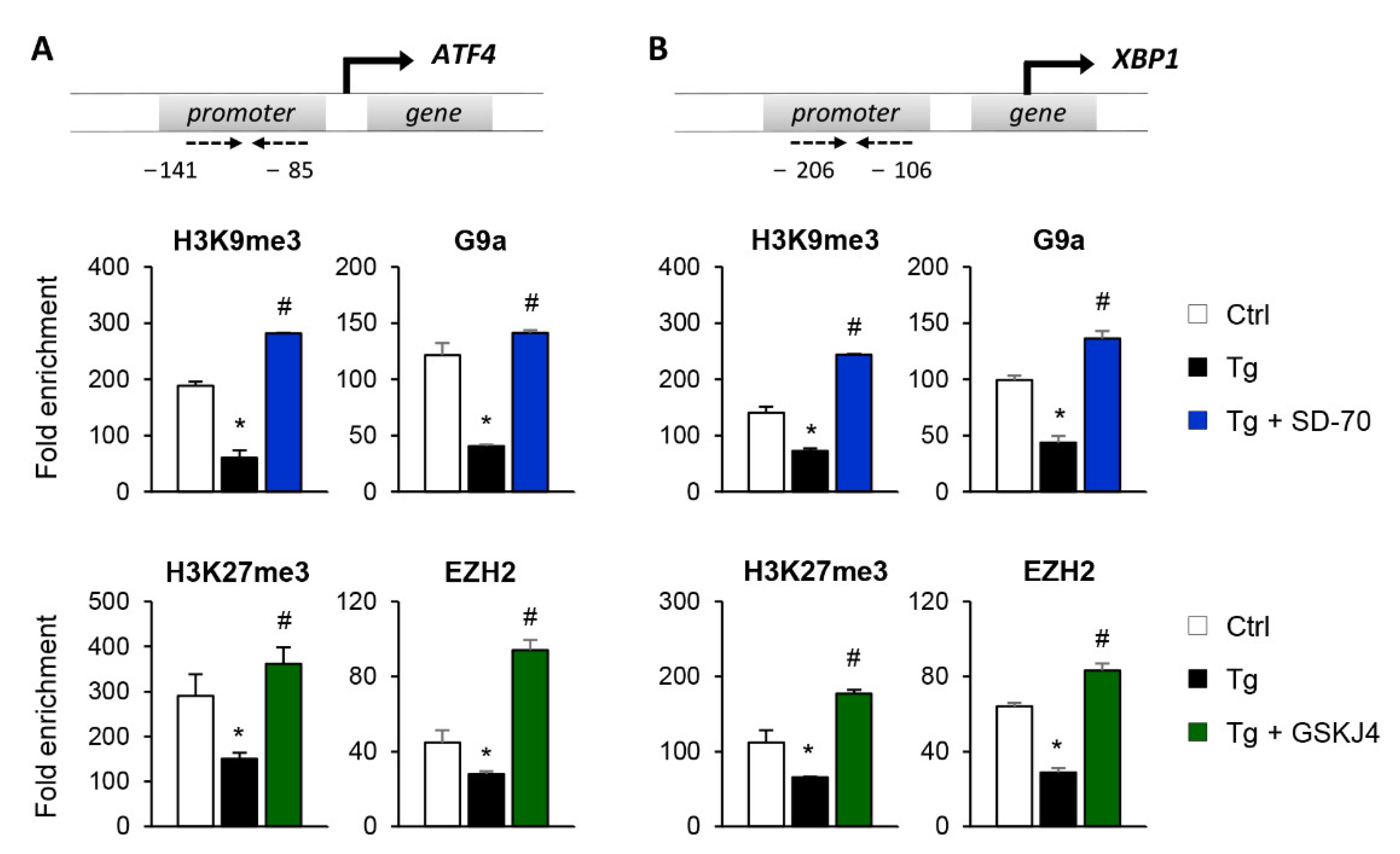
3.4. Blockage of the KDM4C and JMJD3 Histone Demethylases Impairs the ATF4 and XBP1 Expression under UPR Activation
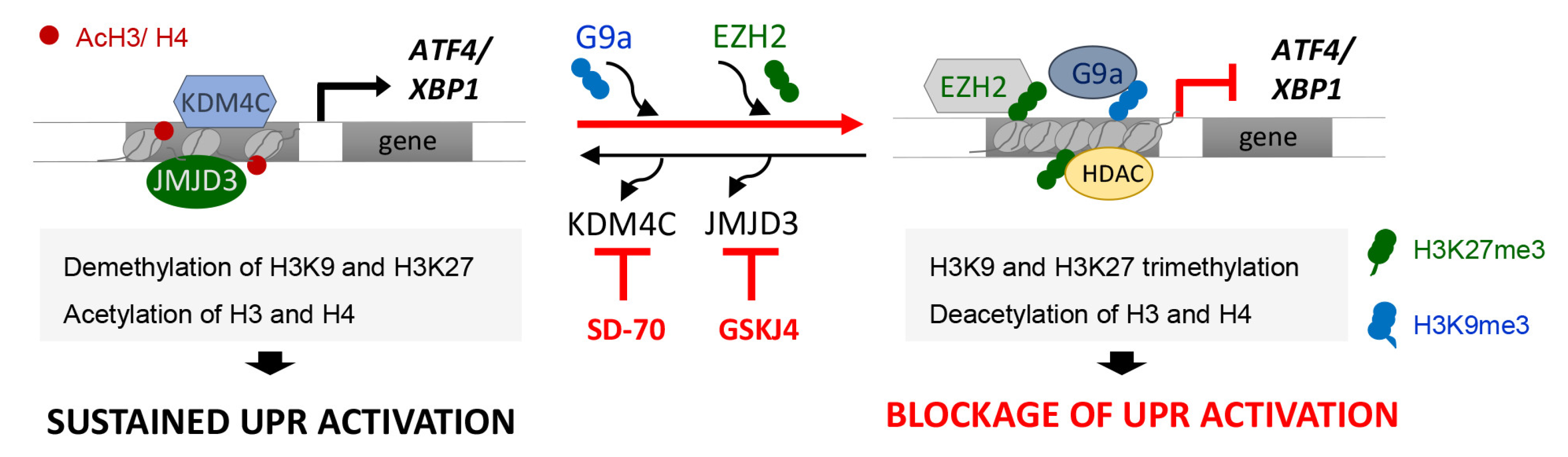
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Wu, C.T.; Huang, K.H.; Wang, C.C.; Guan, S.S.; Chen, L.P.; Chiang, C.K. C/EBP homologous protein (CHOP) deficiency ameliorates renal fibrosis in unilateral ureteral obstructive kidney disease. Oncotarget 2016, 7, 21900–21912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Huang, F.; Wang, Y.; Chen, C.; Wu, S.; Zhou, S.; Hei, Z.; Yuan, D. Connexin32 plays a crucial role in ROS-mediated endoplasmic reticulum stress apoptosis signaling pathway in ischemia reperfusion-induced acute kidney injury. J. Transl. Med. 2018, 16, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Mu, J.; Luo, Z.F.; Zeng, W.; Guo, Y.H.; Pang, Q.; Ye, Z.L.; Liu, L.; Yuan, F.H.; Feng, B. Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metab. Clin. Exp. 2011, 60, 594–603. [Google Scholar] [CrossRef]
- Cao, A.L.; Wang, L.; Chen, X.; Wang, Y.M.; Guo, H.J.; Chu, S.; Liu, C.; Zhang, X.M.; Peng, W. Ursodeoxycholic acid and 4-phenylbutyrate prevent endoplasmic reticulum stress-induced podocyte apoptosis in diabetic nephropathy. Lab. Investig. A J. Tech. Methods Pathol. 2016, 96, 610–622. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Zhang, J.; Xiao, W.; Lee, K.; Li, Z.; Wen, J.; He, L.; Gui, D.; Xue, R.; Jian, G.; et al. Rtn1a-Mediated Endoplasmic Reticulum Stress in Podocyte Injury and Diabetic Nephropathy. Sci. Rep. 2017, 7, 323. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Zhu, J.; Liu, Z.; Tang, C.; Cai, J.; Dong, Z. Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. eBioMedicine 2018, 37, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.H.; Yang, C.C.; Chan, D.C.; Wu, C.T.; Chen, L.P.; Huang, J.W.; Hung, K.Y.; Chiang, C.K. Chemical chaperon 4-phenylbutyrate protects against the endoplasmic reticulum stress-mediated renal fibrosis in vivo and in vitro. Oncotarget 2016, 7, 22116–22127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Fu, L.; Xiao, M.; Xu, C.; Sun, L.; Zhang, T.; Zheng, F.; Mei, C. The nephroprotective effect of tauroursodeoxycholic acid on ischaemia/reperfusion-induced acute kidney injury by inhibiting endoplasmic reticulum stress. Basic Clin. Pharmacol. Toxicol. 2012, 111, 14–23. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.; Weng, X.; Chen, H.; Du, Y.; Diao, C.; Chen, Z.; Liu, X. Inhibition of Brd4 alleviates renal ischemia/reperfusion injury-induced apoptosis and endoplasmic reticulum stress by blocking FoxO4-mediated oxidative stress. Redox Biol. 2019, 24, 101195–101208. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Alvarez, B.; Morgado-Pascual, J.L.; Rayego-Mateos, S.; Rodriguez, R.M.; Rodrigues-Diez, R.; Cannata-Ortiz, P.; Sanz, A.B.; Egido, J.; Tharaux, P.L.; Ortiz, A.; et al. Inhibition of Bromodomain and Extraterminal Domain Family Proteins Ameliorates Experimental Renal Damage. J. Am. Soc. Nephrol. 2017, 28, 504–519. [Google Scholar] [CrossRef] [Green Version]
