
Administration of Glutaredoxin-1 Attenuates Liver Fibrosis Caused by Aging and Non-Alcoholic Steatohepatitis


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Mice
2.3. Collagen Staining, Hematoxylin and Eosin (H&E) Staining
2.4. Gene Expression
2.5. Isolation of Hepatocytes and Primary Culture
2.6. Western Blot
2.7. Adeno-Associated Viruses (AAV) Production and In Vivo Injection
2.8. Liver and Serum Biochemical Measurements
2.9. Microarray
2.10. Statistical Analysis
3. Results
3.1. Glrx Reduces Age-Induced Fibrosis in the Liver
3.2. Modulation of Glrx Levels Controls Fibrosis and Apoptosis in the Aged Liver
3.3. Modulation of Glrx Levels Controls Apoptosis in Hepatocytes in Vitro
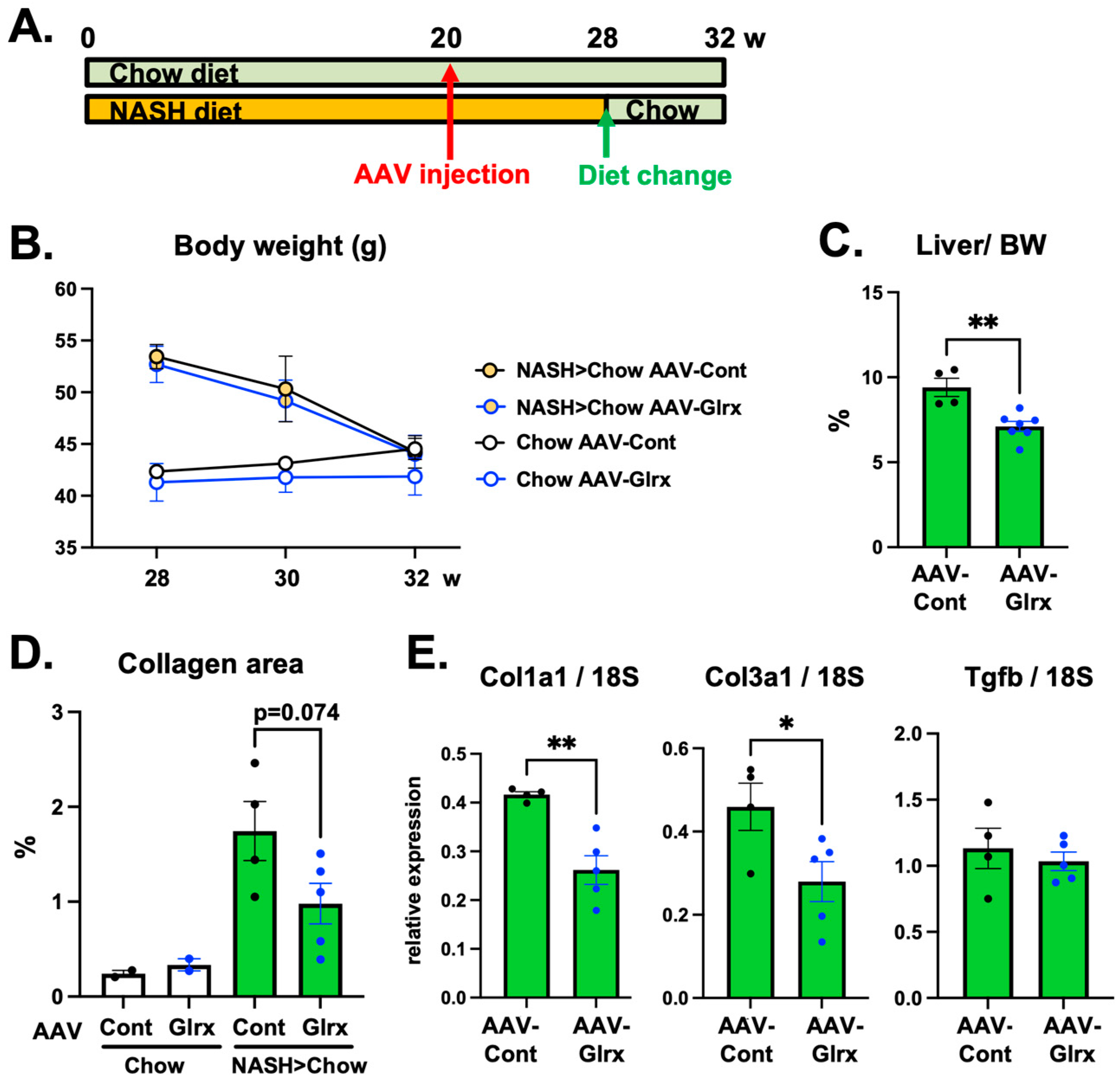
3.4. Overexpression of Glrx Attenuates Liver Fibrosis in Diet-Induced NASH
3.5. AAV-Glrx Provides Anti-Fibrotic Benefits to Dietary Modification in NASH
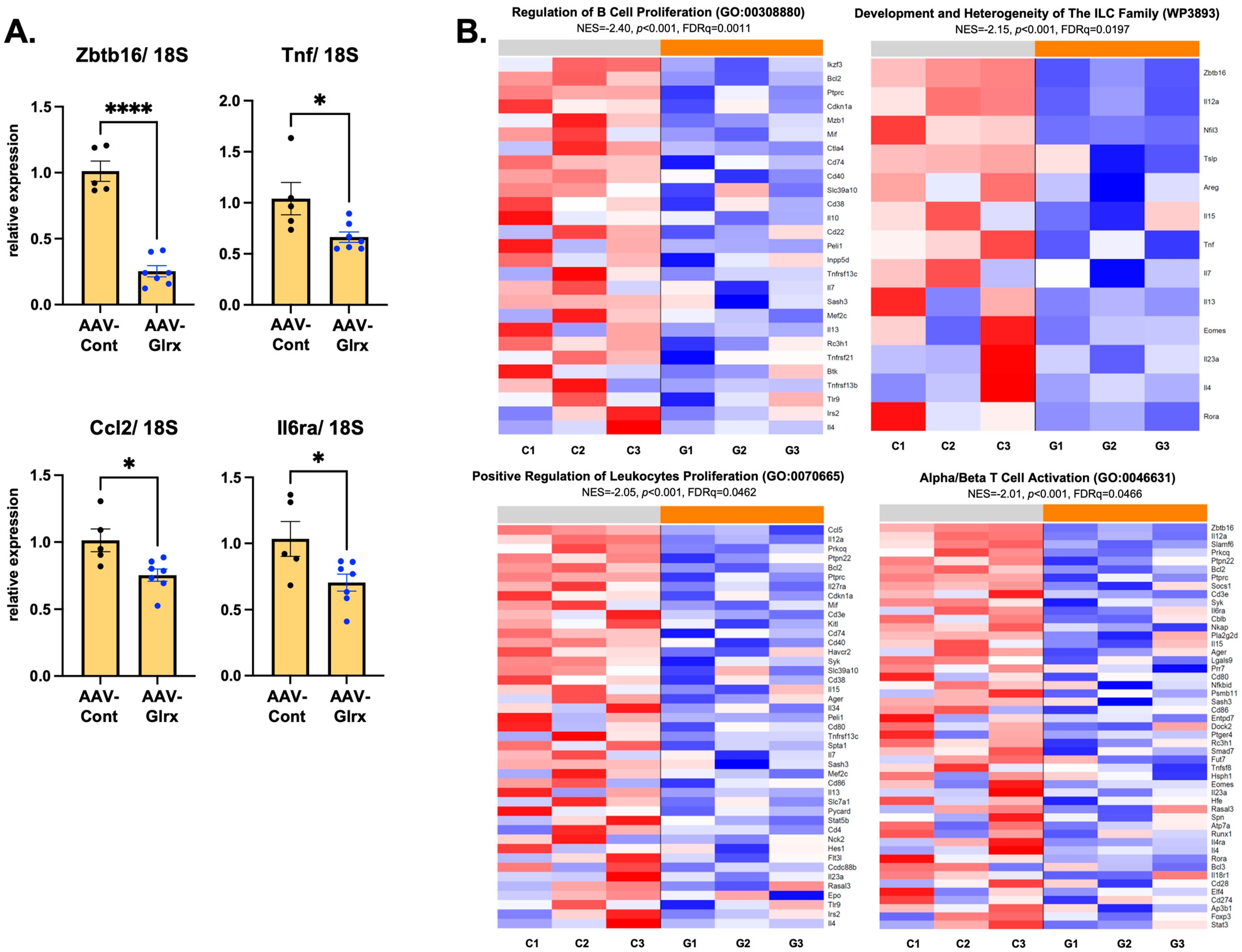
3.6. AAV-Glrx Downregulates Immune-Related Pathways in NASH Liver
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hagström, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stål, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.-W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis affects the progression of non-alcoholic steatohepatitis via the modulation of lipid peroxidation–mediated cell death in mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Walenbergh, S.M.A.; Koek, G.H.; Bieghs, V.; Shiri-Sverdlov, R. Non-alcoholic steatohepatitis: The role of oxidized low-density lipoproteins. J. Hepatol. 2013, 58, 801–810. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Harrison, S.A.; Torgerson, S.; Hayashi, P.; Ward, J.; Schenker, S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2003, 98, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.P.; Sanyal, A.J.; Kowdley, K.V.; Robuck, P.R.; Hoofnagle, J.; Kleiner, D.E.; Unalp, A.; Tonascia, J. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp. Clin. Trials 2009, 30, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Pacana, T.; Sanyal, A.J. Vitamin E and nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 641–648. [Google Scholar] [CrossRef] [PubMed]
