1. Introduction
Skeletal quality and integrity are maintained by a reconstruction process, which is balanced between osteoblastic bone formation and osteoclastic bone resorption [
1]. If the rate of bone resorption is higher than that of bone formation, the volume and mineral density of bone will decrease, resulting in deterioration of bone microarchitecture and weakened mechanical strength such as hardness and elasticity of bone [
2]. As a worldwide common skeletal disease, osteoporosis also causes fractures, physical pain, and disability, seriously affecting the life quality of patients [
3]. Human osteoporosis mainly includes senile osteoporosis, postmenopausal osteoporosis, and glucocorticoid-induced osteoporosis. Besides these, weightlessness and other metabolic diseases such as type 2 diabetes are also important risk factors for osteoporosis [
4]. With the accelerated aging of the population all over the world, the prevalence of senile osteoporosis is increasing rapidly [
5]. In addition, disuse osteoporosis caused by bedridden immobilization, spinal cord injury, and space flight can also induce local or systemic reduction of bone mass [
6]. Prevention and treatment of senile and disuse osteoporosis are not only medical problems worldwide, but also important challenges for the development of the aerospace industry.
At present, osteoporosis is generally prevented by dietary supplementation of calcium and vitamin D, as well as increasing exercise appropriately [
7]. However, these strategies usually take a long time to produce the desired results, and the effects are also limited. Furthermore, exercise is not applicable for disuse osteoporosis. Excessive exercise may even have certain side effects on the bones [
8]. In clinical studies to date, the medical treatments of osteoporosis have similarly exhibited limited curative effects and serious side effects [
9,
10,
11,
12]. Therefore, there is still a lack of therapeutic measures available to safely and effectively prevent or control the progression of osteoporosis.
Daphnetin (7,8-dihydroxycoumarin) is a bioactive molecule isolated from the plant of
Daphne, a genus of deciduous and evergreen shrubs in the family Thymelaeaceae. As a widely studied natural compound, daphnetin has been proven to have various pharmacological activities such as antiinflammation [
13], antioxidation [
14], neuroprotection [
15], and cardiovascular protections [
16]. Previous studies have shown that daphnetin inhibited receptor activator of nuclear factor κB ligand (RANKL)-induced osteoclast formation in vitro through inhibiting reactive oxygen species (ROS) signal transduction, as well as preventing the activation of nuclear factor-kappa B (NF-Κb) and protein kinase B (Akt)/glycogen synthase kinase-3β (GSK-3β) signaling pathways [
17]; ameliorated lipopolysaccharide (LPS)-induced inflammatory osteolysis and RANKL-induced osteoclastogenesis through inhibiting the extracellular signal-regulated kinase (ERK) and nuclear factor of activated T cells 1 (NFATc1) pathways [
18], and played protective role in glucocorticoid-induced osteoporosis through activating the Wnt/GSK-3β/β-catenin signaling pathway [
19]. However, the effects of daphnetin on senile and disuse osteoporosis remain unexplored.
In this study, we investigated the effects of daphnetin on senile and disuse osteoporosis as well as the possible involved mechanisms through in vivo and in vitro experiments. In terms of in vivo experiments, the classic senescence-accelerated mouse prone 8 (SAMP8) model [
20] and hindlimb-unloading mouse model [
21], which correspond to osteoporosis in elderly people, bedridden patients, and astronauts, were used to investigate the effects of daphnetin on senile and disuse osteoporosis. To further explore the possible mechanisms underlying the osteoprotective effects of daphnetin, in vitro experiments were performed with primary osteoblasts and bone marrow monocyte-derived osteoclasts, as well as osteoblast precursor MC3T3-E1 cells and osteoclast precursor RAW264.7 cells.
2. Materials and Methods
2.1. Reagents
Daphnetin (CAS#486-35-1, purity HPLC 98%) was purchased from Nanjing Xinhou Biotechnology Co., Ltd. (Nanjing, China) Minimum Essential Medium α (α-MEM), Trypsin, penicillin, and streptomycin were obtained from Gibco Laboratories (Life Technologies, Inc., Burlington, ON, Canada). Fetal bovine serum (FBS) was obtained from Biological Industries (BioInd, Kibbutz Beit Haemek, Israel). TriPure Isolation Reagent was purchased from Roche (Basel, Switzerland). Antibodies against manganese superoxide dismutase (SOD2), catalase (CAT), heme oxygenase-1 (HO-1), NADPH: quinone oxidoreductase 1 (NQO1), and nuclear respiratory factor 1 (Nrf1) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody against runt related transcription factor 2 (Runx2) was purchased from Abcam (Cambridge, UK). Antibodies against mitochondrial complexes I~V were purchased from Life Technologies (San Diego, CA, USA). Antibodies against optic atrophy 1 (OPA1) and dynamin-related protein 1 (DRP1) were purchased from BD Biosciences (Franklin Lakes, NJ, USA). Antibodies against osteocalcin (OCN), sirtuin 3 (SIRT3), NFATc1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were purchased from Cell Signaling Technology (Danvers, MA, USA). Peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H + L) and peroxidase-conjugated AffiniPure rabbit anti-mouse IgG (H + L) antibodies were purchased from Jackson ImmunoResearch (West Grove, PA, USA). All other chemicals were of analytical grade and obtained from Sigma Aldrich (St. Louis, MO, USA).
2.2. Animals and Treatments
In terms of senile osteoporosis, three-month-old male SAMP8 mice and senescence-accelerated mouse resistance 1 (SAMR1) mice were obtained from the Experimental Animal Center, Peking University. After one week of adaptation, the mice were randomly divided into three groups: SAMR1 group (
n = 9), SAMP8 group (
n = 9), and SAMP8 + daphnetin group (
n = 6). Mice in the daphnetin-treated SAMP8 group were gavaged with daphnetin (100 mg/kg/d) for 3 months, meanwhile, mice in the SAMR1 group and SAMP8 group were gavaged with phosphate-buffered saline (PBS) followed by 4 months of normal feeding. The daphnetin dose and treatment time were chosen based on our preliminary test. Additionally, this dose has been proved to be effective and non-toxic by other in vivo studies [
22,
23].
In terms of disuse osteoporosis, two-month-old male C57BL/6 mice were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). After one week of adaptation, the mice were randomly divided into three groups: control group (n = 9), unloading group (n = 9), and unloading + daphnetin group (n = 9). Mice in the daphnetin-treated unloading group were gavaged with daphnetin (100 mg/kg/d) for 7 days prior to tail suspension and continued with daphnetin gavage for 28 days after tail suspension. Meanwhile, mice in the control group and unloading group were gavaged with PBS.
All animals were housed in a temperature (25~28 °C)- and humidity (60%)-controlled animal room and maintained on a 12 h light/12 h dark cycle with free access to food and water during experiments. Animal procedures were approved by Xi’an Jiaotong University Animal Care and Use Committee (Approval No.: XJTU-2019-21). All animal studies complied with the ARRIVE guidelines, and all efforts were made to minimize the stress and the number of animals used in this study.
2.3. Cell Culture and In Vitro Differentiation
Primary osteoblasts were obtained by enzyme digestion of hindlimb bones from mice following a published protocol with minor modifications [
24]. Briefly, hindlimb bones were isolated from the sacrificed mice and washed with PBS supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin. After removing the soft tissues and bone marrow, the long bones were cut into pieces and digested with 2 mg/mL collagenase II in the culture medium at 37 °C for 2 h with gentle shaking every 5 min. Digestion solution was changed every 30 min. After digestion, the bone chips were washed and suspended in complete cell culture medium, followed by static culture for 11~15 days. Primary osteoblasts growing out of the bone chips were trypsinized and transferred into new dishes or plates for further experiments. When primary osteoblasts reached 90~100% confluence, osteogenic differentiation was initiated with 10 mM β-glycerophosphate and 50 μg/mL ascorbic acid for the indicated time periods.
Bone marrow monocyte-derived osteoclasts were isolated from the marrow of hindlimb bones following a published protocol with minor modifications [
24]. Briefly, hindlimb bones were isolated from the sacrificed mice and washed with PBS. After scraping away the soft tissue, epiphyses were cut off and the bone marrow was flushed out with complete cell culture medium using a 25-gauge needle. The bone marrow was collected in a 50-mL centrifuge tube, and the cells were precipitated by spinning at 300 g for 5 min at room temperature. Cell pellet was resuspended in complete cell culture medium containing 40 ng/mL macrophage colony stimulating factor 1 (M-CSF) (Peprotech, Rocky Hill, NJ, USA) and cultured for 72 h. Then 100 ng/mL RANKL (Peprotech, Rocky Hill, NJ, USA) was added to the medium to induce osteoclastic differentiation, and osteoclasts were acquired after differentiation for 3~4 days.
Primary osteoblasts and bone marrow monocyte-derived osteoclasts, together with murine osteoblast precursor MC3T3-E1 cells and osteoclast precursor RAW264.7 cells purchased from National Collection of Authenticated Cell Cultures (NCACC, Shanghai, China), were all grown in α-MEM supplemented with 10% (v/v) FBS, 0.22% sodium bicarbonate, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified 5% CO2 atmosphere. When MC3T3-E1 cells reached 90~100% confluence, osteogenic differentiation was initiated with 10 mM β-glycerophosphate and 50 μg/mL ascorbic acid, together with 0, 0.1, 0.5, and 1 μM daphnetin treatments for the indicated periods. When RAW264.7 cells reached 60% confluence, osteoclast differentiation was initiated with 100 ng/mL RANKL, together with 0, 0.1, 0.5, and 1 μM daphnetin treatments for the indicated periods. Differentiation medium was changed every 2~3 days.
