
Redox Status Is the Mainstay of SARS-CoV-2 and Host for Producing Therapeutic Opportunities





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
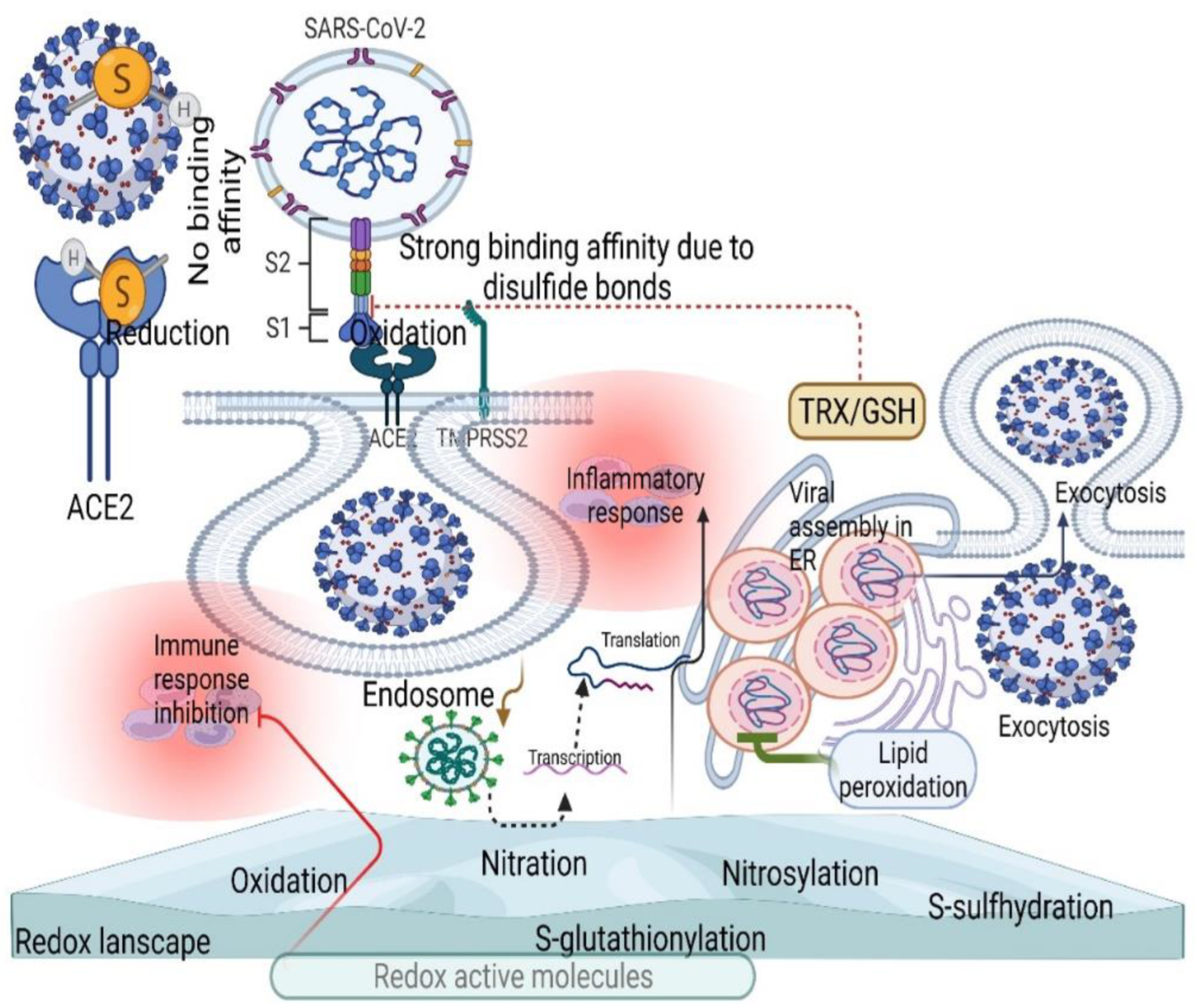
2. Sources of ROS during COVID-19
3. SARS-CoV-2 Controls ROS Levels for Its Survival
4. Redox Chemicals Coordinate Local and Systemic Redox Networks in COVID-19
5. Antioxidant Therapy in COVID-19
6. Challenges Associated with Oxidative Stress during COVID-19 Treatment
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Raymond, J.; Segre, D. The effect of oxygen on biochemical networks and the evolution of complex life. Science 2006, 311, 1764–1767. [Google Scholar] [CrossRef]
- Castresana, J.; Saraste, M. Evolution of energetic metabolism; the respiration early hypothesis. Trends Biochem. Sci. 1995, 20, 443–448. [Google Scholar] [CrossRef]
- Cortese-Krott, M.M.; Pullmann, D.; Feelisch, M. Nitrosopersulfide (SSNO-) targets the Keap-1/Nrf2 redox system. Pharmacol. Res. 2016, 113, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Cortese-Krott, M.M.; Butler, A.R.; Woollins, J.D.; Feelisch, M. Inorganic sulfur-nitrogen compounds: From gunpowder chemistry to the forefront of biological signaling. Dalton Trans. 2016, 45, 5908–5919. [Google Scholar] [CrossRef] [PubMed]
- Luu Trinh, M.D.; Miyazaki, D.; Ono, S.; Nomata, J.; Kono, M.; Mino, H.; Niwa, T.; Okegawa, Y.; Motohashi, K.; Taguchi, H.; et al. The evolutionary conserved iron-sulfur protein TCR controls P700 oxidation in photosystem I. iScience. 2021, 24, 102059. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Shope, R. Global climate change and infectious diseases. Environ. Health Perspect. 1991, 96, 171–174. [Google Scholar] [CrossRef]
- Gorji, S.; Gorji, A. COVID-19 pandemic: The possible influence of the long-term ignorance about climate change. Environ. Sci. Pollut. Res. Int. 2021, 28, 15575–15579. [Google Scholar] [CrossRef]
- Rodó, X.; San-José, A.; Kirchgatter, K.; López, L. Changing climate and the COVID-19 pandemic: More than just heads or tails. Nat. Med. 2021, 27, 576–579. [Google Scholar] [CrossRef]
- Ukhurebor, K.E.; Singh, K.R.; Nayak, V.; Uk-Eghonghon, G. Influence of the SARS-CoV-2 pandemic: A review from the climate change perspective. Environ. Sci. Process Impacts 2021, 23, 1060–1078. [Google Scholar] [CrossRef]
- Jamrozik, E.; Selgelid, M.J. COVID-19 human challenge studies: Ethical issues. Lancet Infect. Dis. 2020, 20, e198–e203. [Google Scholar] [CrossRef]
- Thoradeniya, T.; Jayasinghe, S. COVID-19 and future pandemics: A global systems approach and relevance to SDGs. Global Health 2021, 17, 59. [Google Scholar] [CrossRef]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Cumpstey, A.F.; Clark, A.D.; Santolini, J.; Jackson, A.A.; Feelisch, M. COVID-19: A Redox Disease-What a Stress Pandemic Can Teach Us About Resilience and What We May Learn from the Reactive Species Interactome About Its Treatment. Antioxid. Redox Signal. 2021, 35, 1226–1268. [Google Scholar] [CrossRef] [PubMed]