- El Hadi, H.; Vettor, R.; Rossato, M. Vitamin E as a treatment for nonalcoholic fatty liver disease: Reality or myth? Antioxidants 2018, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Frith, J.; Jones, D.; Newton, J.L. Chronic liver disease in an ageing population. Age Ageing 2009, 38, 11–18. [Google Scholar] [CrossRef]
- Poynard, T.; Mathurin, P.; Lai, C.-L.; Guyader, D.; Poupon, R.; Tainturier, M.-H.; Myers, R.P.; Muntenau, M.; Ratziu, V.; Manns, M.; et al. A comparison of fibrosis progression in chronic liver diseases. J. Hepatol. 2003, 38, 257–265. [Google Scholar] [CrossRef]
- Maeso-Díaz, R.; Ortega-Ribera, M.; Lafoz, E.; JoséLozano, J.; Baiges, A.; Francés, R.; Albillos, A.; Peralta, C.; García-Pagán, J.C.; Bosch, J.; et al. Aging influences hepatic microvascular biology and liver fibrosis in advanced chronic liver disease. Aging Dis. 2019, 10, 684–698. [Google Scholar] [CrossRef]
- Fontana, L.; Zhao, E.; Amir, M.; Dong, H.; Tanaka, K.; Czaja, M.J. Aging promotes the development of diet-induced murine steatohepatitis but not steatosis. Hepatology 2013, 57, 995–1004. [Google Scholar] [CrossRef]
- Kim, I.H.; Xu, J.; Liu, X.; Koyama, Y.; Ma, H.Y.; Diggle, K.; You, Y.H.; Schilling, J.M.; Jeste, D.; Sharma, K.; et al. Aging increases the susceptibility of hepatic inflammation, liver fibrosis and aging in response to high-fat diet in mice. Age (Omaha) 2016, 38, 291–302. [Google Scholar] [CrossRef]
- Delire, B.; Lebrun, V.; Selvais, C.; Henriet, P.; Bertrand, A.; Horsmans, Y.; Leclercq, I.A. Aging enhances liver fibrotic response in mice through hampering extracellular matrix remodeling. Aging (Albany N. Y.). 2017, 9, 98–113. [Google Scholar] [CrossRef]
- Hong, I.-H.; Lewis, K.; Iakova, P.; Jin, J.; Sullivan, E.; Jawanmardi, N.; Timchenko, L.; Timchenko, N. Age-associated Change of C/EBP Family Proteins Causes Severe Liver Injury and Acceleration of Liver Proliferation after CCl4 Treatments. J. Biol. Chem. 2014, 289, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Han, J.; Hou, X.; Fry, J.; Behring, J.B.; Seta, F.; Long, M.T.; Roy, H.K.; Cohen, R.A.; Matsui, R.; et al. Glutaredoxin-1 Deficiency Causes Fatty Liver and Dyslipidemia by Inhibiting Sirtuin-1. Antioxid. Redox Signal. 