- Tejedor-Santamaria, L.; Morgado-Pascual, J.L.; Marquez-Exposito, L.; Suarez-Alvarez, B.; Rodrigues-Diez, R.R.; Tejera-Munoz, A.; Marchant, V.; Mezzano, S.; Lopez-Larrea, C.; Sola, A.; et al. Epigenetic Modulation of Gremlin-1/NOTCH Pathway in Experimental Crescentic Immune-Mediated Glomerulonephritis. Pharmaceuticals 2022, 15, 121. [Google Scholar] [CrossRef] [PubMed]
- Tanemoto, F.; Mimura, I. Therapies Targeting Epigenetic Alterations in Acute Kidney Injury-to-Chronic Kidney Disease Transition. Pharmaceuticals 2022, 15, 123. [Google Scholar] [CrossRef]
- Liu, H.; Wang, W.; Weng, X.; Chen, H.; Chen, Z.; Du, Y.; Liu, X.; Wang, L. The H3K9 histone methyltransferase G9a modulates renal ischemia reperfusion injury by targeting Sirt1. Free Radic. Biol. Med. 2021, 172, 123–135. [Google Scholar] [CrossRef]
- Zhou, X.; Zang, X.; Guan, Y.; Tolbert, T.; Zhao, T.C.; Bayliss, G.; Zhuang, S. Targeting enhancer of zeste homolog 2 protects against acute kidney injury. Cell Death Dis. 2018, 9, 1067. [Google Scholar] [CrossRef]
- Zhou, X.; Zang, X.; Ponnusamy, M.; Masucci, M.V.; Tolbert, E.; Gong, R.; Zhao, T.C.; Liu, N.; Bayliss, G.; Dworkin, L.D.; et al. Enhancer of Zeste Homolog 2 Inhibition Attenuates Renal Fibrosis by Maintaining Smad7 and Phosphatase and Tensin Homolog Expression. J. Am. Soc. Nephrol. 2016, 27, 2092–2108. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Thieme, K.; Batchu, S.N.; Alghamdi, T.A.; Bowskill, B.B.; Kabir, M.G.; Liu, Y.; Advani, S.L.; White, K.E.; Geldenhuys, L.; et al. Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J. Clin. Investig. 2018, 128, 483–499. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, L.; Robin, P.; Mathieu, J.R.; Souidi, M.; Hinaux, H.; Rougeulle, C.; Harel-Bellan, A.; Ameyar-Zazoua, M.; Ait-Si-Ali, S. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol. Cell 2010, 37, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Chen, X.; Xiong, J.; Li, Y.; Li, H.; Ding, X.; Liu, S.; Chen, S.; Gao, S.; Zhu, B. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Res. 2011, 21, 365–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall Troselj, K.; Novak Kujundzic, R.; Ugarkovic, D. Polycomb repressive complex’s evolutionary conserved function: The role of EZH2 status and cellular background. Clin. Epigenet. 2016, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Yu, C.; Zhuang, S. Histone Methyltransferase EZH2: A Potential Therapeutic Target for Kidney Diseases. Front. Physiol. 2021, 12, 640700. [Google Scholar] [CrossRef]
