

Metabolic Rewiring toward Oxidative Phosphorylation Disrupts Intrinsic Resistance to Ferroptosis of the Colon Adenocarcinoma Cells

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Genomic Disruption of FSP1 Using CRISPR-Cas9
2.3. Clonogenicity Assay
2.4. Immunoblotting
2.5. FACS Analysis
2.5.1. Cell Death
2.5.2. Detection of Lipid Hydroperoxides
2.5.3. ROS Level
2.6. Statistical Analysis
3. Results
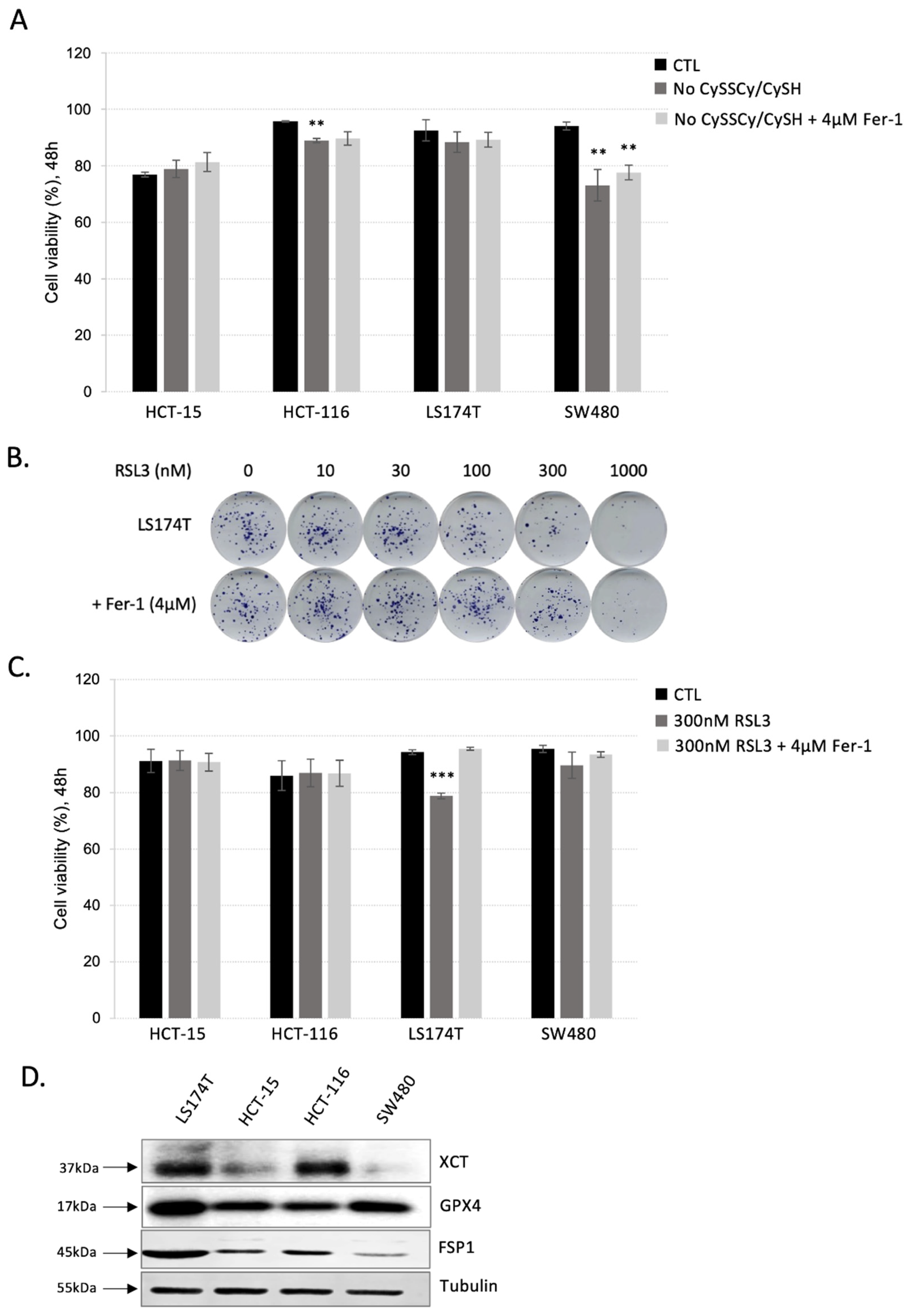
3.1. Colorectal Adenocarcinoma (CRC) Cell Lines Show Great Resistance to Ferroptosis Induction
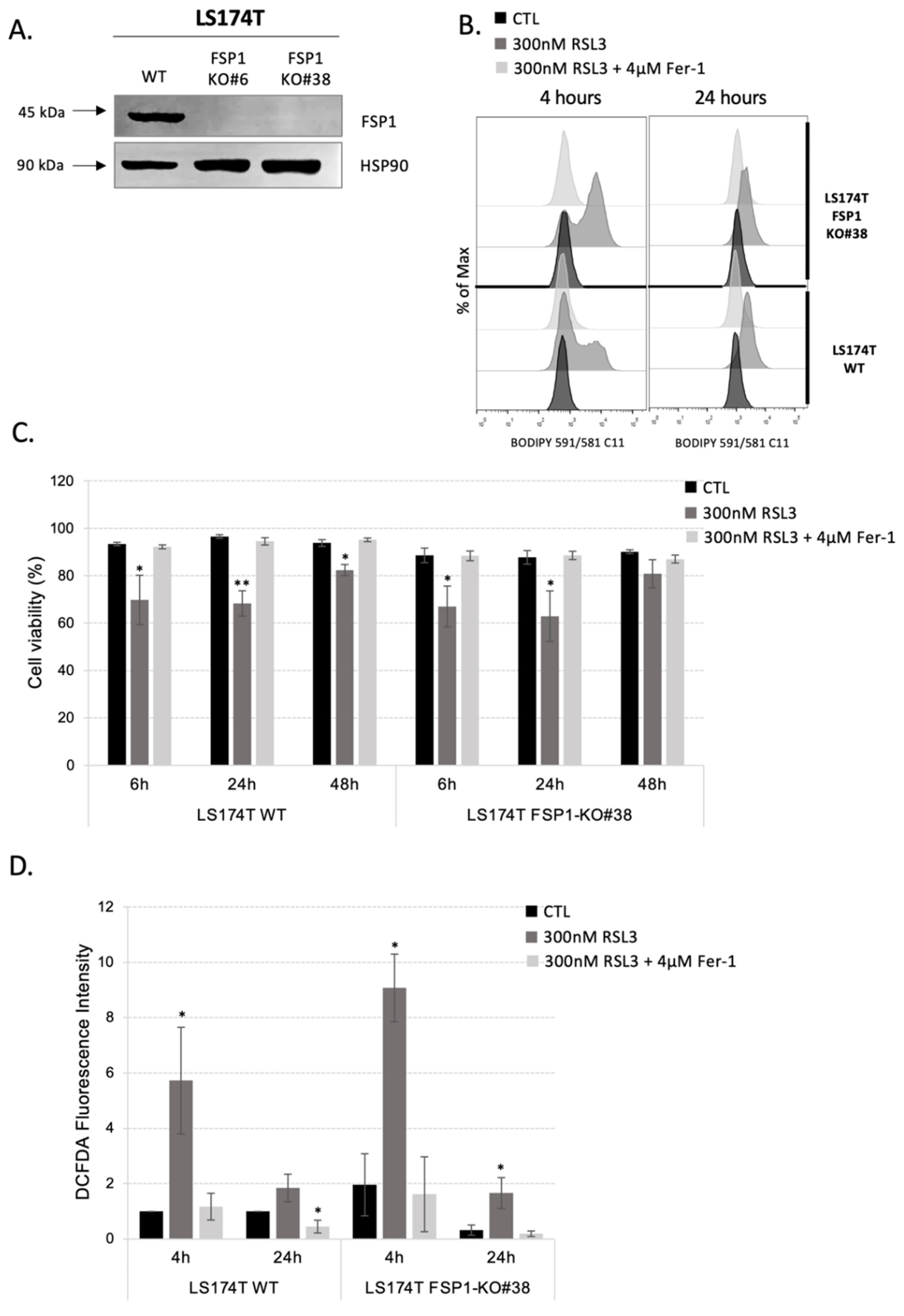
3.2. Inhibition of Both Ferroptosis-Preventing Pathways, GSH-GPX4 and FSP1-CoQ10, Did Not Sensitize CRC to Ferroptosis
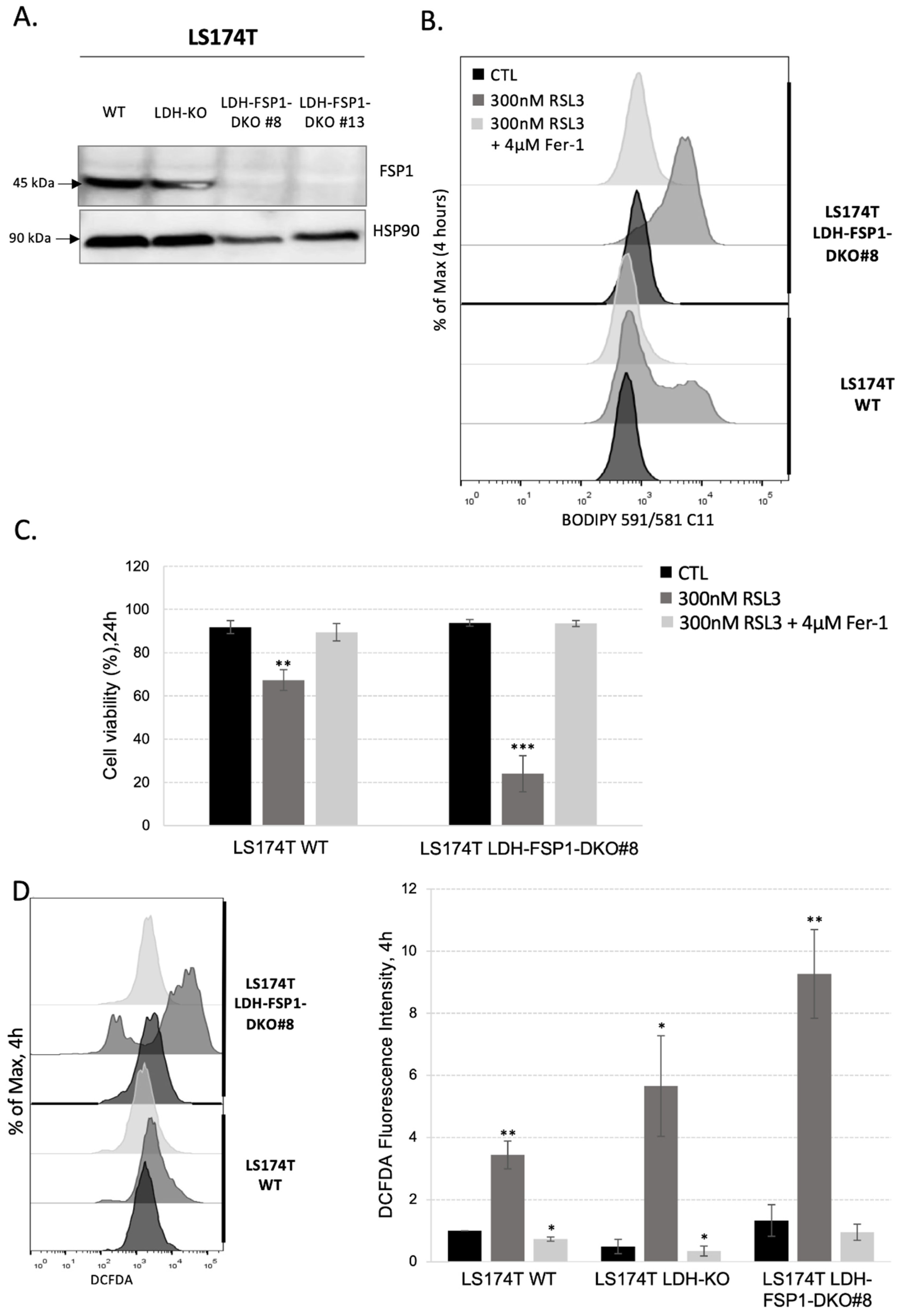
3.3. Rewiring Cellular Metabolism toward OXPHOS Is Not Enough to Sensitize Cells to Ferroptosis
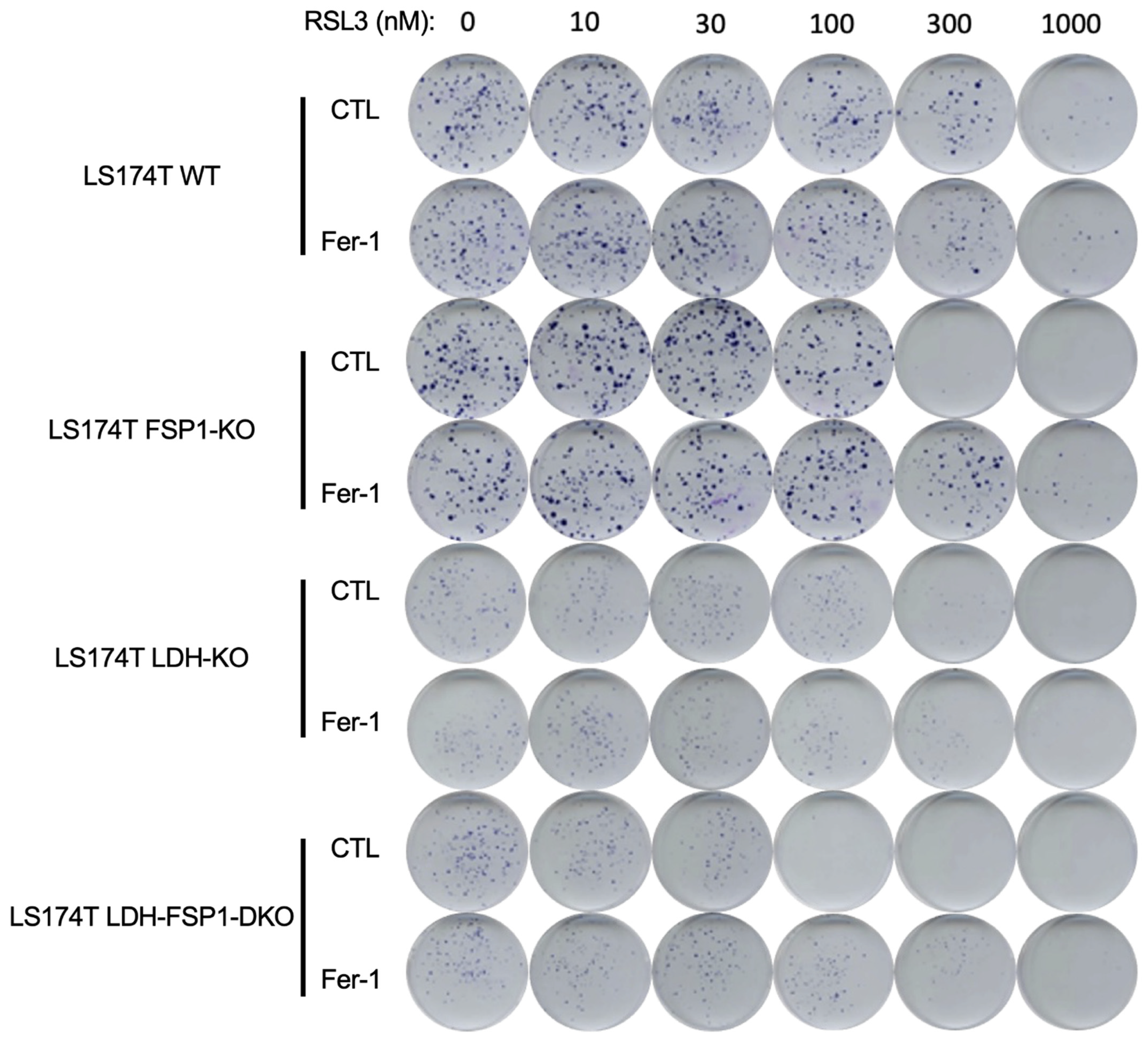
3.4. Combination of Metabolic Rewiring to OXPHOS and Inhibition of Both Ferroptosis-Preventing Pathways Is Necessary to Sensitize CRC to Ferroptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Gregolin, C. The Selenoenzyme Phospholipid Hydroperoxide Glutathione Peroxidase. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Godeas, C.; Tramer, F.; Micali, F.; Soranzo, M.; Sandri, G.; Panfili, E. Distribution and Possible Novel Role of Phospholipid Hydroperoxide Glutathione Peroxidase in Rat Epididymal Spermatozoa. Biol. Reprod. 1997, 57, 1502–1508. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Daher, B.; Meira, W.; Durivault, J.; Gotorbe, C.; Pouyssegur, J.; Vucetic, M. Genetic Disruption of the γ-Glutamylcysteine Ligase in PDAC Cells Induces Ferroptosis-Independent Cell Death In Vitro without Affecting In Vivo Tumor Growth. Cancers 2022, 14, 3154. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Roveri, A.; Benazzi, L.; Bosello, V.; Mauri, P.; Toppo, S.; Tosatto, S.C.E.; Ursini, F. Functional Interaction of Phospholipid Hydroperoxide Glutathione Peroxidase with Sperm Mitochondrion-Associated Cysteine-Rich Protein Discloses the Adjacent Cysteine Motif as a New Substrate of the Selenoperoxidase. J. Biol. Chem. 2005, 280, 38395–38402. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione Depletion Induces Ferroptosis, Autophagy, and Premature Cell Senescence in Retinal Pigment Epithelial Cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef]
- Daher, B.; Vučetić, M.; Pouysségur, J. Cysteine Depletion, a Key Action to Challenge Cancer Cells to Ferroptotic Cell Death. Front. Oncol. 2020, 10, 723. [Google Scholar] [CrossRef]
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouysségur, J.; Vučetić, M. Genetic Ablation of the Cystine Transporter XCT in PDAC Cells Inhibits MTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res. 2019, 79, 3877–3890. [Google Scholar] [CrossRef]
- Vucetic, M.; Daher, B.; Cassim, S.; Meira, W.; Pouyssegur, J. Together We Stand, Apart We Fall: How Cell-to-Cell Contact/Interplay Provides Resistance to Ferroptosis. Cell Death Dis. 2020, 11, 789. [Google Scholar] [CrossRef]
- Meira, W.; Daher, B.; Parks, S.K.; Cormerais, Y.; Durivault, J.; Tambutte, E.; Pouyssegur, J.; Vučetić, M. A Cystine-Cysteine Intercellular Shuttle Prevents Ferroptosis in XCTKO Pancreatic Ductal Adenocarcinoma Cells. Cancers 2021, 13, 1434. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Santofimia-Castaño, P.; Liu, X.; Xia, Y.; Peng, L.; Gotorbe, C.; Neira, J.L.; Tang, D.; Pouyssegur, J.; Iovanna, J. NUPR1 Inhibitor ZZW-115 Induces Ferroptosis in a Mitochondria-Dependent Manner. Cell Death Discov. 2021, 7, 269. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Arroyo, A.; Navarro, F.; Navas, P.; Villalba, J.M. Ubiquinol Regeneration by Plasma Membrane Ubiquinone Reductase. Protoplasma 1998, 205, 107–113. [Google Scholar] [CrossRef]