- Gadotti, A.C.; Lipinski, A.L.; Vasconcellos, F.T.; Marqueze, L.F.; Cunha, E.B.; Campos, A.C.; Oliveira, C.F.; Amaral, A.N.; Baena, C.P.; Telles, J.P.; et al. Susceptibility of the patients infected with Sars-Cov2 to oxidative stress and possible interplay with severity of the disease. Free Radic. Biol. Med. 2021, 165, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Nicotinamide nucleotide compartmentation. In Metabolic Compartmentation; Sies, H., Ed.; Academic: London, UK, 1982; pp. 205–231. [Google Scholar]
- de Oliveira, A.A.; Nunes, K.P. Crosstalk of TLR4, vascular NADPH oxidase, and COVID-19 in diabetes: What are the potential implications? Vasc. Pharmacol. 2021, 139, 106879. [Google Scholar] [CrossRef]
- Pratomo, I.P.; Noor, D.R.; Kusmardi, K.; Rukmana, A.; Paramita, R.I.; Erlina, L.; Fadilah, F.; Gayatri, A.; Fitriani, M.; Purnomo, T.T.H.; et al. Xanthine Oxidase-Induced Inflammatory Responses in Respiratory Epithelial Cells: A Review in Immunopathology of COVID-19. Internat. J. Inflamm. 2021, 2021, 1653392. [Google Scholar] [CrossRef]
- Ryback, R.; Eirin, A. Mitochondria, a Missing Link in COVID-19 Heart Failure and Arrest? Front. Cardiovasc. Med. 2022, 8, 830024. [Google Scholar] [CrossRef]
- Violi, F.; Ceccarelli, G.; Cangemi, R.; Alessandri, F.; D’Ettorre, G.; Oliva, A.; Pastori, D.; Loffredo, L.; Pignatelli, P.; Ruberto, F.; et al. Hypoalbuminemia, Coagulopathy, and Vascular Disease in COVID-19. Circ. Res. 2020, 127, 400–401. [Google Scholar] [CrossRef]
- Violi, F.; Oliva, A.; Cangemi, R.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Alessandri, M.L.F.; Angelis, M.D.; et al. Nox2 activation in COVID-19. Redox Biol. 2020, 36, 101655. [Google Scholar] [CrossRef]
- Werion, A.; Belkhir, L.; Perrot, M.; Schmit, G.; Aydin, S.; Chen, Z.; Penaloza, A.; Greef, J.D.; Yildiz, H.; Pothen, L.; et al. Cliniques universitaires Saint-Luc (CUSL) COVID-19 Research Group. SARS-CoV-2 causes a specific dysfunction of the kidney proximal tubule. Kidney Int. 2020, 98, 296–1307. [Google Scholar] [CrossRef] [PubMed]
- Sekizuka, H. Uric acid, xanthine oxidase, and vascular damage: Potential of xanthine oxidoreductase inhibitors to prevent cardiovascular diseases. Hypertens. Res. 2022, 45, 772–774. [Google Scholar] [CrossRef] [PubMed]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; Junior, C.A.O.D.B.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446. [Google Scholar] [CrossRef]
- Chen, Y.; Gaber, T. Hypoxia/HIF Modulates Immune Responses. Biomedicines 2021, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Knoblach, B.; Ishida, R.; Hobman, T.C.; Rachubinski, R.A. Peroxisomes exhibit compromised structure and matrix protein content in SARS-CoV-2-infected cells. Mol. Biol. Cell 2021, 32, 1273–1282. [Google Scholar] [CrossRef]
- Archambault, A.S.; Zaid, Y.; Rakotoarivelo, V.; Turcotte, C.; Doré, É.; Dubuc, I.; Martin, C.; Flamand, O.; Amar, Y.; Cheikh, A.; et al. High levels of eicosanoids and docosanoids in the lungs of intubated COVID-19 patients. FASEB J. 2021, 35, e21666. [Google Scholar] [CrossRef]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Łos, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S.; et al. Endoplasmic reticulum as a potential therapeutic target for COVID-19 infection management? Eur. J. Pharmacol. 2020, 882, 173288. [Google Scholar] [CrossRef]
