Abstract
Over hundreds of years, humans have faced multiple pandemics and have overcome many of them with scientific advancements. However, the recent coronavirus disease (COVID-19) has challenged the physical, mental, and socioeconomic aspects of human life, which has introduced a general sense of uncertainty among everyone. Although several risk profiles, such as the severity of the disease, infection rate, and treatment strategy, have been investigated, new variants from different parts of the world put humans at risk and require multiple strategies simultaneously to control the spread. Understanding the entire system with respect to the commonly involved or essential mechanisms may be an effective strategy for successful treatment, particularly for COVID-19. Any treatment for COVID-19 may alter the redox profile, which can be an effective complementary method for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry and further replication. Indeed, redox profiles are one of the main barriers that suddenly shift the immune response in favor of COVID-19. Fortunately, several redox components exhibit antiviral and anti-inflammatory activities. However, access to these components as support elements against COVID-19 is limited. Therefore, understanding redox-derived species and their nodes as a common interactome in the system will facilitate the treatment of COVID-19. This review discusses the redox-based perspectives of the entire system during COVID-19 infection, including how redox-based molecules impact the accessibility of SARS-CoV-2 to the host and further replication. Additionally, to demonstrate its feasibility as a viable approach, we discuss the current challenges in redox-based treatment options for COVID-19.
1. Introduction
The selective life-forming elements in the periodic table lose or accept the electrons in earlier life formation which are said to be oxidized or reduced (redox reaction), as oxygen is the final electron acceptor in biology [1]. However, earlier redox reactions used methane and hydrogen as the primary life-forming molecules, which delayed ancient organisms from evolving to aerobic life until the rise of cyanobacteria-like microbes, which split water to produce oxygen [1,2]. Consequently, this scenario introduced oxygen as metabolic waste. Moreover, this produced partially oxidized intermediates such as superoxide, hydrogen peroxide, and hydroxyl radical, which are collectively called reactive oxygen species (ROS) and can cause damage to cellular components, including DNA, proteins, and lipids. Although this review focuses on the oxygen-related redox reaction and its physiopathological functions, ancient anaerobic life depended on sulfur redox chemistry. This generated more S/N hybrid species when interacting with reactive sulfur species (RSS) and reactive nitrogen species (RNS) [3,4], which put the sulfur-based metabolism at high risk for rapid oxidation. All these consequences led the organisms to have controlled electron transfer during the evolutionary process [5]. This occurred within the set point of redox potential inside the same cell at different sites [6]. Unilateral oxidation disrupts the redox process if it is not contained by specialized molecules called antioxidants. The employment of enzymatic and non-enzymatic antioxidants has restored the redox status of the cells, warranting the application of these molecules in treating various redox diseases [6].
Although human populations have effectively overcome several pandemics, anthropogenic activities and the consequent impact on the ecosystem could increase the likelihood of other pandemics [7,8]. Indeed, the perplexities of the ecosystem alter microbial life more deeply than ever by changing the virulence and survival patterns of the microbes [9]. This is pertinent to the novel strain of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which caused a recent pandemic named “coronavirus disease 2019” (COVID-19) [10,11]. Accommodating human life to COVID-19 requires the synchronization of various molecules, including proteins, enzymes, and hormones that have been activated, modified or regulated by redox-active molecules [12]. Nevertheless, humans and microbes, including viruses, are symbiotically co-evolved; both may use these redox-active molecules for fundamental biochemical reactions that regulate cellular redox homeostasis, metabolism, signaling, and mitochondrial function. More ROS and increased oxidative stress disrupt redox-mediated cellular functions in favor of virus survival (Figure 1). For example, at the initial stage of any infection, including viral infection, the host system increases oxidative stress strategically to disrupt redox signaling; at the later stages, the host’s antioxidant system is activated to prevent damage [13]. However, the timeline of this scenario is a dilemma, suggesting that putting forward current knowledge gained in this field can help to establish underlying oxidative stress-induced pathophysiological mechanisms. Otherwise, proposing redox therapy as the most suitable one for COVID-19 may be a daunting challenge. In addition, the risk factors associated with COVID-19 commonly induce oxidative stress, suggesting that COVID-19 can be considered a redox disease [14]. This has been established with increased oxidative stress in COVID-19 patients [15]. However, it is difficult to recommend or formulate redox therapy for COVID-19, as every cell and tissue has a different redox setup in inducing oxidative stress (toward physiological functions) and oxidative damage (toward pathological conditions). For example, the redox potentials of NADH and NADPH are varied in the cytosol and mitochondria [16], which either induce oxidative stress or oxidative damage in the two compartments of the same cell. Therefore, this review addresses how redox-active molecules act as the mainstay of drawing up redox medicine for COVID-19.