- Takahashi, T.; Okamoto, T.; Mori, K.; Sayo, H.; Kishi, T. Distribution of Ubiquinone and Ubiquinol Homologues in Rat Tissues and Subcellular Fractions. Lipids 1993, 28, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.R.; Gong, M.; Wodke, L.; Lamb, J.H.; Jones, D.J.L.; Farmer, P.B.; Scrutton, N.S.; Munro, A.W. The Human Apoptosis-Inducing Protein AMID Is an Oxidoreductase with a Modified Flavin Cofactor and DNA Binding Activity. J. Biol. Chem. 2005, 280, 30735–30740. [Google Scholar] [CrossRef]
- Yao, X.; Li, W.; Fang, D.; Xiao, C.; Wu, X.; Li, M.; Luo, Z. Emerging Roles of Energy Metabolism in Ferroptosis Regulation of Tumor Cells. Adv. Sci. 2021, 8, 2100997. [Google Scholar] [CrossRef]
- Ždralević, M.; Vučetić, M.; Daher, B.; Marchiq, I.; Parks, S.K.; Pouysségur, J. Disrupting the ‘Warburg Effect’ Re-Routes Cancer Cells to OXPHOS Offering a Vulnerability Point via ‘Ferroptosis’-Induced Cell Death. Adv. Biol. Regul. 2018, 68, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Vučetić, M.; Cormerais, Y.; Parks, S.K.; Pouysségur, J. The Central Role of Amino Acids in Cancer Redox Homeostasis: Vulnerability Points of the Cancer Redox Code. Front. Oncol. 2017, 7, 319. [Google Scholar] [CrossRef] [PubMed]
- DeHart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of Voltage Dependent Anion Channels Promotes Reactive Oxygen Species Generation, Mitochondrial Dysfunction and Cell Death in Cancer Cells. Biochem. Pharmacol. 