
Reversal of Lipid Metabolism Dysregulation by Selenium and Folic Acid Co-Supplementation to Mitigate Pathology in Alzheimer’s Disease

 ,
,
Abstract
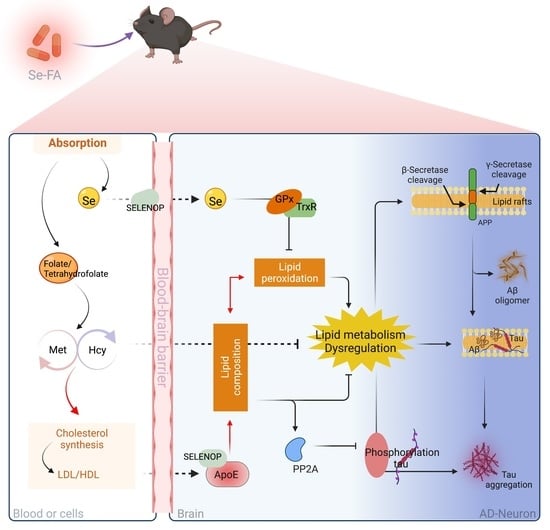
1. Introduction
2. Materials and Methods
2.1. Mice and Diet
2.2. Se Levels
2.3. ELISA and Automated Chemistry Analyzer
2.4. Immunoblotting and Immunostaining
2.5. Untargeted Metabolomics by LC–MS
2.6. Electrophysiological Analysis
2.7. Behavioral Tests
2.8. Statistical Analysis
3. Results
3.1. Se-FA Reduces Hcy Levels by Regulating the Hcy-Met Cycle
3.2. Se-FA Increases Se Levels, Selenoenzyme Activity and Selenoprotein Expression
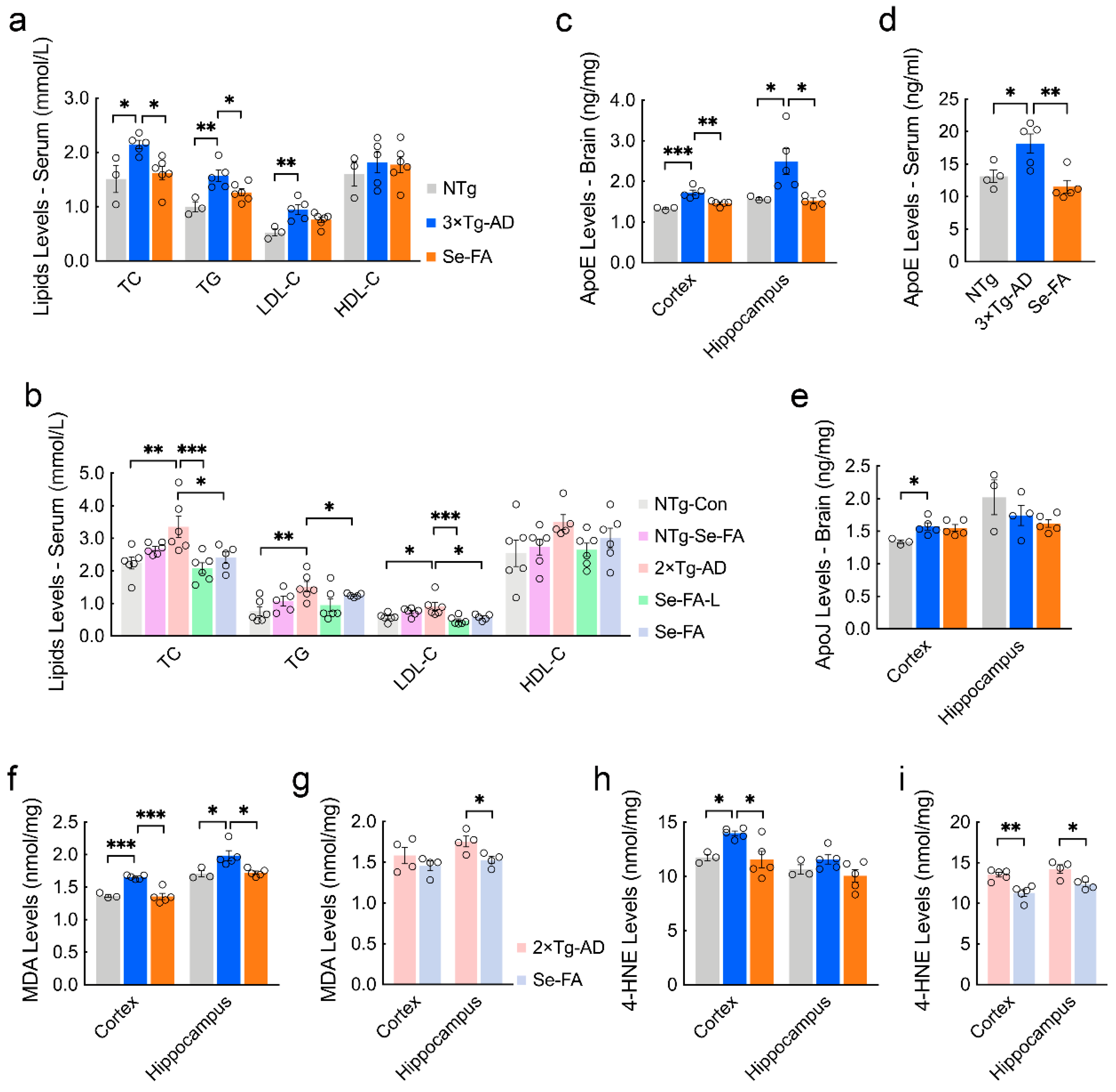
3.3. Se-FA Reverses Pathological Changes in Metabolism in the Brains of AD Mice
3.4. Se-FA Lowers Blood Lipids and Cerebral Lipid Peroxidation Products
3.5. Se-FA Attenuates Aβ and Tau Pathologies
3.6. Se-FA Rescues Synaptic and Behavioral Deficits in AD Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wiley, J. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement 2021, 17, 327–406. [Google Scholar]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement Transl. Res. Clin. Interv. 2021, 7, e12179. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement 2021, 17, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Munoz Fernandez, S.S.; Lima Ribeiro, S.M. Nutrition and Alzheimer Disease. Clin. Geriatr. Med. 2018, 34, 677–697. [Google Scholar] [CrossRef]
- van Eersel, J.; Ke, Y.D.; Liu, X.; Delerue, F.; Kril, J.J.; Gotz, J.; Ittner, L.M. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc. Natl. Acad. Sci. USA 2010, 107, 13888–13893. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Chen, C.; Jia, S.Z.; Cao, X.C.; Liu, M.; Tian, J.; Hoffmann, P.R.; Xu, H.X.; Ni, J.Z.; Song, G.L. Selenium Restores Synaptic Deficits by Modulating NMDA Receptors and Selenoprotein K in an Alzheimer’s Disease Model. Antioxid Redox Signal. 2021, 35, 863–884. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Wu, Q.Y.; Zheng, R.; Chen, C.; Chen, Y.; Liu, Q.; Hoffmann, P.R.; Ni, J.Z.; Song, G.L. Selenomethionine Mitigates Cognitive Decline by Targeting Both Tau Hyperphosphorylation and Autophagic Clearance in an Alzheimer’s Disease Mouse Model. J. Neurosci. 2017, 37, 2449–2462. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schneider, L.S.; Sano, M.; Diaz-Arrastia, R.; van Dyck, C.H.; Weiner, M.F.; Bottiglieri, T.; Jin, S.; Stokes, K.T.; Thomas, R.G.; et al. Alzheimer Disease Cooperative, S.; High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 2008, 300, 1774–1783. [Google Scholar] [CrossRef]
- Li, W.; Liu, H.; Yu, M.; Zhang, X.; Zhang, M.; Wilson, J.X.; Huang, G. Folic acid administration inhibits amyloid beta-peptide accumulation in APP/PS1 transgenic mice. J. Nutr. Biochem. 2015, 26, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Zou, C.; Li, M.; Huang, G.; Gao, Y.; Liu, H. Folic Acid Reduces Tau Phosphorylation by Regulating PP2A Methylation in Streptozotocin-Induced Diabetic Mice. Int. J. Mol. Sci. 2017, 18, 861. [Google Scholar] [CrossRef] [PubMed]