- Hewitson, T.D.; Holt, S.G.; Tan, S.J.; Wigg, B.; Samuel, C.S.; Smith, E.R. Epigenetic Modifications to H3K9 in Renal Tubulointerstitial Cells after Unilateral Ureteric Obstruction and TGF-beta1 Stimulation. Front. Pharmacol. 2017, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Irifuku, T.; Doi, S.; Sasaki, K.; Doi, T.; Nakashima, A.; Ueno, T.; Yamada, K.; Arihiro, K.; Kohno, N.; Masaki, T. Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int. 2016, 89, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Sun, Z. Epigenetic Regulation of KL (Klotho) via H3K27me3 (Histone 3 Lysine [K] 27 Trimethylation) in Renal Tubule Cells. Hypertension 2020, 75, 1233–1241. [Google Scholar] [CrossRef]
- Zhou, X.; Xiong, C.; Tolbert, E.; Zhao, T.C.; Bayliss, G.; Zhuang, S. Targeting histone methyltransferase enhancer of zeste homolog-2 inhibits renal epithelial-mesenchymal transition and attenuates renal fibrosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 5976–5989. [Google Scholar] [CrossRef]
- Zeng, S.; Wu, X.; Chen, X.; Xu, H.; Zhang, T.; Xu, Y. Hypermethylated in cancer 1 (HIC1) mediates high glucose induced ROS accumulation in renal tubular epithelial cells by epigenetically repressing SIRT1 transcription. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 917–927. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Z.; Weng, X.; Chen, H.; Du, Y.; Diao, C.; Liu, X.; Wang, L. Enhancer of zeste homolog 2 modulates oxidative stress-mediated pyroptosis in vitro and in a mouse kidney ischemia-reperfusion injury model. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 835–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Peng, Y.; Fang, Z.; Li, L.; Ming, S.; Dong, H.; Li, R.; Zhu, Y.; Zhang, W.; Zhu, B.; et al. Inhibition of EZH2 ameliorates hyperoxaluria-induced kidney injury through the JNK/FoxO3a pathway. Life Sci. 2022, 291, 120258. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, F.S.; Majumder, S.; Thai, K.; Abdalla, M.; Hu, P.; Advani, S.L.; White, K.E.; Bowskill, B.B.; Guarna, G.; Dos Santos, C.C.; et al. The Histone Methyltransferase Enzyme Enhancer of Zeste Homolog 2 Protects against Podocyte Oxidative Stress and Renal Injury in Diabetes. J. Am. Soc. Nephrol. 2016, 27, 2021–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Liao, R.; Liu, C.; Liu, S.; Huang, H.; Liu, J.; Jin, T.; Guo, H.; Zheng, Z.; Xia, M.; et al. Epigenetic regulation of TXNIP-mediated oxidative stress and NLRP3 inflammasome activation contributes to SAHH inhibition-aggravated diabetic nephropathy. Redox Biol. 2021, 45, 102033. [Google Scholar] [CrossRef]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef]