2018, 148, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, R.; Yu, Q.; Dong, L.; Bi, Y.; Liu, G. Metabolic Reprogramming of Macrophages during Infections and Cancer. Cancer Lett. 2019, 452, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.-M.; et al. Double Genetic Disruption of Lactate Dehydrogenases A and B Is Required to Ablate the “Warburg Effect” Restricting Tumor Growth to Oxidative Metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic Determinants of Cancer Cell Sensitivity to Canonical Ferroptosis Inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef]
- Xi, Y.; Xu, P. Global Colorectal Cancer Burden in 2020 and Projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic Resistance in Colorectal Cancer: A Review. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef]
- Hu, Q.; Wei, W.; Wu, D.; Huang, F.; Li, M.; Li, W.; Yin, J.; Peng, Y.; Lu, Y.; Zhao, Q.; et al. Blockade of GCH1/BH4 Axis Activates Ferritinophagy to Mitigate the Resistance of Colorectal Cancer to Erastin-Induced Ferroptosis. Front. Cell Dev. Biol. 2022, 10, 810327. [Google Scholar] [CrossRef]
- Lee, Y.; Kalimuthu, K.; Seok Park, Y.; Makala, H.; Watkins, S.C.; Choudry, M.H.A.; Bartlett, D.L.; Tae Kwon, Y.; Lee, Y.J. Ferroptotic Agent-Induced Endoplasmic Reticulum Stress Response Plays a Pivotal Role in the Autophagic Process Outcome. J. Cell. Physiol. 2020, 235, 6767–6778. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological Inhibition of Cystine–Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.-J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine Depletion Induces Pancreatic Tumor Ferroptosis in Mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic Compensation: A Phenomenon in Search of Mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ni, H.-M.; Haynes, A.; Manley, S.; Li, Y.; Jaeschke, H.; Ding, W.-X. Chronic Deletion and Acute Knockdown of Parkin Have Differential Responses to Acetaminophen-Induced Mitophagy and Liver Injury in Mice. J. Biol. Chem. 2015, 290, 10934–10946. [Google Scholar] [CrossRef]