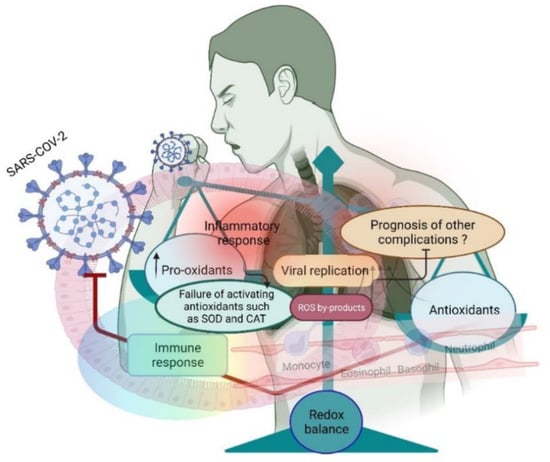
Figure 1.
A small shift in the redox balance facilitates SARS-CoV-2 entry and further replication in the lungs. Entry of the virus induces an inflammatory response and increases the pro-oxidants to facilitate the host’s virus life cycle.
Thus, the review starts with ROS sources during COVID-19. Identifying specific ROS sources may be a prognostic factor in determining COVID-19 disease severity. Next, this review will address how SARS-CoV-2 redesigns the redox system in the host in favor of its survival. This can prevent recommend unsuitable antioxidant therapy as it tilts the redox status. Finally, this review discusses all the challenges associated with oxidative stress-mediated pathophysiological mechanisms during COVID-19.
2. Sources of ROS during COVID-19
Respiratory viruses, including SARS-CoV-2, can induce ROS-generating sources such as nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidases) [17], xanthine oxidase (XO) [18], and mitochondria [19] (Figure 2). For instance, the activation of NOX2 and a consequent increase in ROS promote the setting of thrombotic-linked ischemic events in COVID-19 patients [20,21]. Although there is no direct evidence of XO as a source of ROS in COVID-19, a study showed that COVID-19 patients had hypouricemia (<2.5 mg/dl), which is implicated with the specific dysfunction of the proximal tubule [22]. As uric acid is an antioxidant, a decrease in uric acid might increase ROS and oxidative stress in the renal artery and further dysfunction in the kidney of COVID-19 patients [23], indicating the role of XO as an important source of ROS in COVID-19 [23]. Mitochondria are crucial sources of ROS, and mitochondrial-triggered ROS induces hypoxia-inducible-1 alpha factor (HIF-1 alpha) for improving glycolysis in the monocytes and macrophages [24]. This could directly inhibit the immune response, specifically, T-cell response [25], and decrease epithelial cell survival in COVID-19 [24]. Peroxisomes are another detoxifying source of ROS that have close contact with mitochondria as they are involved in the oxidative metabolism of amino acids and fatty acids [26]. Alterations in the peroxisome structure and loss of matrix content can increase ROS generation [27]. SARS-CoV-2 ORF14 protein altered the morphology of peroxisomes and their biogenesis by interacting with human PEX14 [28]. This could tilt the ROS balance in the host cells and increase lipid peroxidation, consequently inducing an inflammatory lipid storm in the lungs of COVID-19 patients by altering the leukotrienes and prostaglandins [29,30]. A double-center retrospective study reported that COVID-19 patients with severe conditions had hypoxemia [31], which may be from the ER stress response induced by ROS generation in the ER through protein-folding oxidation [32]. Notably, proteins such as serum albumin bind with several transition metals, such as iron and copper, called a “sponge” or a “tramp steamer” [33], to decrease ROS generation. The ligand-binding capacities of albumin are the reason for its antioxidant properties [33,34,35,36]. A low albumin level could increase the ROS level in COVID-19 patients, and retrospective studies have shown that hypoalbuminemia indicates the COVID-19 severity independent of age and co-morbidity [37,38].