- Asleh, R.; Asher, E.; Yagel, O.; Samuel, T.; Elbaz-Greener, G.; Wolak, A.; Durst, R.; Ben-Chetrit, E.; Nir-Paz, R.; Helviz, Y.; et al. Predictors of Hypoxemia and Related Adverse Outcomes in Patients Hospitalized with COVID-19: A Double-Center Retrospective Study. J. Clin. Med. 2021, 10, 3581. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J. All about Albumin; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Roche, M.; Philippe, R.; Singh, N.R.; Tarnus, E.; Bourdon, E. The antioxidant properties of serum albumin. FEBS Lett. 2008, 582, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Stocker, R. Bilirubin attenuates radical-mediated damage to serum albumin. FEBS Lett. 1993, 331, 281–284. [Google Scholar] [CrossRef]
- Cohen, M.P. Intervention strategies to prevent pathogenetic effects of glycated albumin. Arch. Biochem. Biophys. 2003, 419, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cheng, A.; Kumar, R.; Fang, Y.; Chen, G.; Zhu, Y.; Lin, S. Hypoalbuminemia predicts the outcome of COVID-19 independent of age and co-morbidity. J. Med. Virol. 2020, 92, 2152–2158. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Garcia-Rodriguez, V.; Yu, A.; Dutra, B.; Larson, S.; Cash, B.; DuPont, A.; Farooq, A. Elevated transaminases and hypoalbuminemia in COVID-19 are prognostic factors for disease severity. Sci. Rep. 2021, 11, 10308. [Google Scholar] [CrossRef]
- Schwarz, K.B. Oxidative stress during viral infection: A review. Free Radic. biol Med. 1996, 21, 641–649. [Google Scholar] [CrossRef]
- Qadri, I.; Iwahashi, M.; Capasso, J.M.; Hopken, M.W.; Flores, S.; Schaack, J.; Simon, F.R. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: Role of JNK, p38 MAPK and AP-1. Biochem. J. 2004, 378, 919–928. [Google Scholar] [CrossRef]
- Fillebeen, C.; Muckenthaler, M.; Andriopoulos, B.; Bisaillon, M.; Mounir, Z.; Hentze, M.W.; Koromilas, A.E.; Pantopoulos, K. Expression of the subgenomic hepatitis C virus replicon alters iron homeostasis in Huh7 cells. J. Hepatol. 2007, 47, 12–22. [Google Scholar] [CrossRef]
- Mehri, F.; Rahbar, A.H.; Ghane, E.T.; Souri, B.; Esfahani, M. Changes in oxidative markers in COVID-19 patients. Arch. Med. Res. 2021, 52, 843–849. [Google Scholar] [CrossRef]
- Zendelovska, D.; Atanasovska, E.; Petrushevska, M.; Spasovska, K.; Stevanovikj, M.; Demiri, I.; Labachevski, N. Evaluation of oxidative stress markers in hospitalized patients with moderate and severe COVID-19. Rom. J. Intern. Med. 2021, 59, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Lincet, H.; Wu, Z.; Coquerel, A.; Forgez, P.; Alifano, M.; Fournel, L. The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie 2021, 180, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Szarpak, L.; Ruetzler, K.; Safiejko, K.; Hampel, M.; Pruc, M.; Kanczuga-Koda, L.; Filipiak, K.J.; Jaguszewski, M.J. Lactate dehydrogenase level as a COVID-19 severity marker. Am. J. Emerg. Med. 2021, 45, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Jagt, D.L.V.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol. Cell Physiol. 2021, 320, C57–C65. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Liu, Z.; Zhu, Y.; Lu, J.; Ge, C.; Zhang, C.; Li, N.; Jin, N.; Li, Y.; Tian, M.; et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front. Microbiol. 2022, 12, 780768. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus Infection and Death Receptor-Mediated Apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef]