2.4. Micro-CT Analysis of Bone Structure
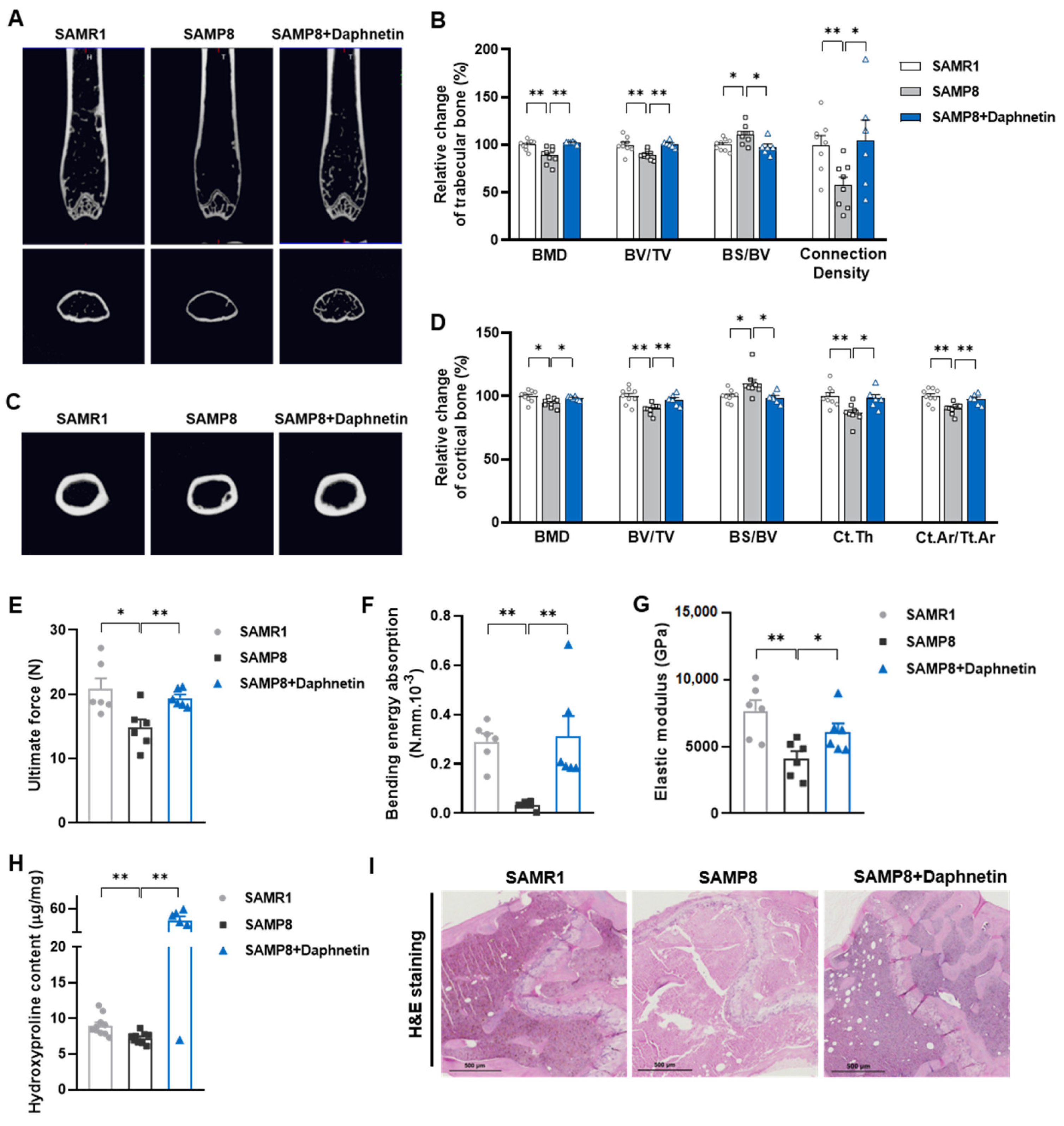
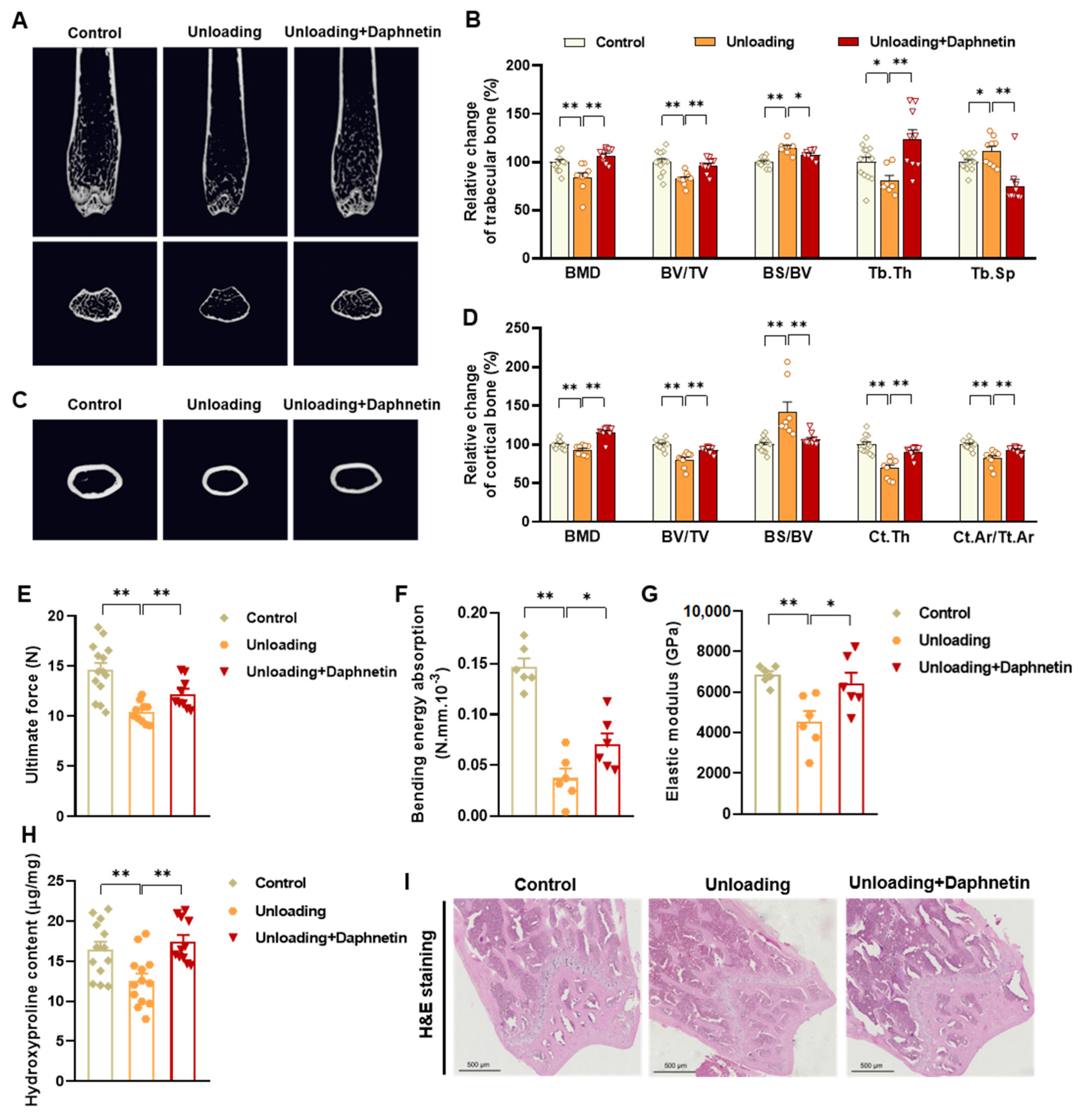
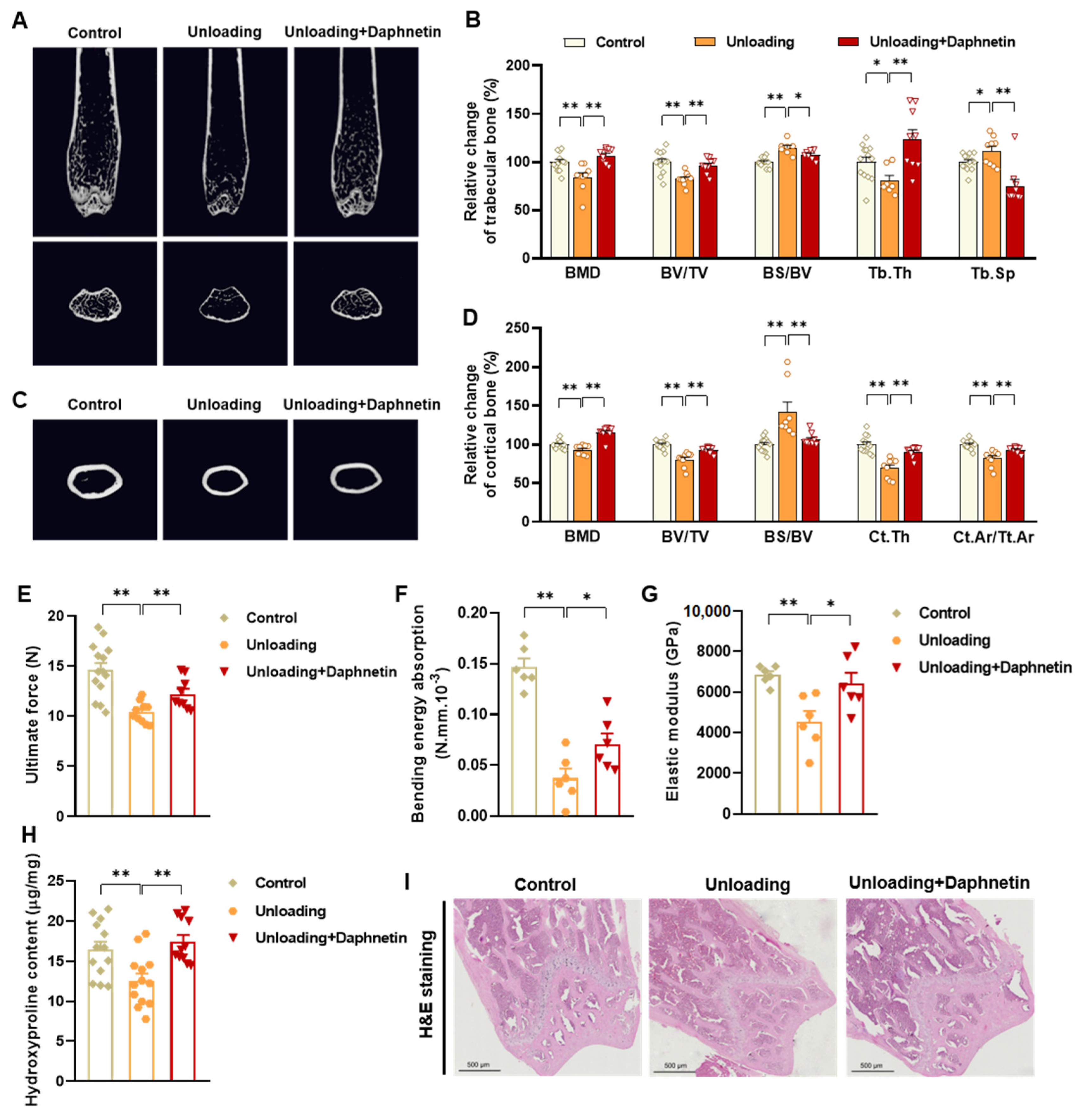
To determine the bone mineral density (BMD) and microarchitecture of bone, the left femurs of mice were scanned using a micro-CT scanner (Quantum GX, PerkinElmer, Antwerp, Belgium). After reconstruction, the micro-CT data were loaded into Analyze 12.0 software (AnalyzeDirect, Overland Park, KS, USA) for calculation. For cancellous bone analysis, trabecular BMD, bone volume fraction (BV/TV), bone surface/bone volume (BS/BV), trabecular thickness (Tb. Th), trabecular separation (Tb. Sp), and connection density were calculated from a 250-slice round region of the distal femur. For cortical bone analysis, cortical BMD, BV/TV, BS/BV, cortical bone thickness (Ct. Th), and cortical area fraction (Ct. Ar/Tt. Ar) were calculated from a 60-slice round region of the mid-diaphysis femur.