- Matés, J.M.; Di Paola, F.J.; Campos-Sandoval, J.A.; Mazurek, S.; Márquez, J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin. Cell Dev. Biol. 2020, 98, 34–43. [Google Scholar] [CrossRef]
- Suzuki, S.; Venkatesh, D.; Kanda, H.; Nakayama, A.; Hosokawa, H.; Lee, E.; Miki, T.; Stockwell, B.R.; Yokote, K.; Tanaka, T.; et al. GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res. 2022, 82, 3209–3222. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; He, X.; Wang, Y.; Hu, Z.; Huang, H.; Zhao, S.; Wei, P.; Li, D. Warburg Effect in Colorectal Cancer: The Emerging Roles in Tumor Microenvironment and Therapeutic Implications. J. Hematol. Oncol. 2022, 15, 160. [Google Scholar] [CrossRef]
- Ludikhuize, M.C.; Gevers, S.; Nguyen, N.T.B.; Meerlo, M.; Roudbari, S.K.S.; Gulersonmez, M.C.; Stigter, E.C.A.; Drost, J.; Clevers, H.; Burgering, B.M.T.; et al. Rewiring Glucose Metabolism Improves 5-FU Efficacy in P53-Deficient/KRASG12D Glycolytic Colorectal Tumors. Commun. Biol. 2022, 5, 1159. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Marchiq, I.; Le Floch, R.; Roux, D.; Simon, M.-P.; Pouyssegur, J. Genetic Disruption of Lactate/H+ Symporters (MCTs) and Their Subunit CD147/BASIGIN Sensitizes Glycolytic Tumor Cells to Phenformin. Cancer Res. 2015, 75, 171–180. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Marchiq, I.; Pouysségur, J. Hypoxia, Cancer Metabolism and the Therapeutic Benefit of Targeting Lactate/H+ Symporters. J. Mol. Med. 2016, 94, 155–171. [Google Scholar] [CrossRef]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.