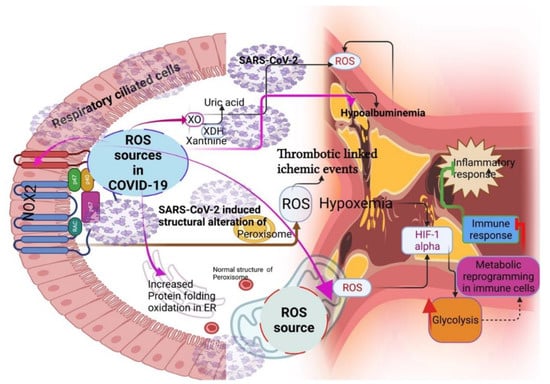
Figure 2.
Sources of ROS during COVID-19 (pink arrow mark). NOX2-induced ROS increases the thrombotic-linked ischemic events in COVID-19. Mitochondrial-triggered ROS reprogram the metabolism for inhibiting immune response through hypoxia-inducible-1 alpha factor (HIF-1 alpha). SARS-CoV-2 altered the peroxisome morphology, consequently increasing ROS generation.
3. SARS-CoV-2 Controls ROS Levels for Its Survival
An increase in ROS could cause damaging outcomes to SARS-CoV-2. To overcome this, viruses could naturally evolve to counteract ROS-induced oxidative damage in the cellular environment [39]. Possibly, this could increase the antioxidant capacity of the host to control the ROS environment. This concept was established with Huh7 cells, which underwent genetic reprogramming to permit hepatitis C virus (HCV) subgenomic replicon, induce oxidative stress by altering iron homeostasis, and activate manganese superoxide dismutase (MnSOD) and glutathione peroxidase 4 (GPx4) [40,41]. Regarding SARS-CoV-2, an observational study showed an increase in antioxidant capacity by inducing SOD and CAT in COVID-19 patients [42]. However, this was inconsistent with other COVID-19 patients [43]. This warrants additional studies to establish the link between SARS-CoV-2 and the host antioxidant system’s activation. Next, SARS-CoV-2 probably controls the excess ROS for survival through a metabolic switch. For example, SARS-CoV-2 may increase glycolysis to divert fuel to generate anabolic intermediates, similar to the Warburg effect [44,45]. This scenario prevents mitochondrial ROS. Supporting this idea, increased lactate dehydrogenase activity (LDH) was reported in COVID-19 patients [45]. Additionally, the knockdown of LDH increases pyruvate and promotes oxidative stress [46], suggesting that SARS-CoV-2 controls ROS using metabolic shifts [37]. Establishing this concept could form a new therapeutic approach to support redox-based treatment options. Next, SARS-CoV-2 induces mitochondrial dysfunction by decreasing mitochondrial membrane potential and causing mitochondrial permeability transition pore opening (MPTP), resulting in increased ROS release [47,48]. Studies have shown that viruses can induce apoptosis mechanisms for tissue injury or disease progression [49,50,51]. For example, SARS-CoV-2 ORF3a can induce an extrinsic apoptotic pathway by activating/cleaving caspase-8 [52]; targeting SARS-CoV-2-induced apoptosis could offer a promising target for SARS-CoV-2 treatment. Furthermore, SARS-CoV-2 fabricates intracellular signaling for survival, mainly through increasing H2O2. For instance, SARS-CoV-2 may decrease the accumulation of selenoprotein transcripts that regulate the phospholipid hydroperoxide glutathione peroxidase (GPX4) and mitochondrial functions [53], resulting in an increase in H2O2 in the host cells. This scenario alters redox-sensitive proteins such as mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription proteins (STATs), Toll-like receptors (TLRs), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), and nuclear factor erythroid 2-related factor 2 (Nrf-2) [53]. In addition, the hyperactivation of TLR 3 and 4 could induce pro-inflammatory cytokines, including IL-1 and IL-6, and cause a cytokine storm for SARS-CoV-2 survival [54,55,56].
4. Redox Chemicals Coordinate Local and Systemic Redox Networks in COVID-19
Redox-active molecules coordinate the systemic redox network (nonlinearly) locally first (beginning of the redox disequilibrium), and then turn to the entire system [57], which collapses the systemic redox equilibrium. Consequently, changes in the redox tones in the cells induce an “oxidative storm” in place of the “cytokine storm” [14], which could allow SARS-CoV-2 (probably a secondary exposure) to bypass redox-mediated immune vigilance (respiratory burst). This scenario can encourage virus fusion and viral loads to increase within the infection (Figure 3) [58,59].