2017, 27, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-S.; Xiong, Y.; Ho, D.S.; Gao, J.; Chua, B.H.L.; Pai, H.; Mieyal, J.J. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic. Biol. Med. 2007, 43, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Bachschmid, M.M.; Xu, S.; Maitland-Toolan, K.A.; Ho, Y.-S.; Cohen, R.A.; Matsui, R. Attenuated cardiovascular hypertrophy and oxidant generation in response to angiotensin II infusion in glutaredoxin-1 knockout mice. Free Radic. Biol. Med. 2010, 49, 1221–1229. [Google Scholar] [CrossRef][Green Version]
- Shao, D.; Fry, J.L.; Han, J.; Hou, X.; Pimentel, D.R.; Matsui, R.; Cohen, R.A.; Bachschmid, M.M. A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 2014, 289, 7293–7306. [Google Scholar] [CrossRef]
- Anathy, V.; Aesif, S.W.; Guala, A.S.; Havermans, M.; Reynaert, N.L.; Ho, Y.S.; Budd, R.C.; Janssen-Heininger, Y.M.W. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J. Cell Biol. 2009, 184, 241–252. [Google Scholar] [CrossRef]
- Anathy, V.; Lahue, K.G.; Chapman, D.G.; Chia, S.B.; Casey, D.T.; Aboushousha, R.; van der Velden, J.L.J.; Elko, E.; Hoffman, S.M.; McMillan, D.H.; et al. Reducing protein oxidation reverses lung fibrosis. Nat. Med. 2018, 24, 1128–1135. [Google Scholar] [CrossRef]
- Murdoch, C.E.; Shuler, M.; Haeussler, D.J.F.; Kikuchi, R.; Bearelly, P.; Han, J.; Watanabe, Y.; Fuster, J.J.; Walsh, K.; Ho, Y.-S.; et al. Glutaredoxin-1 Up-regulation Induces Soluble Vascular Endothelial Growth Factor Receptor 1, Attenuating Post-ischemia Limb Revascularization. J. Biol. Chem. 2014, 289, 8633–8644. [Google Scholar] [CrossRef]
- Boland, M.L.; Oró, D.; Tølbøl, K.S.; Thrane, S.T.; Nielsen, J.C.; Cohen, T.S.; Tabor, D.E.; Fernandes, F.; Tovchigrechko, A.; Veidal, S.S.; et al. Towards a standard diet-induced and biopsy-confirmed mouse model of non-alcoholic steatohepatitis: Impact of dietary fat source. World J. Gastroenterol. 2019, 25, 4904–4920. [Google Scholar] [CrossRef]
- Clapper, J.R.; Hendricks, M.D.; Gu, G.; Wittmer, C.; Dolman, C.S.; Herich, J.; Athanacio, J.; Villescaz, C.; Ghosh, S.S.; Heilig, J.S.; et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol.-Gastrointest. Liver Physiol. 2013, 305, 483–495. [Google Scholar] [CrossRef]