2.5. Three-Point Bending Test
To evaluate bone biomechanical properties, the right femurs of sacrificed mice were removed and cleaned of soft tissues. Then three-point bending test was performed using a high precision material test system Instron Microtester 5848 (Instron, Norwood, MA, USA). The Instron machine was set to a loading rate of 1.2 mm/min. Each femur was placed in the same orientation on supports placed 1 cm apart. The Loading-Displacement curve was recorded as the ultimate breaking force required to break the bone, and the ultimate force, bending energy absorption and elastic modulus were calculated.
2.6. Hydroxyproline Determination
To evaluate collagen metabolism in bone tissues, hydroxyproline as the main component of collagen was measured with Hydroxyproline assay kit (Lot No.: A030-2-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to manufacturer’s instructions. The content of hydroxyproline was calculated according to OD550 nm measured with a spectrometer (FlexStation 3, Molecular Devices, Sunnyvale, CA, USA).
2.7. Bone Tissue Section Staining
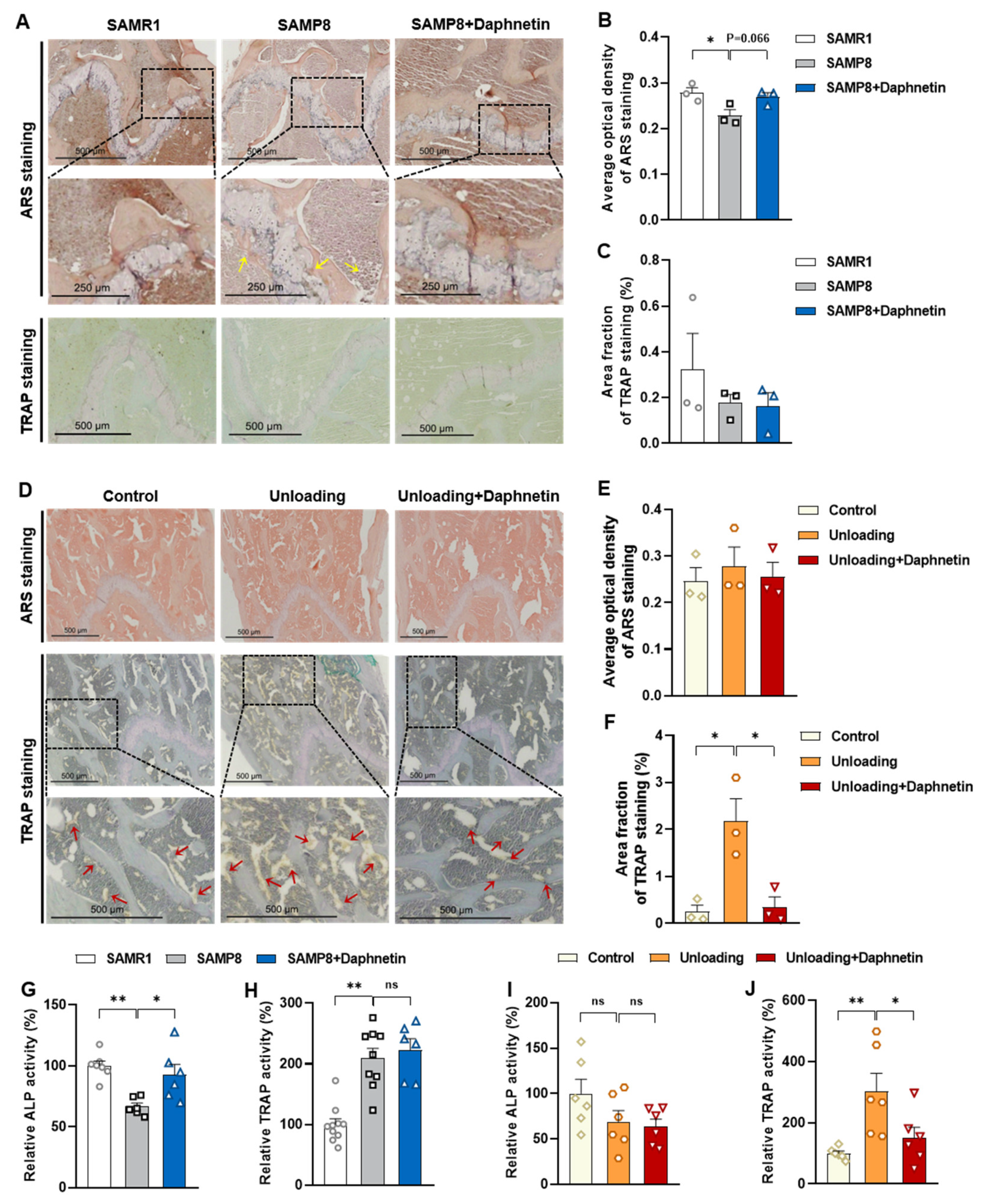
Femur tissues dissected from mice were fixed with 10% formalin for 48 h and decalcified in 14% (w/v) ethylenediaminetetraacetic acid (EDTA) in PBS (pH 7.4) for 21 days at room temperature. Sections were cut from paraffin wax embedded tissues, then, hematoxylin-eosin (H & E) staining was performed to observe the pathomorphological changes of bone tissues. Alizarin Red S (ARS) staining was performed using Alizarin Red S solution (1%, pH 4.2) (Solarbio, Beijing, China) to detect the matrix mineralization deposition of osteoblasts. Tartrate resistant acid phosphatase (TRAP) staining was performed using Acid Phosphatase, Leukocyte (TRAP) Kit (Sigma-Aldrich, St. Louis, MO, USA) to evaluate osteoclastogenesis.
2.8. Determination of Bone Turnover Parameters
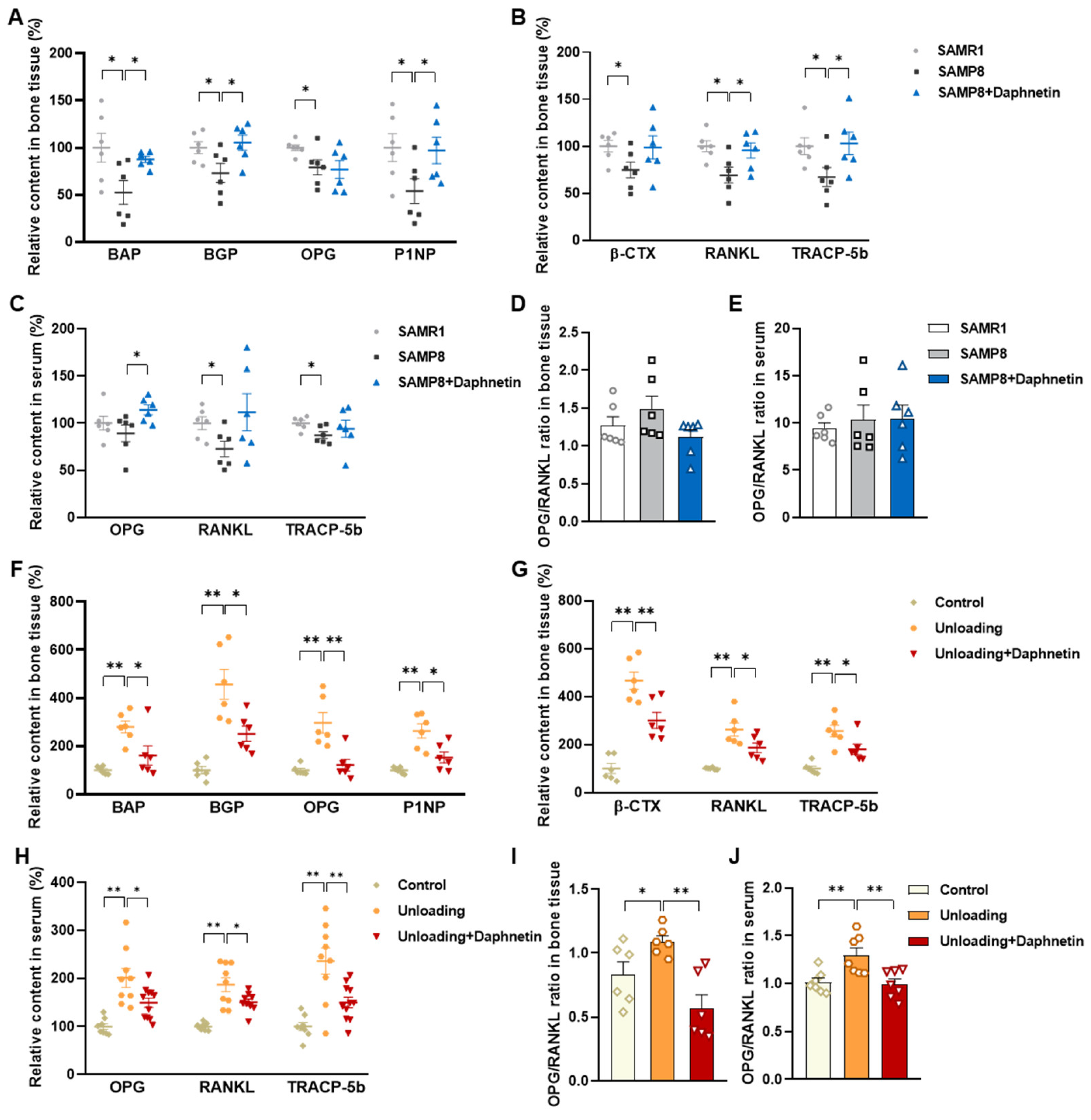
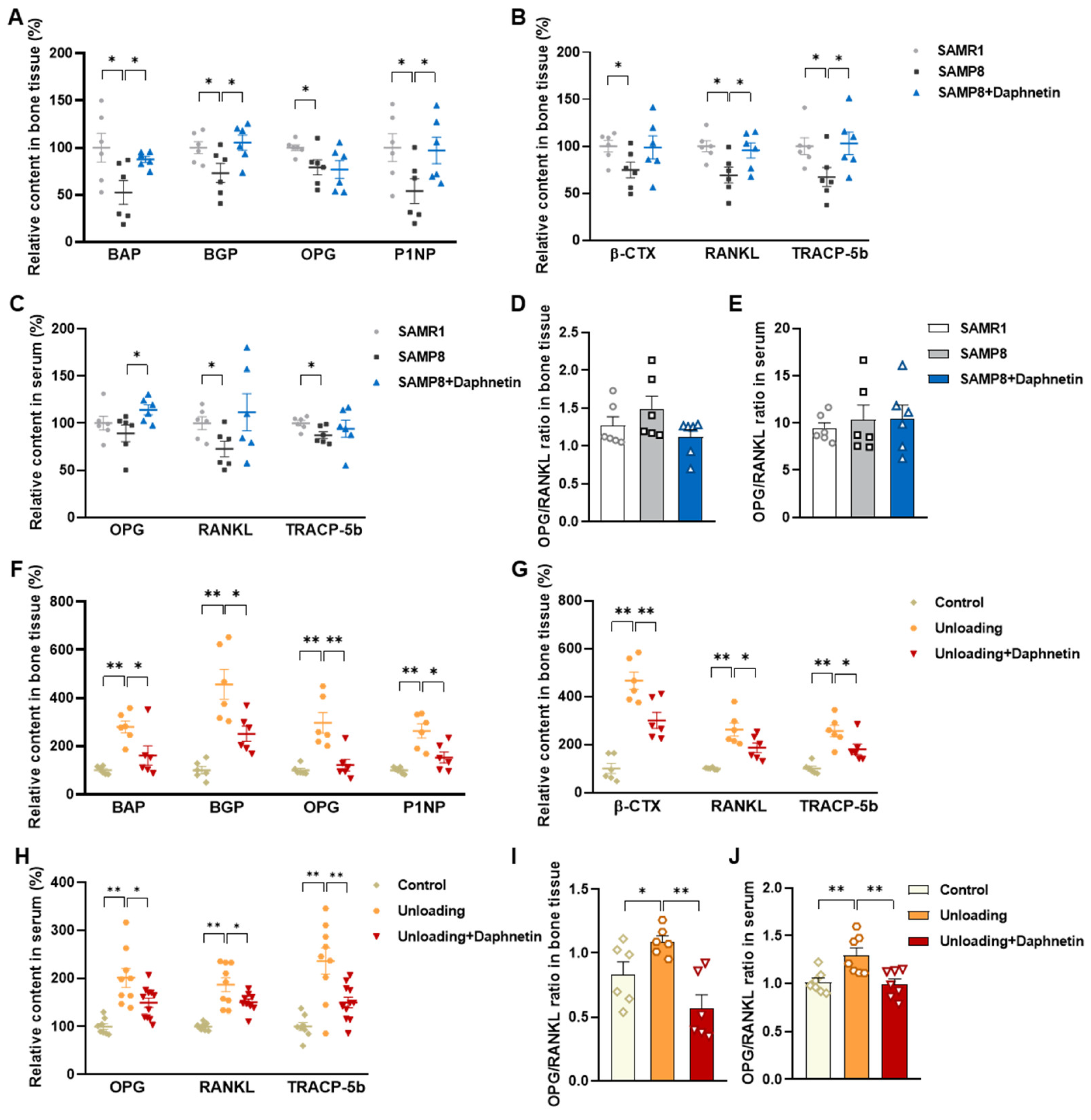