- Soininen, H.; Solomon, A.; Visser, P.J.; Hendrix, S.B.; Blennow, K.; Kivipelto, M.; Hartmann, T. LipiDiDiet clinical study, g., 24-month intervention with a specific multinutrient in people with prodromal Alzheimer’s disease (LipiDiDiet): A randomised, double-blind, controlled trial. Lancet Neurol. 2017, 16, 965–975. [Google Scholar] [CrossRef]
- Scheltens, P.; Kamphuis, P.J.; Verhey, F.R.; Olde Rikkert, M.G.; Wurtman, R.J.; Wilkinson, D.; Twisk, J.W.; Kurz, A. Efficacy of a medical food in mild Alzheimer’s disease: A randomized, controlled trial. Alzheimers Dement 2010, 6, 1–10.e1. [Google Scholar] [CrossRef] [PubMed]
- Tamtaji, O.R.; Heidari-Soureshjani, R.; Mirhosseini, N.; Kouchaki, E.; Bahmani, F.; Aghadavod, E.; Tajabadi-Ebrahimi, M.; Asemi, Z. Probiotic and selenium co-supplementation, and the effects on clinical, metabolic and genetic status in Alzheimer’s disease: A randomized, double-blind, controlled trial. Clin. Nutr. 2019, 38, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhou, X.; Li, Q.; Zhao, J.; Song, A.; An, P.; Du, Y.; Xu, W.; Huang, G. Effects of Folic Acid and Vitamin B12, Alone and in Combination on Cognitive Function and Inflammatory Factors in the Elderly with Mild Cognitive Impairment: A Single-blind Experimental Design. Curr. Alzheimer Res. 2019, 16, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Kim, T.K.; Lee, J.E.; Park, S.K.; Lee, K.W.; Seo, J.S.; Im, J.Y.; Kim, S.T.; Lee, J.Y.; Kim, Y.H.; Lee, J.K.; et al. Analysis of differential plaque depositions in the brains of Tg2576 and Tg-APPswe/PS1dE9 transgenic mouse models of Alzheimer disease. Exp. Mol. Med. 2012, 44, 492–502. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Chen, C.; Wu, Q.Y.; Zheng, R.; Liu, Q.; Ni, J.Z.; Hoffmann, P.R.; Song, G.L. Selenomethionine reduces the deposition of beta-amyloid plaques by modulating beta-secretase and enhancing selenoenzymatic activity in a mouse model of Alzheimer’s disease. Metallomics 2016, 8, 782–789. [Google Scholar] [CrossRef]
- Zhu, Z.; Chen, Y.; Shi, G.; Zhang, X. Selenium delays tomato fruit ripening by inhibiting ethylene biosynthesis and enhancing the antioxidant defense system. Food Chem. 2017, 219, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Fujisaka, S.; Avila-Pacheco, J.; Soto, M.; Kostic, A.; Dreyfuss, J.M.; Pan, H.; Ussar, S.; Altindis, E.; Li, N.; Bry, L.; et al. Diet, Genetics, and the Gut Microbiome Drive Dynamic Changes in Plasma Metabolites. Cell Rep. 2018, 22, 3072–3086. [Google Scholar] [CrossRef] [PubMed]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol. Biol. 2019, 1916, 105–111. [Google Scholar]