- Wang, S.; Guo, H.; Zhu-Salzman, K.; Ge, F.; Sun, Y. PEBP balances apoptosis and autophagy in whitefly upon arbovirus infection. Nat. Commun. 2022, 13, 846. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 8, 881–883. [Google Scholar] [CrossRef]
- Kültz, D. Evolution of cellular stress response mechanisms. J. Exp. Zool. A Ecol. Integr. Physiol. 2020, 333, 359–378. [Google Scholar] [CrossRef]
- Taylor, E.W.; Radding, W. Understanding selenium and glutathione as antiviral factors in COVID-19: Does the viral Mpro protease target host selenoproteins and glutathione synthesis? Front. Nutr. 2020, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.F.; Lung, K.C.; Tso, E.Y.; Liu, R.; Chung, T.W.; Chu, M.Y.; Ng, Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef] [PubMed]
- Jamison, D.A., Jr.; Anand Narayanan, S.; Trovão, N.S.; Guarnieri, J.W.; Topper, M.J.; Moraes-Vieira, P.M.; Zaksas, V.; Singh, K.K.; Wurtele, E.S.; Beheshti, A.; et al. A comprehensive SARS-CoV-2 and COVID-19 review, Part 1: Intracellular overdrive for SARS-CoV-2 infection. Eur. J. Hum. Genet. 2022, 30, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Chiang, C.C.; Korinek, M.; Cheng, W.J.; Hwang, T.L. Targeting Neutrophils to Treat Acute Respiratory Distress Syndrome in Coronavirus Disease. Front. Pharmacol. 2020, 11, 572009. [Google Scholar] [CrossRef]
- Wang, S.; Guo, F.; Liu, K.; Wang, H.; Rao, S.; Yang, P.; Jiang, C. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008, 136, 8–15. [Google Scholar] [CrossRef]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef]
- Kumar, P.; Osahon, O.; Vides, D.B.; Hanania, N.; Minard, C.G.; Sekhar, R.V. Severe Glutathione Deficiency, Oxidative Stress and Oxidant Damage in Adults Hospitalized with COVID-19: Implications for GlyNAC (Glycine and N-Acetylcysteine) Supplementation. Antioxidants 2021, 11, 50. [Google Scholar] [CrossRef]
- Santolini, J.; Wootton, S.A.; Jackson, A.A.; Feelisch, M. The Redox architecture of physiological function. Curr. Opin. Physiol. 2019, 9, 34–47. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Shi, Z.; Puyo, C.A. N-Acetylcysteine to Combat COVID-19: An Evidence Review. Ther. Clin. Risk Manag. 2020, 16, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; DiNicolantonio, J.J. Nutraceuticals have potential for boosting the type 1 interferon response to RNA viruses including influenza and coronavirus. Prog. Cardiovasc. Dis. 2020, 63, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Jorge-Aarón, R.M.; Rosa-Ester, M.P. N-acetylcysteine as a potential treatment for COVID-19. Future Microbiol. 2020, 15, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Fraternale, A.; Zara, C.; De Angelis, M.; Nencioni, L.; Palamara, A.T.; Retini, M.; Mambro, T.D.; Magnani, M.; Crinelli, R. Intracellular Redox-Modulated Pathways as Targets for Effective Approaches in the Treatment of Viral Infection. Int. J. Mol. Sci. 2021, 22, 3603. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Geicu, O.I.; Bilteanu, L.; Serban, A.I. Antioxidant, anti-inflammatory and immunomodulatory roles of vitamins in COVID-19 therapy. Eur. J. Med. Chem. 2022, 232, 114175. [Google Scholar] [CrossRef]