- Mederacke, I.; Dapito, D.H.; Affò, S.; Uchinami, H.; Schwabe, R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015, 10, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Yan, H.; Ou, H. Human thyroxine binding globulin (TBG) promoter directs efficient and sustaining transgene expression in liver-specific pattern. Gene 2012, 506, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Ferran, B.; Tsukahara, Y.; Shang, Q.; Desai, S.; Fedoce, A.; Pimentel, D.R.; Luptak, I.; Adachi, T.; Ido, Y.; et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Sci. Rep. 2019, 9, 13601. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Carrasco, N.; Chandler, R.J.; Chandrasekaran, S.; Venditti, C.P. Liver-directed recombinant adeno-associated viral gene delivery rescues a lethal mouse model of methylmalonic acidemia and provides long-term phenotypic correction. Hum. Gene Ther. 2010, 21, 1147–1154. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.-S.; Matthews, D.E.; et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef]
- Shelton, M.D.; Kern, T.S.; Mieyal, J.J. Glutaredoxin regulates nuclear factor kappa-B and intercellular adhesion molecule in Müller cells: Model of diabetic retinopathy. J. Biol. Chem. 2007, 282, 12467–12474. [Google Scholar] [CrossRef]
- Zee, R.S.; Yoo, C.B.; Pimentel, D.R.; Perlman, D.H.; Burgoyne, J.R.; Hou, X.; McComb, M.E.; Costello, C.E.; Cohen, R.A.; Bachschmid, M.M. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid. Redox Signal. 2010, 13, 1023–1032. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, P.; Chen, Z.; Xia, H.; Li, S.; Zhang, Y.; Kobberup, S.; Zou, W.; Lin, J.D. Hepatic neuregulin 4 signaling defines an endocrine checkpoint for steatosis-to-NASH progression. J. Clin. Investig. 2017, 127, 4449–4461. [Google Scholar] [CrossRef]
- Witek, R.P.; Stone, W.C.; Karaca, F.G.; Syn, W.K.; Pereira, T.A.; Agboola, K.M.; Omenetti, A.; Jung, Y.; Teaberry, V.; Choi, S.S.; et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1421–1430. [Google Scholar] [CrossRef]