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A.; Pedersen, W.A.; Cutler, R.G.; Kruman, Y.; Haughey, N.; Lee, J.; Evans, M.; Mattson, M.P. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Teng, W.; Li, J.; Hao, F.; Wang, N. Homocysteine and Alzheimer’s Disease: Evidence for a Causal Link from Mendelian Randomization. J. Alzheimers Dis. 2016, 52, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Mikoda, N.; Kitazawa, M.; LaFerla, F.M. Treatment of Alzheimer’s disease with anti-homocysteic acid antibody in 3xTg-AD male mice. PLoS ONE 2010, 5, e8593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Wu, Q.Y.; Chen, C.; Zheng, R.; Chen, Y.; Ni, J.Z.; Song, G.L. Comparison of the effects of selenomethionine and selenium-enriched yeast in the triple-transgenic mouse model of Alzheimer’s disease. Food Funct. 2018, 9, 3965–3973. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Song, G.L. Roles of Selenoproteins in Brain Function and the Potential Mechanism of Selenium in Alzheimer’s Disease. Front Neurosci. 2021, 15, 646518. [Google Scholar] [CrossRef]
- Xu, J.; Begley, P.; Church, S.J.; Patassini, S.; Hollywood, K.A.; Jullig, M.; Curtis, M.A.; Waldvogel, H.J.; Faull, R.L.; Unwin, R.D.; et al. Graded perturbations of metabolism in multiple regions of human brain in Alzheimer’s disease: Snapshot of a pervasive metabolic disorder. Biochim. Biophys. Acta 2016, 1862, 1084–1092. [Google Scholar] [CrossRef]
- Leduc, V.; Jasmin-Belanger, S.; Poirier, J. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med. 2010, 16, 469–477. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacol. Ther. 2019, 195, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Frey, U.; Huang, Y.Y.; Kandel, E.R. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 1993, 260, 1661–1664. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef]
- Fajardo, V.; Varela-Moreiras, G. Efficacy of adding folic acid to foods. Int. J. Vitam. Nutr. Res. 2012, 82, 177–186. [Google Scholar] [CrossRef]
- Foley, P. Lipids in Alzheimer’s disease: A century-old story. Biochim. Biophys Acta 2010, 1801, 750–753. [Google Scholar] [CrossRef]
- Toledo, J.B.; Arnold, M.; Kastenmuller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimers Dement 2017, 13, 965–984. [Google Scholar] [CrossRef]
- He, X.; Huang, Y.; Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef]
- Kao, Y.C.; Ho, P.C.; Tu, Y.K.; Jou, I.M.; Tsai, K.J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- El Gaamouch, F.; Jing, P.; Xia, J.; Cai, D. Alzheimer’s Disease Risk Genes and Lipid Regulators. J. Alzheimers. Dis. 2016, 53, 15–29. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Thinakaran, G. Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim. Biophys. Acta 2010, 1801, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Hattori, C.; Asai, M.; Onishi, H.; Sasagawa, N.; Hashimoto, Y.; Saido, T.C.; Maruyama, K.; Mizutani, S.; Ishiura, S. BACE1 interacts with lipid raft proteins. J. Neurosci. Res. 2006, 84, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, I.Y.; Prager, K.; Barth, E.; Heneka, M.; Sandhoff, K.; Walter, J. Inhibition of glycosphingolipid biosynthesis reduces secretion of the beta-amyloid precursor protein and amyloid beta-peptide. J. Biol. Chem. 2005, 280, 28110–28117. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, N.; Ko, M.; Yu, W.; Zou, K.; Hanada, K.; Suzuki, T.; Gong, J.S.; Yanagisawa, K.; Michikawa, M. Modulation of amyloid precursor protein cleavage by cellular sphingolipids. J. Biol. Chem. 2004, 279, 11984–11991. [Google Scholar] [CrossRef]