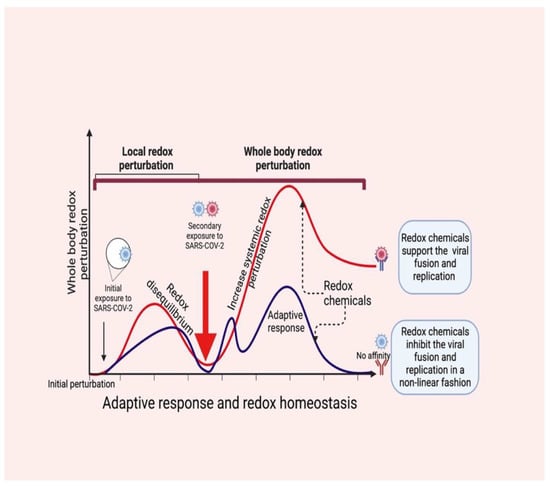
Figure 3.
SARS-CoV-2 infection perturbs redox homeostasis in supporting viral fusion and replication (red color). Local redox disequilibrium causes systemic redox disequilibrium, which favors the entry of SARS-CoV-2 and consequent viral loads (secondary exposure). Redox molecules act as redox codes for proper redox communication, which induces an adaptive response by causing redox equilibrium in the host (blue color). This supports viral inhibition.
Although redox byproducts such as H2S, nitroxyl, beta-hydroxybutyrate, and different mixed sulfide compounds rebalance the entire redox network by acting as redox codes [4,59], active disulfides in the host contribute to spike protein binding and ACE2 by allosteric regulation (Figure 4) [60]. Perhaps GSH could negatively mediate this effective binding process by reducing active disulfides, which is confirmed by the decrease in GSH and increase in oxidative stress in severe COVID-19 patients [61,62], showing the therapeutic value of GSH in COVID-19. This scenario could also activate various redox molecules in the redox landscape, allowing the virus to enter the host cells by endocytosis, where ACE2 translocates to the endosomal lumen [63,64].
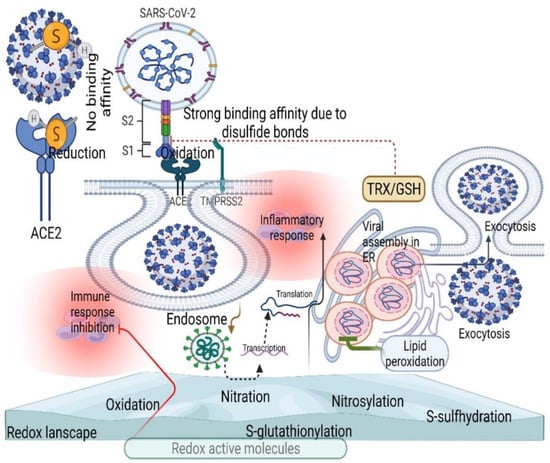
Figure 4.
Changes in the redox landscape (thiol-based redox mechanisms) facilitate viral fusion and replication. Disulfide bonds induce a stronger affinity between SAS-COV-2 and ACE2, while thiol compounds do not induce binding affinity between SARS-CoV-2 and ACE2 (red inhibitory mark). Disulfide activation by the allosteric mechanism facilitates initial viral fusion by activating various redox molecules (endocytosis). In contrast, redox imbalance induces lipid peroxidation to inhibit viral replication in the ER (green inhibitory mark).