- Bujisic, B.; De Gassart, A.; Tallant, R.; Demaria, O.; Zaffalon, L.; Chelbi, S.; Gilliet, M.; Bertoni, F.; Martinon, F. Impairment of both IRE1 expression and XBP1 activation is a hallmark of GCB DLBCL and contributes to tumor growth. Blood 2017, 129, 2420–2428. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.Y.; Lo, H.L.; Yang, P.M. EZH2 inhibitors transcriptionally upregulate cytotoxic autophagy and cytoprotective unfolded protein response in human colorectal cancer cells. Am. J. Cancer Res. 2016, 6, 1661–1680. [Google Scholar]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Mozzetta, C.; Pontis, J.; Fritsch, L.; Robin, P.; Portoso, M.; Proux, C.; Margueron, R.; Ait-Si-Ali, S. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell 2014, 53, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, K.; Kitajima, H.; Niinuma, T.; Maruyama, R.; Nishiyama, N.; Ohtani, H.; Sudo, G.; Toyota, M.; Sasaki, H.; Yamamoto, E.; et al. Dual EZH2 and G9a inhibition suppresses multiple myeloma cell proliferation by regulating the interferon signal and IRF4-MYC axis. Cell Death Discov. 2021, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Hu, Y.; Guo, D.; Zhang, A.; Gu, Y.; Zhang, S.; Zhao, C.; Gong, P.; Shen, X.; Li, Y.; et al. DNA methyltransferase 3A isoform b contributes to repressing E-cadherin through cooperation of DNA methylation and H3K27/H3K9 methylation in EMT-related metastasis of gastric cancer. Oncogene 2018, 37, 4358–4371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Shang, M.; Li, H.; Wu, L.; Dong, M.; Huang, B.; Lu, J.; Zhang, Y. Dual Inhibition of H3K9me2 and H3K27me3 Promotes Tumor Cell Senescence without Triggering the Secretion of SASP. Int. J. Mol. Sci. 2022, 23, 3911. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.R.; Brand, O.J.; Pasini, A.; Jenkins, G.; Knox, A.J.; Pang, L. Interplay between EZH2 and G9a Regulates CXCL10 Gene Repression in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef] [Green Version]
- Jagsi, R. Quality of life after multicatheter breast brachytherapy. Lancet Oncol. 2018, 19, 726–727. [Google Scholar] [CrossRef]
- Yilmaz, D.E.; Kirschner, K.; Demirci, H.; Himmerkus, N.; Bachmann, S.; Mutig, K. Immunosuppressive calcineurin inhibitor cyclosporine A induces proapoptotic endoplasmic reticulum stress in renal tubular cells. J. Biol. Chem. 2022, 298, 101589. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Zhao, E.; Ding, J.; Xia, Y.; Liu, M.; Ye, B.; Choi, J.H.; Yan, C.; Dong, Z.; Huang, S.; Zha, Y.; et al. KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep. 2016, 14, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Huang, Y.; Zhu, X.; Liu, C.; Yuan, Y.; Su, H.; Zhang, C.; Liu, C.; Xiong, M.; Qu, Y.; et al. Histone demethylase UTX is a therapeutic target for diabetic kidney disease. J. Physiol. 