-N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150.e9. [Google Scholar] [CrossRef]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- Granja, S.; Marchiq, I.; Floch, R.L.; Moura, C.S.; Baltazar, F.; Pouysségur, J. Disruption of BASIGIN Decreases Lactic Acid Export and Sensitizes Non-Small Cell Lung Cancer to Biguanides Independently of the LKB1 Status. Oncotarget 2015, 6, 6708–6721. [Google Scholar] [CrossRef] [PubMed]
- Le Floch, R.; Chiche, J.; Marchiq, I.; Naiken, T.; Ilc, K.; Murray, C.M.; Critchlow, S.E.; Roux, D.; Simon, M.-P.; Pouysségur, J. CD147 Subunit of Lactate/H+ Symporters MCT1 and Hypoxia-Inducible MCT4 Is Critical for Energetics and Growth of Glycolytic Tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 16663–16668. [Google Scholar] [CrossRef] [PubMed]
- de Padua, M.C.; Delodi, G.; Vučetić, M.; Durivault, J.; Vial, V.; Bayer, P.; Noleto, G.R.; Mazure, N.M.; Ždralević, M.; Pouysségur, J. Disrupting Glucose-6-Phosphate Isomerase Fully Suppresses the “Warburg Effect” and Activates OXPHOS with Minimal Impact on Tumor Growth except in Hypoxia. Oncotarget 2017, 8, 87623–87637. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Marchiq, I.; Parks, S.K.; Durivault, J.; Ždralević, M.; Vucetic, M. ‘Warburg Effect’ Controls Tumor Growth, Bacterial, Viral Infections and Immunity—Genetic Deconstruction and Therapeutic Perspectives. Semin. Cancer Biol. 2022, 86, 334–346. [Google Scholar] [CrossRef]
- Ždralević, M.; Marchiq, I.; de Padua, M.M.C.; Parks, S.K.; Pouysségur, J. Metabolic Plasiticy in Cancers—Distinct Role of Glycolytic Enzymes GPI, LDHs or Membrane Transporters MCTs. Front. Oncol. 2017, 7, 313. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Ždralević, M. Reply to Beltinger: Double Genetic Disruption of Lactate Dehydrogenases A and B Is Required to Ablate the “Warburg Effect” Restricting Tumor Growth to Oxidative Metabolism. J. Biol. Chem. 2019, 294, 67. [Google Scholar] [CrossRef] [PubMed]