5. Antioxidant Therapy in COVID-19
The link between redox metabolism and viruses has been considered within the field of antioxidant intervention for several years. Although the purpose of recommending antioxidant therapy is not new, the recent pandemic brought antioxidant therapy to the frontline, as COVID-19 lacks specific antiviral drugs. N-acetyl cysteine (NAC) is considered to be the best-known antioxidant for alleviating SARS-CoV-2 infection [65]. There are some possible mechanisms proposed thus far, such as interfering with angiotensin II cleavage to angiotensin 1–7 via ACE2 and attenuating oxidative damage by increasing TLR-7, restoring type-IFN production [66], and antagonizing proteasome inhibitors to reduce the accumulation of the viral proteins [66,67]. NAC possibly interacts with the SARS-CoV-2 E protein by cleaving disulfide bonds of the triple cysteine motif (NH2- … L-Cys-A-Y-Cys-Cys-N … -COOH) [66]. This protein regulates the cellular polarity and cell–cell junctions in the epithelial cells by binding the PALS1 PDZ domain [67]. This could reduce the infection rate. NAC prevents the glycosylation events in SARS-CoV-2 by restoring platelet GSH, suggesting an alteration in the GSH level could be a therapeutic approach for fighting against SARS-CoV-2 [67,68]. Higher activation of proinflammatory cytokines is common in patients with a level of TNF-alpha [69]. Therefore, regulating these molecules ensures the reinstatement of the immune system. Antioxidant therapy could be the major antagonist to these pro-inflammatory cytokines which alleviate hyperinflammation in patients with severe COVID-19 [70]. A recent in vitro and ex vivo study reported that nanocoated CAT downregulates pro-inflammatory cytokines to regulate immune homeostasis [71]. However, these methods are unreliable in assessing ROS and oxidative damage, as they are vulnerable to artifacts. Nitric oxide (NO) has another potential molecule that inhibits SARS-CoV-2 replication and enhances oxygenation in COVID-19 patients. The administration of H2S inhibits oxidative stress by preventing platelet activation and neutrophil extracellular trap (NET) formation. The ingestion of H2S may be an effective treatment for COVID-19. Other antioxidants, including CoQ10 with NADH, curcumin, vitamins C, D, and E, selenium, melatonin with pentoxifylline, ebselen, and disulfiram, can target redox imbalance in COVID-19 [72,73,74,75,76,77,78,79]. For example, the use of antioxidants such as vitamin C, E, and NAC with pentoxifylline decreased the lipid peroxidation and total antioxidant capacity in COVID-19 patients at the end of the hospital stay, while pentoxifylline alone did not decrease the oxidative stress markers [80], suggesting the use of antioxidants as a possible adjuvant therapy for improving survival prognosis in COVID-19 patients. However, further studies are warranted to prove the reproducibility of this data. Because noncatalytic cysteine residues of the 3C-like protease (3CLpro) in SARS-CoV-2 can protect the virus from oxidative damage, as current drugs such as nirmatrelvir are mainly inhibiting the cysteine residue of 3CLpro, the use of these antioxidants may interfere with this process and aggravate SARS-CoV-2 replication.
6. Challenges Associated with Oxidative Stress during COVID-19 Treatment
Although various vaccines and drugs are a major part of treatment support during COVID-19, the risk profile of patients is ambiguous. Rapidly spreading variants from different parts of the world is an additional concern that effectively overcomes the available treatments [81,82]. Furthermore, targeting localized oxidative damage (specific tissue environment) with antioxidants perturbs the systemic redox network [14]. Furthermore, treatment options, either with vaccines or drugs, perturb redox homeostasis and induce oxidative stress. For example, tocilizumab and hydrocortisone provide better protection against COVID-19 [83]. However, these drugs with ventilatory support induce oxidative damage and alter the immune response while protecting the endothelial glycocalyx, whose function is to maintain the redox balance in COVID-19 [83]. Dexamethasone is another drug that can activate redox-active molecules and increase oxidative stress, enabling antioxidant defense through KEAP1/NRF-2 activation, affecting electron transport complexes, and increasing NOX-2, all of which dominate the redox system [84,85]. Capivasertib could inhibit SARS-CoV-2 entry by improving glycolysis and oxidative phosphorylation through ROS-mediated AKT inhibition [86]. Hydroxychloroquine and ivermectin can also induce ROS production in vitro and in vivo [85,87,88]. Other drugs, such as anakinra, sotrovimab, and ruxolitinib exacerbate ROS pathways to control COVID-19 [87,88]. Treatment with these drugs increases oxidative stress and compromises their clinical efficacy for COVID-19 treatment [89]. Therefore, implementing redox-based therapies requires a deeper knowledge of oxidative stress research. However, recent advancements in this field, from redox imaging to redox metabolomics, will facilitate the revealing of the key molecule’s functions in biological systems. Furthermore, advanced omics methods can support characterizing redox molecules and their functions. This will provide an opportunity to overcome current challenges associated with COVID-19 treatment. Otherwise, using nonspecific kits to measure oxidative damage will not provide any definite results to treat COVID-19, because using these kits can partially reflect the oxidative stress status within the cells. For instance, ROS probes such as 2′,7′-dichlorodihydrofluorescein (DCFH) can be oxidized with several ROS molecules and are not specific for any particular ROS [90]. Therefore, monitoring real-time changes in the intracellular redox-active molecules will help to develop a high-throughput global profiling methodology for finding ROS molecules.