2019, 597, 1643–1660. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Reddy, M.A.; Das, S.; Oh, H.J.; Abdollahi, M.; Yuan, H.; Zhang, E.; Lanting, L.; Wang, M.; Natarajan, R. Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-beta1-induced gene expression in mesangial cells and diabetic kidney. J. Biol. Chem. 2019, 294, 12695–12707. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Xiong, C.; Tang, J.; Hou, X.; Liu, N.; Bayliss, G.; Zhuang, S. Histone demethylase JMJD3 protects against renal fibrosis by suppressing TGFbeta and Notch signaling and preserving PTEN expression. Theranostics 2021, 11, 2706–2721. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhuang, S. Histone Methyltransferases as Therapeutic Targets for Kidney Diseases. Front. Pharmacol. 2019, 10, 1393. [Google Scholar] [CrossRef] [PubMed]
- Duan, A.; Wang, H.; Zhu, Y.; Wang, Q.; Zhang, J.; Hou, Q.; Xing, Y.; Shi, J.; Hou, J.; Qin, Z.; et al. Chromatin architecture reveals cell type-specific target genes for kidney disease risk variants. BMC Biol. 2021, 19, 38. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-Bulnes, P.; Saiz, M.L.; Corte-Iglesias, V.; Rodrigues-Diez, R.R.; Bernardo Florez, A.; Ruiz Bernet, C.; Martin Martin, C.; Ruiz-Ortega, M.; Suarez-Alvarez, B.; López-Larrea, C. Demethylation of H3K9 and H3K27 Contributes to the Tubular Renal Damage Triggered by Endoplasmic Reticulum Stress. Antioxidants 2022, 11, 1355. https://doi.org/10.3390/antiox11071355
Diaz-Bulnes P, Saiz ML, Corte-Iglesias V, Rodrigues-Diez RR, Bernardo Florez A, Ruiz Bernet C, Martin Martin C, Ruiz-Ortega M, Suarez-Alvarez B, López-Larrea C. Demethylation of H3K9 and H3K27 Contributes to the Tubular Renal Damage Triggered by Endoplasmic Reticulum Stress. Antioxidants. 2022; 11(7):1355. https://doi.org/10.3390/antiox11071355
Chicago/Turabian StyleDiaz-Bulnes, Paula, Maria Laura Saiz, Viviana Corte-Iglesias, Raúl R Rodrigues-Diez, Aida Bernardo Florez, Cristian Ruiz Bernet, Cristina Martin Martin, Marta Ruiz-Ortega, Beatriz Suarez-Alvarez, and Carlos López-Larrea. 2022. "Demethylation of H3K9 and H3K27 Contributes to the Tubular Renal Damage Triggered by Endoplasmic Reticulum Stress" Antioxidants 11, no. 7: 1355. https://doi.org/10.3390/antiox11071355
APA StyleDiaz-Bulnes, P., Saiz, M. L., Corte-Iglesias, V., Rodrigues-Diez, R. R., Bernardo Florez, A., Ruiz Bernet, C., Martin Martin, C., Ruiz-Ortega, M., Suarez-Alvarez, B., & López-Larrea, C. (2022). Demethylation of H3K9 and H3K27 Contributes to the Tubular Renal Damage Triggered by Endoplasmic Reticulum Stress. Antioxidants, 11(7), 1355. https://doi.org/10.3390/antiox11071355