- Franchi, A.; Silvestre, P.; Pouysségur, J. A Genetic Approach to the Role of Energy Metabolism in the Growth of Tumor Cells: Tumorigenicity of Fibroblast Mutants Deficient Either in Glycolysis or in Respiration. Int. J. Cancer 1981, 27, 819–827. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gotorbe, C.; Durivault, J.; Meira, W.; Cassim, S.; Ždralević, M.; Pouysségur, J.; Vučetić, M. Metabolic Rewiring toward Oxidative Phosphorylation Disrupts Intrinsic Resistance to Ferroptosis of the Colon Adenocarcinoma Cells. Antioxidants 2022, 11, 2412. https://doi.org/10.3390/antiox11122412
Gotorbe C, Durivault J, Meira W, Cassim S, Ždralević M, Pouysségur J, Vučetić M. Metabolic Rewiring toward Oxidative Phosphorylation Disrupts Intrinsic Resistance to Ferroptosis of the Colon Adenocarcinoma Cells. Antioxidants. 2022; 11(12):2412. https://doi.org/10.3390/antiox11122412
Chicago/Turabian StyleGotorbe, Célia, Jérôme Durivault, Willian Meira, Shamir Cassim, Maša Ždralević, Jacques Pouysségur, and Milica Vučetić. 2022. "Metabolic Rewiring toward Oxidative Phosphorylation Disrupts Intrinsic Resistance to Ferroptosis of the Colon Adenocarcinoma Cells" Antioxidants 11, no. 12: 2412. https://doi.org/10.3390/antiox11122412
APA StyleGotorbe, C., Durivault, J., Meira, W., Cassim, S., Ždralević, M., Pouysségur, J., & Vučetić, M. (2022). Metabolic Rewiring toward Oxidative Phosphorylation Disrupts Intrinsic Resistance to Ferroptosis of the Colon Adenocarcinoma Cells. Antioxidants, 11(12), 2412. https://doi.org/10.3390/antiox11122412