- Park, J.Y.; DiPalma, D.T.; Kwon, J.; Fink, J.; Park, J.H. Quantitative Difference in PLZF Protein Expression Determines iNKT Lineage Fate and Controls Innate CD8 T Cell Generation. Cell Rep. 2019, 27, 2548–2557.e4. [Google Scholar] [CrossRef]
- Šeda, O.; Šedová, L.; Vcelák, J.; Vanková, M.; Liška, F.; Bendlová, B. ZBTB16 and metabolic syndrome: A network perspective. Physiol. Res. 2017, 66, S357–S365. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Kumar, A.; Chang, C.-H.; Pyaram, K. Reactive Oxygen Species Regulate the Inflammatory Function of NKT Cells through Promyelocytic Leukemia Zinc Finger. J. Immunol. 2017, 199, 3478–3487. [Google Scholar] [CrossRef]
- Maricic, I.; Marrero, I.; Eguchi, A.; Nakamura, R.; Johnson, C.D.; Dasgupta, S.; Hernandez, C.D.; Nguyen, P.S.; Swafford, A.D.; Knight, R.; et al. Differential Activation of Hepatic Invariant NKT Cell Subsets Plays a Key Role in Progression of Nonalcoholic Steatohepatitis. J. Immunol. 2018, 201, 3017–3035. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimizu, Y. Role of NKT Cells in the Pathogenesis of NAFLD. Int. J. Hepatol. 2012, 2012, 850836. [Google Scholar] [CrossRef]
- Barrow, F.; Khan, S.; Wang, H.; Revelo, X.S. The Emerging Role of B Cells in the Pathogenesis of NAFLD. Hepatology 2021, 74, 2277–2286. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, M.J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.; An, M.R.; Papaconstantinou, J. Differential expression of three C/EBP isoforms in multiple tissues during the acute phase response. J. Biol. Chem. 1992, 267, 5021–5024. [Google Scholar] [CrossRef]
- Watanabe, Y.; Watanabe, K.; Fujioka, D.; Nakamura, K.; Nakamura, T.; Uematsu, M.; Bachschmid, M.M.; Matsui, R.; Kugiyama, K. Protein S-glutathionylation stimulate adipogenesis by stabilizing C/EBPβ in 3T3L1 cells. FASEB J. 2020, 34, 5827–5837. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Cervera, A.; Vinué, Á.; Burks, D.J.; González-Navarro, H. Genetic inactivation of the LIGHT (TNFSF14) cytokine in mice restores glucose homeostasis and diminishes hepatic steatosis. Diabetologia 2019, 62, 2143–2157. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Accession | Forward Primer | Reverse Primer |