- Benjannet, S.; Elagoz, A.; Wickham, L.; Mamarbachi, M.; Munzer, J.S.; Basak, A.; Lazure, C.; Cromlish, J.A.; Sisodia, S.; Checler, F.; et al. Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J. Biol. Chem. 2001, 276, 10879–10887. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef]
- Oaks, J.; Ogretmen, B. Regulation of PP2A by Sphingolipid Metabolism and Signaling. Front. Oncol. 2014, 4, 388. [Google Scholar] [CrossRef]
- Pratico, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef]
- Grimm, M.O.; Haupenthal, V.J.; Mett, J.; Stahlmann, C.P.; Blumel, T.; Mylonas, N.T.; Endres, K.; Grimm, H.S.; Hartmann, T. Oxidized Docosahexaenoic Acid Species and Lipid Peroxidation Products Increase Amyloidogenic Amyloid Precursor Protein Processing. Neurodegener. Dis. 2016, 16, 44–54. [Google Scholar] [CrossRef]
- Chen, L.; Na, R.; Gu, M.; Richardson, A.; Ran, Q. Lipid peroxidation up-regulates BACE1 expression in vivo: A possible early event of amyloidogenesis in Alzheimer’s disease. J. Neurochem. 2008, 107, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ramos, A.; Diaz-Nido, J.; Smith, M.A.; Perry, G.; Avila, J. Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. J. Neurosci. Res. 2003, 71, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Kuhla, B.; Haase, C.; Flach, K.; Luth, H.J.; Arendt, T.; Munch, G. Effect of pseudophosphorylation and cross-linking by lipid peroxidation and advanced glycation end product precursors on tau aggregation and filament formation. J. Biol. Chem. 2007, 282, 6984–6991. [Google Scholar] [CrossRef]
- Lu, C.; Chan, S.L.; Haughey, N.; Lee, W.T.; Mattson, M.P. Selective and biphasic effect of the membrane lipid peroxidation product 4-hydroxy-2,3-nonenal on N-methyl-D-aspartate channels. J. Neurochem. 2001, 78, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Walker, A.J.; Berk, M.; Maes, M.; Puri, B.K. Cell Death Pathways: A Novel Therapeutic Approach for Neuroscientists. Mol. Neurobiol. 2018, 55, 5767–5786. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Aissa, A.F.; Tryndyak, V.; de Conti, A.; Melnyk, S.; Gomes, T.D.; Bianchi, M.L.; James, S.J.; Beland, F.A.; Antunes, L.M.; Pogribny, I.P. Effect of methionine-deficient and methionine-supplemented diets on the hepatic one-carbon and lipid metabolism in mice. Mol. Nutr. Food Res. 2014, 58, 1502–1512. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar]
- Liao, D.; Yang, X.; Wang, H. Hyperhomocysteinemia and high-density lipoprotein metabolism in cardiovascular disease. Clin. Chem. Lab. Med. 2007, 45, 1652–1659. [Google Scholar] [CrossRef]