Blood samples were allowed to clot undisturbed for 30 min at room temperature. Serum was separated through centrifugation at 3000× g for 10 min. Bone tissues were homogenized with the aid of a Polytron homogenizer in ice-cold PBS, and bone homogenates were prepared through centrifugation at 4 °C, 1000× g for 10 min. Both serum and bone homogenate were used for subsequent analysis. The levels of biochemical markers of bone turnover such as bone alkaline phosphatase (BAP), bone Gla-protein (BGP), osteoprotegerin (OPG), procollagen type I N-terminal propeptide (P1NP), β isomer of C-terminal telopeptide of type I collagen (β-CTX), RANKL, and tartrate-resistant acid phosphatase 5b (TRACP-5b) were measured by enzyme-linked immunosorbent assay (ELISA).
2.9. Alkaline Phosphatase (ALP) Activity Assay
ALP activity of bone tissue and osteoblast homogenate was measured with a commercially available assay kit (Lot No.: A059-2, Nanjing Jiancheng Bioengineering Institute) according to manufacturer’s instructions. ALP activity was calculated according to OD520 nm measured with spectrometer (FlexStation 3, Molecular Devices, Sunnyvale, CA, USA).
2.10. TRAP Activity Assay
TRAP activity of bone tissue and osteoclast homogenates was measured with a commercially available assay kit (Lot No.: TE0043, Leagene Biotechnology, Inc., Beijing, China) according to manufacturer’s instructions. The activity of TRAP was calculated according to OD 400~415 nm measured with spectrometer (FlexStation 3, Molecular Devices, Sunnyvale, CA, USA).
2.11. ALP Staining of Osteoblasts
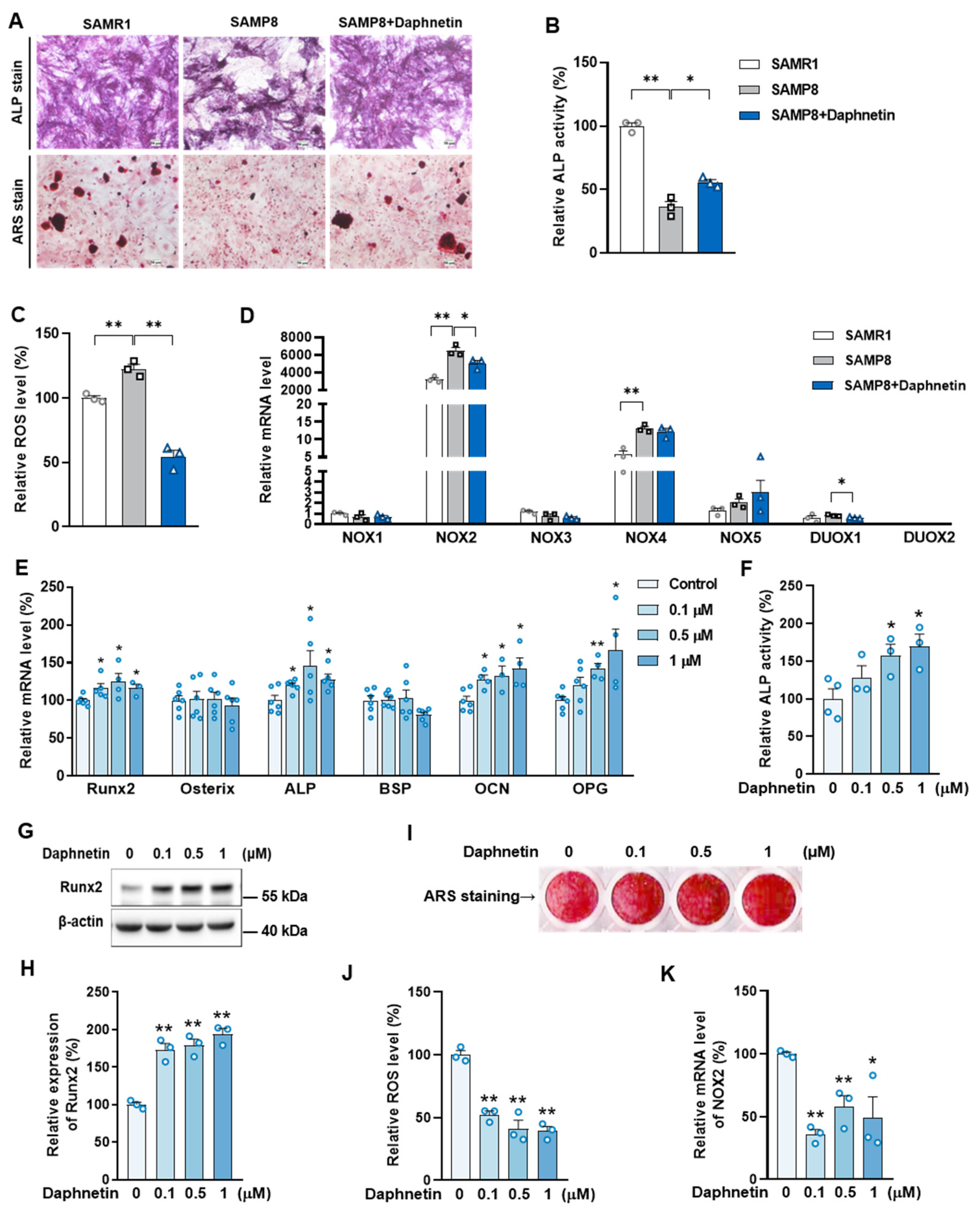
Primary osteoblasts isolated from SAMP8 mice were induced to differentiate for 7 days, then the intracellular ALP level was determined by ALP staining using BCIP/NBT alkaline phosphatase color development kit (Lot No.: C3206, Beyotime, Haimen, China) according to the manufacturer’s instructions, then the stained matrix was observed with light microscope.