|---|---|---|---|
| Ccl2 | NM_011333 | 5′-TTAAAAACCTGGATCGGAACCAA-3′ | 5′-GCATTAGCTTCAGATTTACGGGT-3′ |
| Col1a1 | NM_007742 | 5′-CCCGAACCCCAAGGAAAAGA-3′ | 5′-CGTACTCGAACGGGAATCCA-3′ |
| Col3a1 | NM_009930 | 5′-CAGAGGAGAAACTGGCCCTG-3′ | 5′-TTGTCACCTCGTGGACCTTG-3′ |
| Ctgf | NM_010217 | 5′-CTAGCTGCCTACCGACTGGA-3′ | 5′-GTAACTCGGGTGGAGATGCC-3′ |
| Il1b | NM_008361 | 5′-GCAACTGTTCCTGAACTCAACT-3′ | 5′-ATCTTTTGGGGTCCGTCAACT-3′ |
| Il4 | NM_021283 | 5′-GGTCTCAACCCCCAGCTAGT-3′ | 5′-GCCGATGATCTCTCTCAAGTGAT-3′ |
| Il6ra | NM_010559 | 5′-CCTGAGACTCAAGCAGAAATGG-3′ | 5′-AGAAGGAAGGTCGGCTTCAGT-3′ |
| Tgfb1 | NM_011577 | 5′-GCTGAACCAAGGAGACGGAA-3′ | 5′-ATGTCATGGATGGTGCCCAG-3′ |
| Tnf | NM_013693 | 5′-ATGGCCTCCCTCTCATCAGT-3′ | 5′-CTTGGTGGTTTGCTACGACG-3′ |
| Zbtb16 | NM_001033324 | 5′-CTGGGACTTTGTGCGATGTG-3′ | 5′-CGGTGGAAGAGGATCTCAAACA-3′ |
| 18S RNA | NR_003278.3 | 5′-CCGCCGCCATGTCTCTAGT-3′ | 5′-GCCCATCGATGTTGGTGTTG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsukahara, Y.; Ferran, B.; Minetti, E.T.; Chong, B.S.H.; Gower, A.C.; Bachschmid, M.M.; Matsui, R. Administration of Glutaredoxin-1 Attenuates Liver Fibrosis Caused by Aging and Non-Alcoholic Steatohepatitis. Antioxidants 2022, 11, 867. https://doi.org/10.3390/antiox11050867
Tsukahara Y, Ferran B, Minetti ET, Chong BSH, Gower AC, Bachschmid MM, Matsui R. Administration of Glutaredoxin-1 Attenuates Liver Fibrosis Caused by Aging and Non-Alcoholic Steatohepatitis. Antioxidants. 2022; 11(5):867. https://doi.org/10.3390/antiox11050867
Chicago/Turabian StyleTsukahara, Yuko, Beatriz Ferran, Erika T. Minetti, Brian S. H. Chong, Adam C. Gower, Markus M. Bachschmid, and Reiko Matsui. 2022. "Administration of Glutaredoxin-1 Attenuates Liver Fibrosis Caused by Aging and Non-Alcoholic Steatohepatitis" Antioxidants 11, no. 5: 867. https://doi.org/10.3390/antiox11050867
APA StyleTsukahara, Y., Ferran, B., Minetti, E. T., Chong, B. S. H., Gower, A. C., Bachschmid, M. M., & Matsui, R. (2022). Administration of Glutaredoxin-1 Attenuates Liver Fibrosis Caused by Aging and Non-Alcoholic Steatohepatitis. Antioxidants, 11(5), 867. https://doi.org/10.3390/antiox11050867