- Obeid, R.; Herrmann, W. Homocysteine and lipids: S-adenosyl methionine as a key intermediate. FEBS Lett. 2009, 583, 1215–1225. [Google Scholar] [CrossRef]
- Le, N.A. Lipoprotein-associated oxidative stress: A new twist to the postprandial hypothesis. Int. J. Mol. Sci. 2014, 16, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, S.; Hill, K.E.; McDonald, W.H.; Burk, R.F. Long isoform mouse selenoprotein P (Sepp1) supplies rat myoblast L8 cells with selenium via endocytosis mediated by heparin binding properties and apolipoprotein E receptor-2 (ApoER2). J. Biol. Chem. 2012, 287, 28717–28726. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E.; Motley, A.K.; Winfrey, V.P.; Kurokawa, S.; Mitchell, S.L.; Zhang, W. Selenoprotein P and apolipoprotein E receptor-2 interact at the blood-brain barrier and also within the brain to maintain an essential selenium pool that protects against neurodegeneration. FASEB J. 2014, 28, 3579–3588. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chung, Y.W.; Jung, M.K.; Lee, J.H.; Ko, K.Y.; Jang, J.K.; Ham, M.; Kang, H.; Pack, C.G.; Mihara, H.; et al. Apolipoprotein E-mediated regulation of selenoprotein P transportation via exosomes. Cell Mol. Life Sci. 2020, 77, 2367–2386. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Concentration | Unit | Method |
|---|---|---|---|
| Vitamin B12 | Not detected, detection limit 1.0 | μg/g | GB/T 5009.217-2008 |
| Vitamin B6 | Not detected, detection limit 0.02 | mg/100g | GB 5009.154-2016 |
| Vitamin E | <0.12 | mg/100g | GB 5009.82-2016 |
| Item | Concentration | Unit |
|---|---|---|
| Ca | 1.19 | % |
| P | 0.77 | % |
| Lys | 0.93 | % |
| Met + Cys | 0.67 | % |
| Arg | 1.02 | % |
| His | 0.50 | % |
| Trp | 0.22 | % |
| Phe + Tyr | 1.50 | % |
| Thr | 0.78 | % |
| Leu | 1.59 | % |
| Ile | 0.76 | % |
| Val | 0.90 | % |
| VA | 10.70 | KIU/kg |
| VD | 1.50 | KIU/kg |
| VE | 103.05 | IU/kg |
| VK | 6.14 | mg/kg |
| VB1 | 16.00 | mg/kg |
| VB2 | 16.03 | mg/kg |
| VB6 | 10.43 | mg/kg |
| VB3 | 89.00 | mg/kg |
| VB5 | 30.09 | mg/kg |
| Folate | 7.45 | mg/kg |
| Biotin | 0.28 | mg/kg |
| VB12 | 0.03 | mg/kg |
| Choline | 1900.00 | mg/kg |
| Mg | 0.26 | % |
| K | 0.64 | % |
| Na | 0.32 | % |
| Fe | 180.00 | mg/kg |
| Mn | 123.49 | mg/kg |
| Cu | 17.80 | mg/kg |
| Zn | 56.20 | mg/kg |
| I | 0.61 | mg/kg |
| Se | 0.16 | mg/kg |
| ANTIBODIES | |||
|---|---|---|---|
| Reagent | Source | Applications | Identifier |
| Rabbit anti-BACE1 | CST | 1:1000 | 5606 |
| Mouse anti-Aβ | Biolegend | 1:2000 | SIG-39300 |
| Rabbit anti-APP | Abcam | 1:1000 | ab32136 |
| Mouse anti-Tau | Invitrogen | 1:500 | MN1000 |
| Mouse anti-pSTau (Ser202) | Invitrogen | 1:500 | MN1020 |
| Mouse anti-pSTau (Ser214) | Invitrogen | 1:500 | MN1060 |
| Rabbit anti-PP2A | CST | 1:1000 | 2038 |
| Rabbit anti-p-PP2A (Tyr307) | ThermoFisher | 1:1000 | AA1910N |