2.12. Alizarin Red S (ARS) Staining of Osteoblasts
Primary osteoblasts isolated from SAMP8 mice, and MC3T3-E1 cells were induced to differentiate for 7 or 14 days, then mineralization of cell matrix was determined by the deposition of crystalline hydroxyapatite and visualized by ARS staining. Cells were gently washed twice with PBS and fixed with ice-cold 70% (v/v) ethanol for 30 min at 4 °C. Then, cells were washed with PBS for three times, and stained with 5% ARS solution (pH 4.2) for 30 min at 37 °C. Cells were finally washed with PBS to remove excess dye, and the stained matrix was photographed and observed with light microscope.
2.13. RNA Isolation and Quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
After various treatments, cells were lysed using TRIzol reagent, then total RNA was separated by chloroform, precipitated by isopropanol, and washed with 75% (
v/
v) ethanol. After quantification, reverse transcription was performed using the PrimeScript RT-PCR Kit (Lot No.: RR036A, Takara, Otsu, Shiga, Japan) followed by quantitative real-time PCR using real-time PCR Master Mix (Lot No.: RR820A, Takara, Otsu, Shiga, Japan) following the manufacturer’s protocol. Melting curves were assessed over the range 55~99 °C to ensure specific DNA amplification. The cycle number at which the various transcripts were detected (Ct) was compared with that of GAPDH, referred to as ΔCt. Results were presented as a percentage of the control. Sequences of primers used in the real-time PCR were listed in
Table S1. The primers were derived from GenBank, verified by Primer-BLAST of NCBI, and synthesized by Qingke Biotechnology Co., Ltd. (Beijing, China).
2.14. Protein Extraction and Western Blot Analysis
After various treatments, cells were washed twice with ice-cold PBS and lysed with Western and IP lysis buffer (Beyotime, Haimen, China) containing 1 mM phenylmethanesulfonylfluoride on ice. The lysates were centrifuged at 13,000× g for 6 min at 4 °C. Protein concentrations of the collected supernatants were quantified with BCA kit (Lot No.: 23225, Pierce Biotechnology, Rockford, IL, USA). Equal aliquots of protein samples were subjected to SDS-PAGE, and then transferred to NC membranes. After blocking with 5% (w/v) skimmed milk for 1 h at room temperature, membranes were incubated with primary antibodies (1:1000 dilution) at 4 °C overnight. After washing with TBST three times (15 min each), the membranes were incubated with horseradish-peroxidase-conjugated secondary antibodies (1:4000 dilution) for 1 h at room temperature. After washing with TBST three times, chemiluminescent detection was performed using ECL western blotting substrate (Bio-Rad, Hercules, CA, USA) and quantified by scanning densitometry.
2.15. Cell Viability Assay
MC3T3-E1 and RAW264.7 cells were seeded in 96-well plates. After being treated with indicated concentrations of daphnetin for 48 h, cells were incubated with 0.5 mg/mL 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) dissolved in FBS-free DMEM for 4 h at 37 °C. Then, the supernatants were removed, and MTT-formazan products were solubilized with 200 mL DMSO each well. The optical densities were measured at 550 nm using a microplate spectrophotometer (FlexStation 3, Molecular Devices, Sunnyvale, CA, USA).
2.16. Determination of Intracellular ROS
After various treatments, cells were incubated with 10 μM H
2DCF-DA (Life Technologies, San Diego, CA, USA) in cell culture medium for 30 min at 37 °C and lysed with solution containing 10 mM Tris-base, 150 mM NaCl, 0.1 mM EDTA-Na
2, and 0.5% (
v/
v) Triton X-100. Intracellular ROS level was determined by the formation of fluorescent 2′,7′-dichlorofluorescein upon oxidation of non-fluorescent, reduced DCFH [
25]. The fluorescence intensity of the supernatant was measured with a fluorescence spectrometer (FlexStation 3, Molecular Devices, Sunnyvale, CA, USA) at 485 ex/538 em. Cellular oxidant levels were expressed as the relative fluorescence per microgram of protein.
2.17. Determination of Total Antioxidant Capacity (T-AOC)
T-AOC of cell homogenates was measured with a commercially available assay kit (Lot No.: A015, Nanjing Jiancheng Bioengineering Institute) according to the manufacturer’s instructions. T-AOC was calculated according to OD520 nm measured with a spectrometer (FlexStation 3, Molecular Devices, Sunnyvale, CA, USA).
2.18. Determination of GSH
GSH levels were measured with the 2,3-naphthalenedicarboxyaldehyde (NDA) method [
26]. Briefly, 20 μL of cell homogenate and 180 μL of NDA derivatization solution [50 mM Tris (pH 10), 0.5 N NaOH and 10 mM NDA in Me2SO,
v/
v/
v 1.4:0.2:0.2] were added to each well of a 96-well plate. The plate was covered to protect the wells from light and incubated at room temperature for 30 min. The NDA-GSH fluorescence intensity was measured (485 ex/538 em) with a fluorescence spectrometer (FlexStation 3, Molecular Devices, Sunnyvale, CA, USA).
2.19. Superoxide Dismutase 2 (SOD2) Activity Assay
SOD2 activity in cell homogenate was measured with a commercially available assay kit (Lot No.: A001-2-2, Nanjing Jiancheng Bioengineering Institute) according to the manufacturer’s instructions. Activities of total SOD and CuZn SOD were calculated according to OD550 nm measured with a spectrometer (FlexStation 3, Molecular Devices, Sunnyvale, CA, USA). SOD2 activity was equal to total SOD activity minus CuZn SOD activity.
2.20. Tartrate-Resistant Acid Phosphatase (TRAP) Staining of Osteoclasts
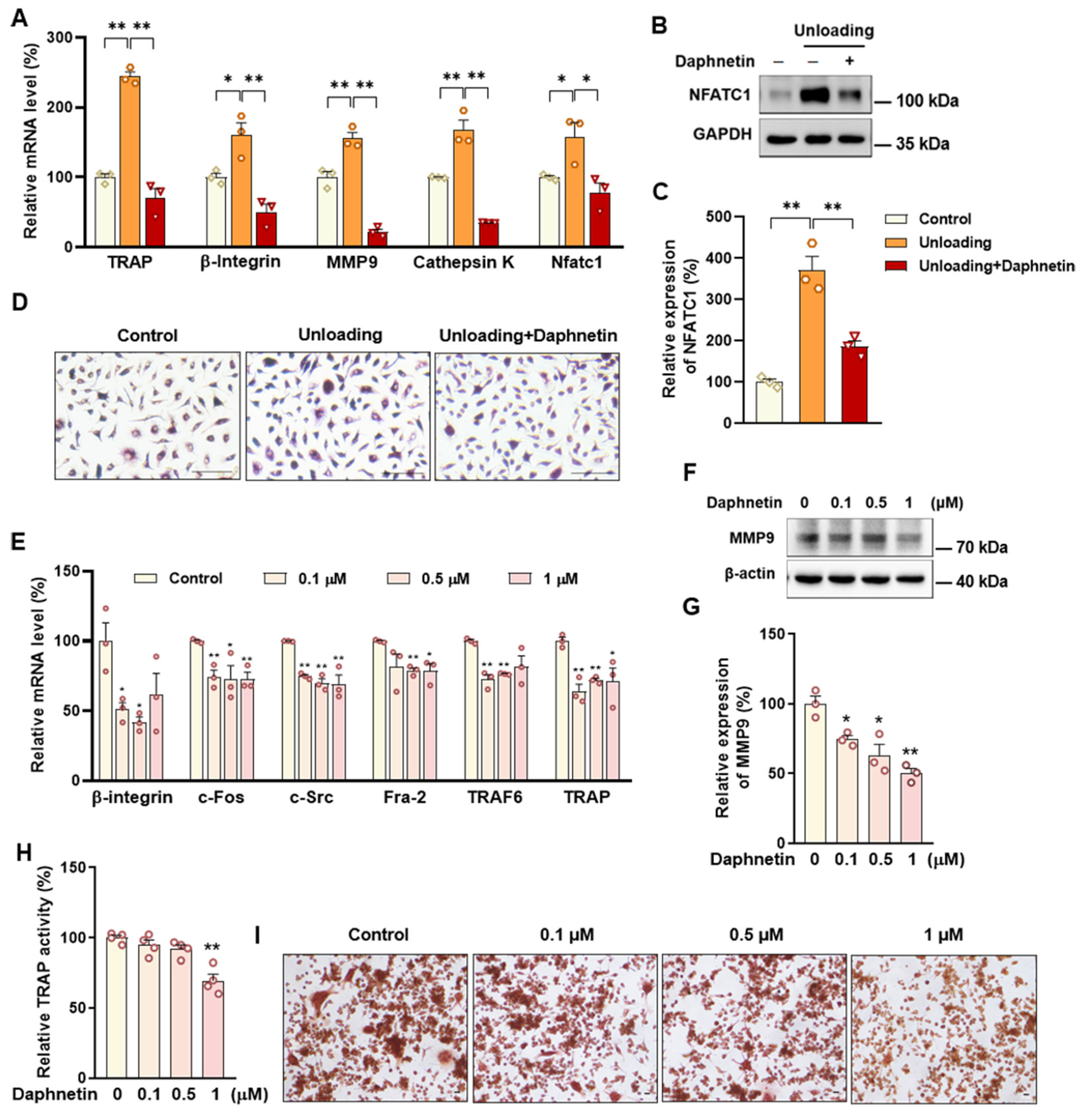
Bone marrow monocyte-derived osteoclasts isolated from hindlimb unloading mice, and RAW264.7 cells were induced to differentiate for 6 or 4 days, then TRAP staining was performed using Acid Phosphatase, Leukocyte (TRAP) Kit (Lot No.: 387A, Sigma-Aldrich, St. Louis, MO, USA) to evaluate osteoclastogenesis. Cells were gently washed twice with PBS and successively fixed with 10% (v/v) formaldehyde and fixative solution (ethanol: acetone 1:1) for 1 min at room temperature. Then, cells were washed with deionized water for three times, and stained with prewarmed staining solution (prepared according to the instructions) for 1 h at 37 °C protected from light. Cells were finally washed with deionized water to remove excess dye, and the stained matrix was photographed and observed with light microscope.