7. Conclusions
This review discussed several ROS sources that are perhaps activated by SARS-CoV-2 for its survival. This may increase local and systemic oxidative stress. However, why this scenario does not affect SARS-CoV-2 survival in the host is unknown. Instead, it can escape from the host oxidative stress response using its noncatalytic cysteine residues. Using any currently available drugs with redox-based adjuvant therapies or as a main therapy may interfere with this cysteine-mediated oxidation and could support SARS-CoV-2 replication. Therefore, careful evaluation is required to design an integrated approach to understand the whole body’s systemic redox status. From this perspective, we have discussed the possible reasons for this scenario, starting from ROS sources to therapeutical challenges in treating COVID-19. This may help to consider COVID-19 as a redox disease and support redox-based therapies as the possible preference for COVID-19 treatment. However, before implementing redox-based therapies, every COVID-19 patient should be interrogated with oxidative stress parameters, as they can show different redox statuses. Moreover, personalized approaches will require appropriate redox-based intervention, as redox homeostasis is more susceptible to a specific intervention, which further provokes oxidative stress. Consequently, disconnecting the redox-mediated communication between the organs can reduce the chances of regaining COVID-19 control. Therefore, the routine collection of clinical information with regard to the redox status of an individual may provide new therapeutic opportunities to restore the redox balance and decrease COVID-19 mortality and possibly manage long-COVID symptoms.
Author Contributions
Conceptualization, A.T. and R.A.P.; writing, A.T.; R.A.P. gave critical issues of the manuscript; Z.R. and Y.G. edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Raymond, J.; Segre, D. The effect of oxygen on biochemical networks and the evolution of complex life. Science 2006, 311, 1764–1767. [Google Scholar] [CrossRef]
- Castresana, J.; Saraste, M. Evolution of energetic metabolism; the respiration early hypothesis. Trends Biochem. Sci. 1995, 20, 443–448. [Google Scholar] [CrossRef]
- Cortese-Krott, M.M.; Pullmann, D.; Feelisch, M. Nitrosopersulfide (SSNO-) targets the Keap-1/Nrf2 redox system. Pharmacol. Res. 2016, 113, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Cortese-Krott, M.M.; Butler, A.R.; Woollins, J.D.; Feelisch, M. Inorganic sulfur-nitrogen compounds: From gunpowder chemistry to the forefront of biological signaling. Dalton Trans. 2016, 45, 5908–5919. [Google Scholar] [CrossRef] [PubMed]
- Luu Trinh, M.D.; Miyazaki, D.; Ono, S.; Nomata, J.; Kono, M.; Mino, H.; Niwa, T.; Okegawa, Y.; Motohashi, K.; Taguchi, H.; et al. The evolutionary conserved iron-sulfur protein TCR controls P700 oxidation in photosystem I. iScience. 2021, 24, 102059. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Shope, R. Global climate change and infectious diseases. Environ. Health Perspect. 1991, 96, 171–174. [Google Scholar] [CrossRef]
- Gorji, S.; Gorji, A. COVID-19 pandemic: The possible influence of the long-term ignorance about climate change. Environ. Sci. Pollut. Res. Int. 2021, 28, 15575–15579. [Google Scholar] [CrossRef]
- Rodó, X.; San-José, A.; Kirchgatter, K.; López, L. Changing climate and the COVID-19 pandemic: More than just heads or tails. Nat. Med. 2021, 27, 576–579. [Google Scholar] [CrossRef]