| Rabbit anti-PSD95 | Abcam | 1:5000 | ab76115 |
| Rabbit anti-Synaptophysin | CST | 1:1000 | 4329s |
| Mouse anti-TrxR1 | Santa cruz | 1:500 | sc-28321 |
| Rabbit anti-GPX4 | Abcam | 1:1000 | ab125066 |
| Rabbit anti-GPX1 | GeneTex | 1:1000 | GTX116040 |
| Rabbit anti-SELENOP | Abcam | 1:1000 | ab109514 |
| Rabbit anti-MSRB1 (SELENOR) | Invitrogen | 1:1000 | Cat# PA5-77009 |
| Rabbit anti-SELENOK | Sigma | 1:1000 | HPA-008196 |
| Mouse anti-SELENOM | Santa cruz | 1:1000 | sc-514952 |
| Mouse anti-SELENON | Santa cruz | 1:1000 | sc-365824 |
| Rabbit anti-SELENOS | Proteintech | 1:1000 | 15591 |
| Rabbit anti-SELENOT | Acris | 1:1000 | AP53842PU |
| Mouse anti-beta-actin | Proteintech | 1:10000 | 66009 |
| Mouse anti-α-tubulin | Proteintech | 1:10000 | 11224 |
| Rabbit anti-GAPDH | Proteintech | 1:10000 | 10494 |
| Goat anti-Rabbit IgG | CST | 1:10000 | 7074 |
| Goat anti-Mouse IgG | CST | 1:10000 | 7076 |
| Anti-mouse Alexa Fluor 594 | Abcam | 1:1000 | ab150116 |
| Anti-rabbit Alexa Fluor 488 | Abcam | 1:1000 | ab150077 |
| KITS | |||
| Reagent | Source | Identifier | |
| HCY ELISA | MLBIO | ml037451 | |
| VB6 ELISA | MLBIO | ml001803 | |
| VB12 ELISA | MLBIO | ml057867 | |
| Folate ELISA | MLBIO | ml002074 | |
| Met ELISA | MLBIO | ml058151 | |
| MS ELISA | MLBIO | Ml672003 | |
| MDA ELISA | MLBIO | ml002001 | |
| Aβ1-42 ELISA | MLBIO | ml002201 | |
| ApoE ELISA | MLBIO | ml002179 | |
| ApoJ ELISA | MLBIO | Ml880235 | |
| GPx activity Kit | Beyotime | S0058 | |
| TrxR activity Kit | COMIN | TRXR-1-W | |
| TC Assay Kit | iCubio | CHOD-PAP | |
| TG Assay Kit | iCubio | GPO-PAP | |
| HDL-C Assay Kit | iCubio | - | |
| LDL-C Assay Kit | iCubio | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-H.; Cao, X.-C.; Peng, J.-Y.; Huang, S.-L.; Chen, C.; Jia, S.-Z.; Ni, J.-Z.; Song, G.-L. Reversal of Lipid Metabolism Dysregulation by Selenium and Folic Acid Co-Supplementation to Mitigate Pathology in Alzheimer’s Disease. Antioxidants 2022, 11, 829. https://doi.org/10.3390/antiox11050829
Zhang Z-H, Cao X-C, Peng J-Y, Huang S-L, Chen C, Jia S-Z, Ni J-Z, Song G-L. Reversal of Lipid Metabolism Dysregulation by Selenium and Folic Acid Co-Supplementation to Mitigate Pathology in Alzheimer’s Disease. Antioxidants. 2022; 11(5):829. https://doi.org/10.3390/antiox11050829
Chicago/Turabian StyleZhang, Zhong-Hao, Xian-Chun Cao, Jia-Ying Peng, Shao-Ling Huang, Chen Chen, Shi-Zheng Jia, Jia-Zuan Ni, and Guo-Li Song. 2022. "Reversal of Lipid Metabolism Dysregulation by Selenium and Folic Acid Co-Supplementation to Mitigate Pathology in Alzheimer’s Disease" Antioxidants 11, no. 5: 829. https://doi.org/10.3390/antiox11050829
APA StyleZhang, Z.-H., Cao, X.-C., Peng, J.-Y., Huang, S.-L., Chen, C., Jia, S.-Z., Ni, J.-Z., & Song, G.-L. (2022). Reversal of Lipid Metabolism Dysregulation by Selenium and Folic Acid Co-Supplementation to Mitigate Pathology in Alzheimer’s Disease. Antioxidants, 11(5), 829. https://doi.org/10.3390/antiox11050829