2.21. Statistical Analysis
The in vitro experiments were performed in triplicate and repeated at least three times. Data were presented as the mean ± S.E.M. and were analyzed using the GraphPad Prism version 8 software (San Diego, CA, USA). Statistical significance was evaluated using one-way analysis of variance (ANOVA) followed by Tukey’s HSD test. Value of p < 0.05 was considered as significant.
4. Discussion
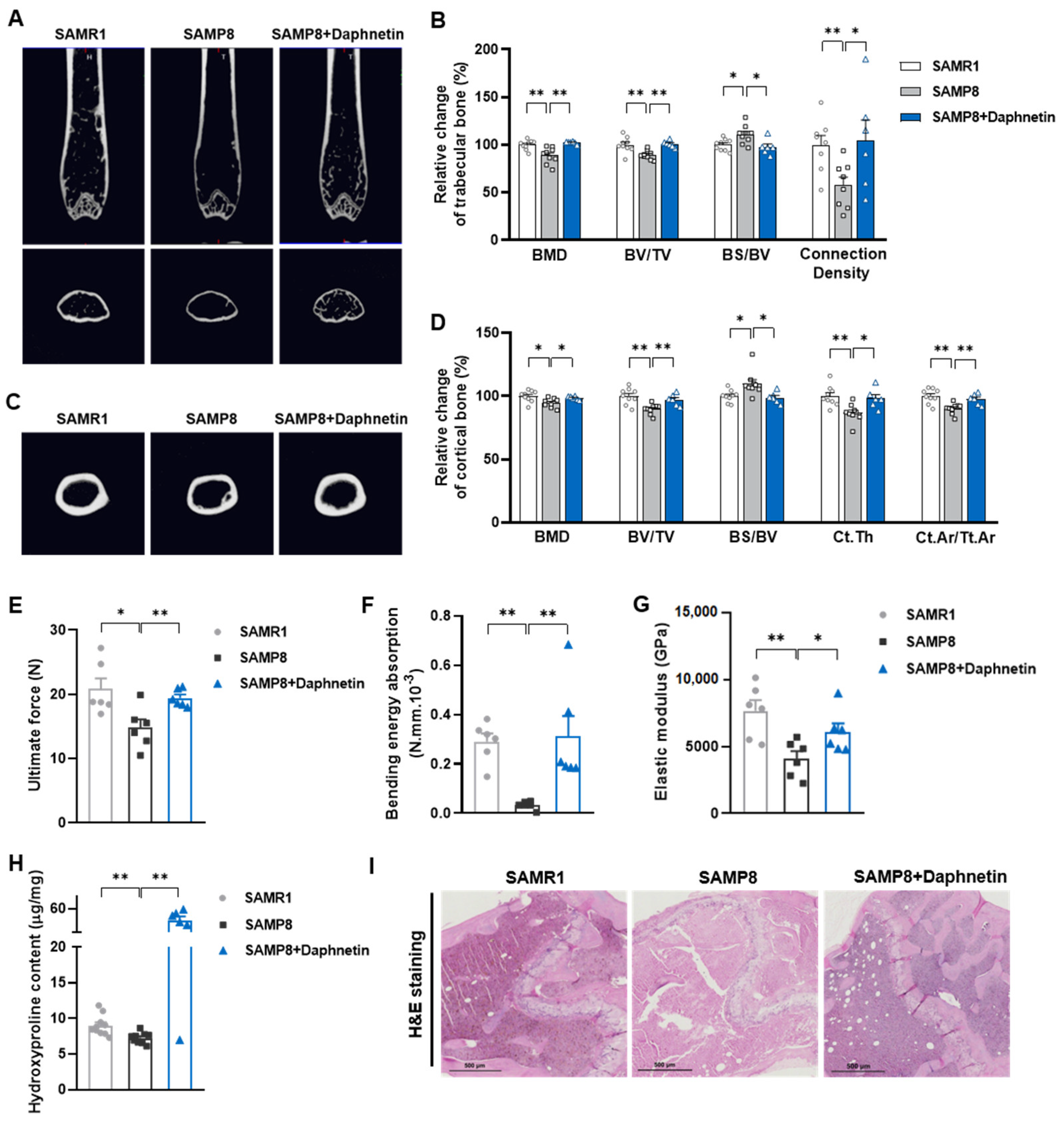
In the present study, SAMP8 and hindlimb unloading mice were adopted as animal models to evaluate the effects of daphnetin on senile and disuse osteoporosis, respectively. Through micro-CT scanning and H&E staining, we found significant loss of bone mass and deteriorated bone microstructure in both models. Studies have shown that when osteoporosis occurs, cortical bone bears more strength load [
27]. In this study, degenerative changes of cortical bone accompanied with that of trabecular bone also occurred, reflecting the comprehensive changes of bone metabolism in SAMP8 and hindlimb unloading mice. Three-point bending tests also showed weakened bone strength in these two models, which were consistent with the results of previous studies [
28,
29]. It is known that bone strength is reflected in the hardness and toughness of bone. Inorganic minerals determine the density and hardness of bone, while bone organic matter dominated by collagen determines the toughness of bone. Therefore, we detected the content of hydroxyproline, the main representative component of collagen, and the results confirmed the decrease of bone organic matter in both SAMP8 and hindlimb unloading mice. Interestingly, these changes were all effectively reversed by daphnetin administration, indicating that daphnetin has effective osteoprotective effects against senile and disuse osteoporosis.
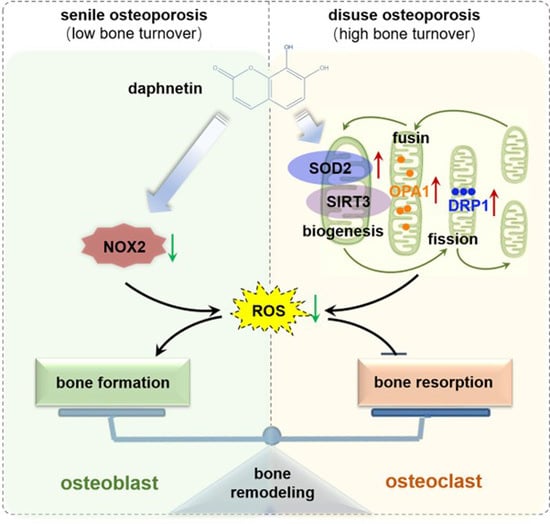
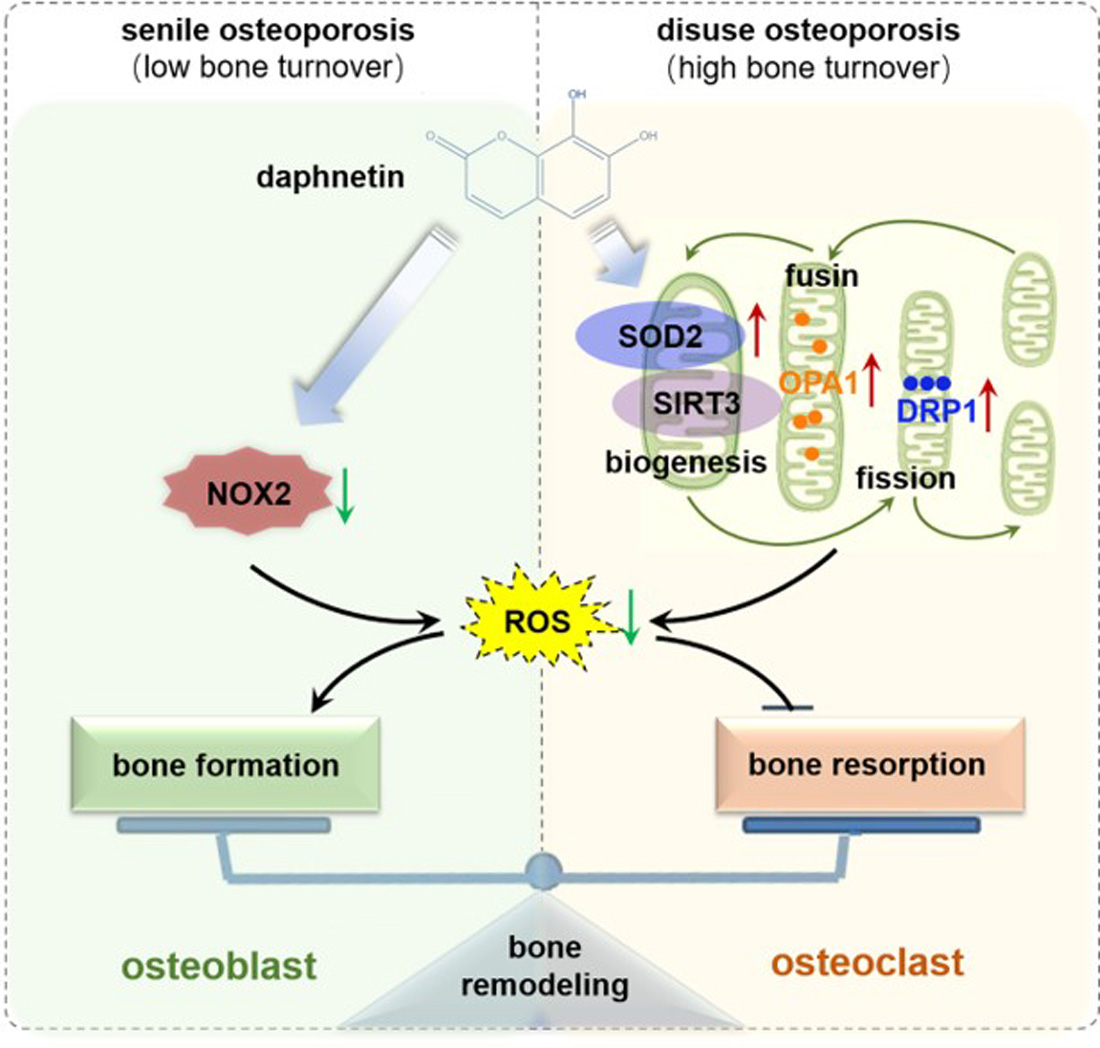
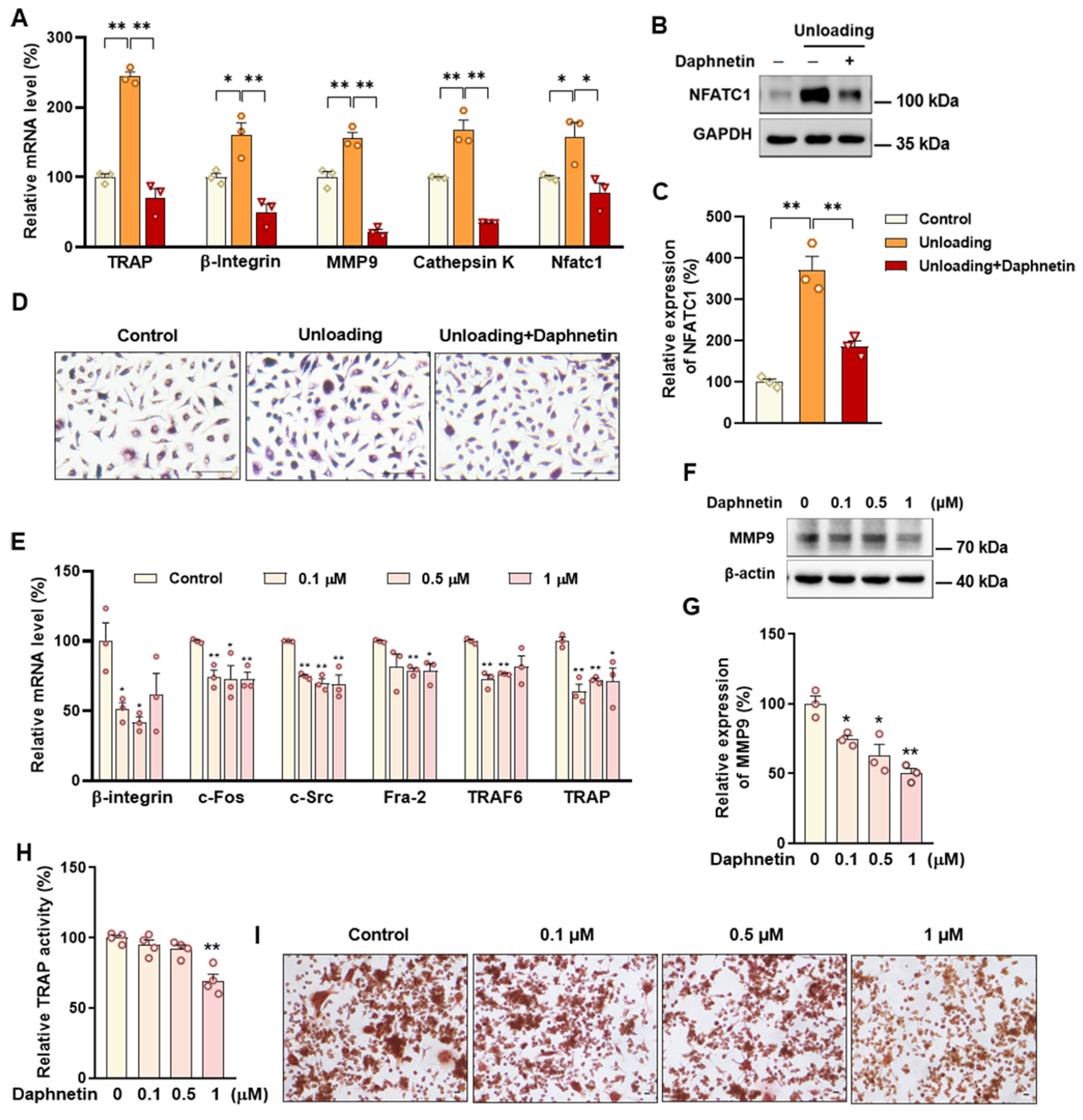
We further found that the contents of bone remodeling markers related to osteoblastic bone formation and osteoclastic bone resorption in SAMP8 mice were all decreased in bone homogenate, suggesting a low bone turnover status in senile osteoporosis. However, there was a marked increase in serum level of both bone formation- and resorption-related markers in hindlimb unloading mice, indicating a high bone turnover status in disuse osteoporosis. OPG/RANKL pathway, which mediates the molecular correlation between osteoblasts and osteoclasts, plays a vital role in bone remodeling regulation. Up-regulating OPG/RANKL ratio is beneficial to promote osteogenesis and particularly inhibit the formation and activation of osteoclasts [
30]. Since osteoporosis is usually accompanied by the decrease of OPG/RANKL, we speculated that the abnormal increase of OPG/RANKL may be an accompanying compensatory effect, and OPG/RANKL may not be the main pathway affecting disuse osteoporosis. Subsequently, the potential mechanisms underlying the protective effects of daphnetin against senile and disuse osteoporosis were further explored. Through activity assays of ALP and TRAP, which are key enzymes of osteogenesis and osteoclastogenesis, as well as ARS and TRAP staining, we found that daphnetin specifically preserved osteoblastic bone formation, but did not reverse the accelerated bone resorption in SAMP8 mice. While in hindlimb unloading mice, daphnetin specifically inhibited osteoclastic bone resorption rather than preserved bone formation. These results further confirmed that the distinct roles of daphnetin in senile and disuse osteoporosis may be dependent on different bone turnover statuses. To our knowledge, this is the first study to explore the osteoprotective effects of daphnetin against senile osteoporosis and disuse osteoporosis, and to reveal that daphnetin alleviates bone loss by distinctly modulating bone formation and bone resorption according to different bone turnover statuses.
It has been demonstrated that oxidative stress and mitochondrial dysfunction are the key pathophysiological characteristics of osteoporosis [
31]. Excessive ROS will induce oxidative damage and mitochondrial dysfunction, while mitochondrial dysfunction further leads to more ROS generation. Studies have reported that unregulated accumulation of ROS is a major contributor to aging and age-associated diseases including osteoporosis and other bone diseases [