- Soto, M.; di Zerega, G.; Rodgers, K.E. Countermeasure and therapeutic: A(1-7) to treat acute respiratory distress syndrome due to COVID-19 infection. J. Renin-Angiotensin-Aldosterone Syst. 2020, 21, 1470320320972018. [Google Scholar] [CrossRef]
- Qin, M.; Cao, Z.; Wen, J.; Yu, Q.; Liu, C.; Wang, F.; Zhang, J.; Yang, F.; Li, Y.; Fishbein, G.; et al. An Antioxidant Enzyme Therapeutic for COVID-19. Adv. Mater. 2020, 32, e2004901. [Google Scholar] [CrossRef]
- Peng, M.Y.; Liu, W.C.; Zheng, J.Q.; Lu, C.L.; Hou, Y.C.; Zheng, C.M.; Song, J.Y.; Lu, K.C.; Chao, Y.C. Immunological Aspects of SARS-CoV-2 Infection and the Putative Beneficial Role of Vitamin-D. Int. J. Mol. Sci. 2021, 22, 5251. [Google Scholar] [CrossRef]
- Mercola, J.; Grant, W.B.; Wagner, C.L. Evidence Regarding Vitamin D and Risk of COVID-19 and Its Severity. Nutrients 2020, 12, 3361. [Google Scholar] [CrossRef] [PubMed]
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: Matched cohort study. BMJ 2021, 372, n579. [Google Scholar] [CrossRef] [PubMed]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 And Novel Therapeutics Against Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022; Volume 2022. [Google Scholar]
- Childs, A.; Jacobs, C.; Kaminski, T.; Halliwell, B.; Leeuwenburgh, C. Supplementation with vitamin C and N-acetyl-cysteine increases oxidative stress in humans after an acute muscle injury induced by eccentric exercise. Free Radic. Biol. Med. 2001, 31, 745–753. [Google Scholar] [CrossRef]
- Fukuda, S.; Nojima, J.; Kajimoto, O.; Yamaguti, K.; Nakatomi, Y.; Kuratsune, H.; Watanabe, Y. Ubiquinol-10 supplementation improves autonomic nervous function and cognitive function in chronic fatigue syndrome. Biofactors 2016, 42, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Castro-Marrero, J.; Cordero, M.D.; Segundo, M.J.; Sáez-Francàs, N.; Calvo, N.; Román-Malo, L.; Aliste, L.; Sevilla, T.F.D.; Alegre, J. Does oral coenzyme Q10 plus NADH supplementation improve fatigue and biochemical parameters in chronic fatigue syndrome? Antioxid. Redox Signal. 2015, 22, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Faller, S.; Seiler, R.; Donus, R.; Engelstaedter, H.; Hoetzel, A.; Spassov, S.G. Pre- and posttreatment with hydrogen sulfide prevents ventilator-induced lung injury by limiting inflammation and oxidation. PLoS ONE 2017, 12, e0176649. [Google Scholar]
- Chavarría, A.P.; Vázquez, R.R.V.; Cherit, J.G.D.; Bello, H.H.; Suastegui, H.C.; Moreno-Castañeda, L.; Estrada, G.A.; Hernández, F.; González-Marcos, O.; Saucedo-Orozco, H.; et al. Antioxidants and pentoxifylline as coadjuvant measures to standard therapy to improve prognosis of patients with pneumonia by COVID-19. Comput. Struct. Biotechnol. J. 2021, 19, 1379–1390. [Google Scholar] [CrossRef]