32]. Excessive ROS is known to inhibit osteogenic differentiation and mineralization deposition [
33]. Our previous study has demonstrated that SIRT3/SOD2 pathway mediated enhancement of antioxidant capacity and mitochondrial function are essential to osteogenic differentiation [
34]. In this study, we consistently found that, the hindered osteogenic differentiation in SAMP8 mice was accompanied with aging-induced oxidative stress and loss of mitochondrial homeostasis in osteoblasts, characterized by increased intracellular ROS level, down-regulation of SIRT3 as well as decreased expressions of mitochondrial biogenesis- and dynamic-related proteins. These data suggest that daphnetin administration may preserve osteogenesis by eliminating excessive ROS of osteoblasts in senile osteoporosis.
To further investigate the mechanisms involved in daphnetin ameliorating oxidative stress in osteoblasts of SAMP8 mice, we evaluated the ability of antioxidant defense system. Because of the vital role of SOD2, CAT, HO-1, and NQO1 as antioxidant enzymes, their expressions, T-AOC, GSH level, and SOD2 activity were determined. There were no significant changes of T-AOC, GSH level, SOD2 activity or its expression in osteoblasts in SAMP8 mice. Although the expression of HO-1 was significantly decreased, daphnetin administration did not reverse this change, but counteracted the excessive ROS generation by up-regulating the expression of enzymatic scavengers CAT and NQO1. Therefore, the role of daphnetin in eliminating oxidative stress of osteoblasts may attribute to inhibiting ROS production rather than enhancing antioxidant capacity and ROS scavenging in senile osteoporosis.
It is known that mitochondrial homeostasis is closely related to the production of mitochondrial ROS. Since daphnetin showed no effect on preserving mitochondrial biogenesis or dynamics of osteoblasts, and ROS produced from NOX participates in regulating various physiological processes including cell differentiation [
35], we speculated that the protective effects of daphnetin against oxidative stress may not rely on the improvement of antioxidant defense, or the restoration of mitochondrial homeostasis, but on inhibiting the non-mitochondrial ROS production. Then, we investigated the transcription of NOX family genes, the important source of non-mitochondrial ROS. The transcription of NOX2, the most abundant isoform in osteoblasts, was significantly increased in osteoblasts of SAMP8 mice, while daphnetin administration effectively inhibited the transcription of NOX2, which may result in elimination of ROS. In addition, results of in vitro experiments performed on pre-osteoblast MC3T3-E1 cells also confirmed that daphnetin promoted osteogenic differentiation, and this effect was accompanied with NOX2 inhibition and ROS elimination. Taken together, all these data suggest that daphnetin may preserve bone formation of SAMP8 mice through inhibiting NOX2-mediated ROS production in osteoblast, leading to alleviation of senile osteoporosis.
Accumulating evidence has indicated that intracellular ROS is highly involved in bone homeostasis not only by influencing osteogenesis, but also by intervening osteoclast differentiation [
36,
37,
38,
39]. Excessive ROS can down-regulate OPG and simultaneously up-regulate RANKL, then RANKL recruits the linker molecule TRAF6 to the receptor activator of NF-κB (RANK), activates mitogen-activated protein kinase (MAPK) signaling pathway, and promotes nuclear translocation of key transcription factors such as NFATc1 [
36], thereby stimulating the expression of downstream osteoclast differentiation marker genes, such as TRAP, Cathepsin K, and MMP9, resulting in activation of osteoclasts and bone resorption, ultimately leading to bone microstructure deterioration and bone density reduction [
40,
41,
42,
43]. In this study, we found that daphnetin administration significantly decreased intracellular ROS of osteoclasts, and effectively hindered osteoclast differentiation in hindlimb unloading mice, indicated by inhibited transcription of bone resorption marker genes, decreased expression of NFATC1 and TRAP activity, as well as weakened TRAP staining after daphnetin administration.