- Wise, J. COVID-19: Critically ill patients treated with arthritis drug tocilizumab show improved outcomes, researchers report. BMJ 2020, 371, m4530. [Google Scholar] [CrossRef]
- Hu, Y.P.; Peng, Y.B.; Zhang, Y.F.; Wang, Y.; Yu, W.R.; Yao, M.; Fu, X.J. Reactive Oxygen Species Mediated Prostaglandin E2 Contributes to Acute Response of Epithelial Injury. Oxidative Med. Cell. Longev. 2017, 2017, 4123854. [Google Scholar] [CrossRef]
- Biagiotti, S.; Menotta, M.; Orazi, S.; Spapperi, C.; Brundu, S.; Fraternale, A.; Bianchi, M.; Rossi, L.; Chessa, L.; Magnani, M.. Dexamethasone improves redox state in ataxia telangiectasia cells by promoting an NRF2-mediated antioxidant response. FEBS J. 2016, 283, 3962–3978. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. After 62 years of regulating immunity, dexamethasone meets COVID-19. Nat. Rev. Immunol. 2020, 20, 587–588. [Google Scholar]
- Beltran Gonzalez, J.L.; González Gámez, M.; Mendoza Enciso, E.A.; Esparza Maldonado, R.J.; Hernández Palacios, D. Efficacy and Safety of Ivermectin and Hydroxychloroquine in Patients with Severe COVID-19: A Randomized Controlled Trial. Infect. Dis. Rep. 2022, 14, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Mu, C.; Kwok, H.F.; Xu, J.; Wu, Y.; Liu, W.; Sabatier, J.M.; Annweiler, C.; Li, X.; Cao, Z.; et al. Capivasertib restricts SARS-CoV-2 cellular entry: A potential clinical application for COVID-19. Int. J. Biol. Sci. 2021, 17, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Patrì, A.; Fabbrocini, G. Hydroxychloroquine and ivermectin: A synergistic combination for COVID-19 chemoprophylaxis and treatment? J. Am. Acad. Dermatol. 2020, 82, e221. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Choi, K.; Jeong, E.; Kwon, D.; Benveniste, E.N.; Choi, C. Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia 2004, 47, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Devrim-Lanpir, A.; Hill, L.; Knechtle, B. How N-Acetylcysteine Supplementation Affects Redox Regulation, Especially at Mitohormesis and Sarcohormesis Level: Current Perspective. Antioxidants 2021, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thirupathi, A.; Gu, Y.; Radak, Z.; Pinho, R.A. Redox Status Is the Mainstay of SARS-CoV-2 and Host for Producing Therapeutic Opportunities. Antioxidants 2022, 11, 2061. https://doi.org/10.3390/antiox11102061
Thirupathi A, Gu Y, Radak Z, Pinho RA. Redox Status Is the Mainstay of SARS-CoV-2 and Host for Producing Therapeutic Opportunities. Antioxidants. 2022; 11(10):2061. https://doi.org/10.3390/antiox11102061
Chicago/Turabian StyleThirupathi, Anand, Yaodong Gu, Zsolt Radak, and Ricardo A Pinho. 2022. "Redox Status Is the Mainstay of SARS-CoV-2 and Host for Producing Therapeutic Opportunities" Antioxidants 11, no. 10: 2061. https://doi.org/10.3390/antiox11102061
APA StyleThirupathi, A., Gu, Y., Radak, Z., & Pinho, R. A. (2022). Redox Status Is the Mainstay of SARS-CoV-2 and Host for Producing Therapeutic Opportunities. Antioxidants, 11(10), 2061. https://doi.org/10.3390/antiox11102061