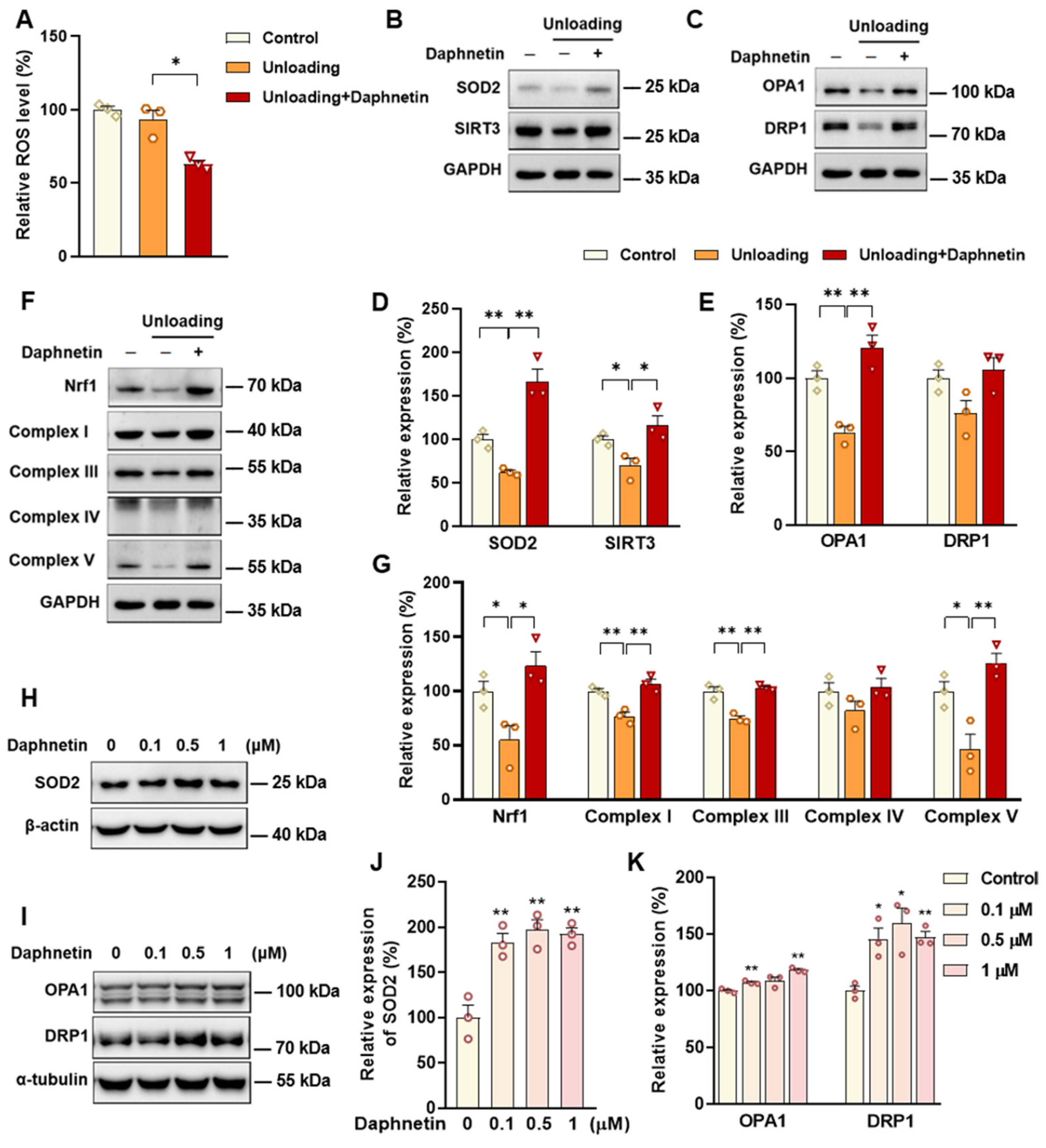
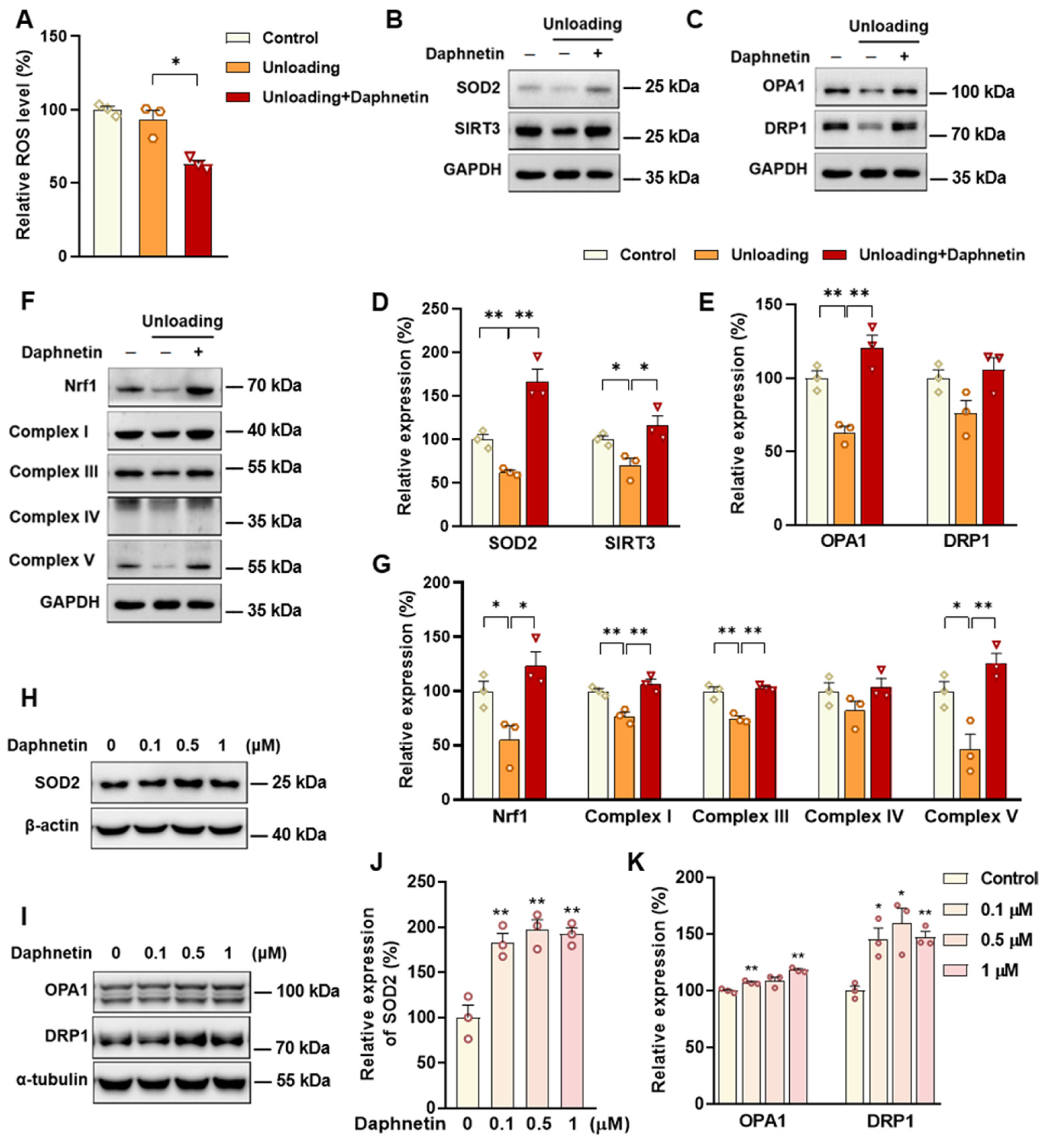
SOD2, an important enzyme responsible for reducing ROS in mitochondria, plays an inhibitory role in osteoclastogenesis [
44]. In this study, we also found that daphnetin significantly up-regulated SOD2 expression, resulting in enhanced antioxidant capacity and elimination of ROS in osteoclasts of hindlimb unloading mice. In addition, SIRT3 has been confirmed to regulate osteoclastogenesis through activating SOD2 and regulating AMPK-peroxisome proliferator-activated receptor-coactivator 1-beta (PGC-1β) axis [
44,
45]. Mitochondrial homeostasis including the dynamics of mitochondrial biogenesis, fusion/fission and mitophagy, is also essential for redox state of cells. In this study, we found decreased SIRT3 expression and mitochondrial homeostasis failure in osteoclasts of unloading mice, indicated by significantly decreased expressions of mitochondrial biogenesis- and dynamic-related proteins, while daphnetin administration effectively restored SIRT3 expression and mitochondrial homeostasis of osteoclasts. These results suggest that SIRT3 deficiency and mitochondrial dysfunction in osteoclasts may play an important role in the pathological process of disuse osteoporosis.
Similar to the study performed on osteoblasts of SAMP8 mice, we evaluated the changes of NOX/ROS cascade in osteoclasts of hindlimb unloading mice. It is interesting to note that neither disuse osteoporosis nor daphnetin administration influenced NOX-mediated ROS production in osteoclasts. These results suggest that accompanied with up-regulating SOD2, restoring SIRT3 expression and mitochondrial homeostasis of osteoclasts may be another potential mechanism underlying the protective effects of daphnetin against disuse osteoporosis. Experiments performed on osteoclast precursor RAW264.7 cells further confirmed that daphnetin stimulated the expressions of SOD2 and mitochondrial dynamic-related proteins OPA1 and DRP1 in normal conditions. Perhaps only under pathological conditions can daphnetin show the effects of reducing ROS, preserving SIRT3 and mitochondrial homeostasis. The results of in vitro experiments similarly indicated that daphnetin can enhance antioxidant capacity and is beneficial to quantity- and quality-control of mitochondria in osteoclasts. Although further efforts should be made to illuminate the deep mechanisms underlying the above phenomena, in the current study, we have provided new evidence of the effects of daphnetin on SIRT3/SOD2 pathway and mitochondrial homeostasis of osteoclasts.
In conclusion, we demonstrated that daphnetin alleviated both senile osteoporosis and disuse osteoporosis with different mechanisms. In terms of senile osteoporosis, daphnetin mainly reduced NOX2-mediated ROS production in osteoblasts, resulting in promotion of osteogenic differentiation and bone formation. While in terms of disuse osteoporosis, daphnetin up-regulated SOD2, eliminated ROS, restored SIRT3 expression and mitochondrial homeostasis in osteoclasts, resulting in inhibition of bone resorption. Combined with the results of previous studies, daphnetin has shown a wide range of efficacy and application potential in the prevention and treatment of osteoporosis, although the underlying mechanisms may vary with different types of osteoporosis. In this study, we focused our investigations on the changes of NOX/ROS cascade, SIRT3/SOD2 pathway, and mitochondrial homeostasis involved in osteoprotective effects of daphnetin. However, the deep mechanisms underlying these changes deserve further exploration. Nevertheless, our findings have provided the basis for the application of daphnetin in prevention and treatment of osteoporosis, and also suggest that it may be of great importance to adopt targeted prevention and therapy strategies according to distinct bone turnover statuses of osteoporosis.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}