- Ukhurebor, K.E.; Singh, K.R.; Nayak, V.; Uk-Eghonghon, G. Influence of the SARS-CoV-2 pandemic: A review from the climate change perspective. Environ. Sci. Process Impacts 2021, 23, 1060–1078. [Google Scholar] [CrossRef]
- Jamrozik, E.; Selgelid, M.J. COVID-19 human challenge studies: Ethical issues. Lancet Infect. Dis. 2020, 20, e198–e203. [Google Scholar] [CrossRef]
- Thoradeniya, T.; Jayasinghe, S. COVID-19 and future pandemics: A global systems approach and relevance to SDGs. Global Health 2021, 17, 59. [Google Scholar] [CrossRef]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Cumpstey, A.F.; Clark, A.D.; Santolini, J.; Jackson, A.A.; Feelisch, M. COVID-19: A Redox Disease-What a Stress Pandemic Can Teach Us About Resilience and What We May Learn from the Reactive Species Interactome About Its Treatment. Antioxid. Redox Signal. 2021, 35, 1226–1268. [Google Scholar] [CrossRef] [PubMed]
- Gadotti, A.C.; Lipinski, A.L.; Vasconcellos, F.T.; Marqueze, L.F.; Cunha, E.B.; Campos, A.C.; Oliveira, C.F.; Amaral, A.N.; Baena, C.P.; Telles, J.P.; et al. Susceptibility of the patients infected with Sars-Cov2 to oxidative stress and possible interplay with severity of the disease. Free Radic. Biol. Med. 2021, 165, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Nicotinamide nucleotide compartmentation. In Metabolic Compartmentation; Sies, H., Ed.; Academic: London, UK, 1982; pp. 205–231. [Google Scholar]
- de Oliveira, A.A.; Nunes, K.P. Crosstalk of TLR4, vascular NADPH oxidase, and COVID-19 in diabetes: What are the potential implications? Vasc. Pharmacol. 2021, 139, 106879. [Google Scholar] [CrossRef]
- Pratomo, I.P.; Noor, D.R.; Kusmardi, K.; Rukmana, A.; Paramita, R.I.; Erlina, L.; Fadilah, F.; Gayatri, A.; Fitriani, M.; Purnomo, T.T.H.; et al. Xanthine Oxidase-Induced Inflammatory Responses in Respiratory Epithelial Cells: A Review in Immunopathology of COVID-19. Internat. J. Inflamm. 2021, 2021, 1653392. [Google Scholar] [CrossRef]
- Ryback, R.; Eirin, A. Mitochondria, a Missing Link in COVID-19 Heart Failure and Arrest? Front. Cardiovasc. Med. 2022, 8, 830024. [Google Scholar] [CrossRef]
- Violi, F.; Ceccarelli, G.; Cangemi, R.; Alessandri, F.; D’Ettorre, G.; Oliva, A.; Pastori, D.; Loffredo, L.; Pignatelli, P.; Ruberto, F.; et al. Hypoalbuminemia, Coagulopathy, and Vascular Disease in COVID-19. Circ. Res. 2020, 127, 400–401. [Google Scholar] [CrossRef]
- Violi, F.; Oliva, A.; Cangemi, R.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Alessandri, M.L.F.; Angelis, M.D.; et al. Nox2 activation in COVID-19. Redox Biol. 2020, 36, 101655. [Google Scholar] [CrossRef]
- Werion, A.; Belkhir, L.; Perrot, M.; Schmit, G.; Aydin, S.; Chen, Z.; Penaloza, A.; Greef, J.D.; Yildiz, H.; Pothen, L.; et al. Cliniques universitaires Saint-Luc (CUSL) COVID-19 Research Group. SARS-CoV-2 causes a specific dysfunction of the kidney proximal tubule. Kidney Int. 2020, 98, 296–1307. [Google Scholar] [CrossRef] [PubMed]
- Sekizuka, H. Uric acid, xanthine oxidase, and vascular damage: Potential of xanthine oxidoreductase inhibitors to prevent cardiovascular diseases. Hypertens. Res. 2022, 45, 772–774. [Google Scholar] [CrossRef] [PubMed]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; Junior, C.A.O.D.B.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446. [Google Scholar] [CrossRef]
- Chen, Y.; Gaber, T. Hypoxia/HIF Modulates Immune Responses. Biomedicines 2021, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Knoblach, B.; Ishida, R.; Hobman, T.C.; Rachubinski, R.A. Peroxisomes exhibit compromised structure and matrix protein content in SARS-CoV-2-infected cells. Mol. Biol. Cell 2021, 32, 1273–1282. [Google Scholar] [CrossRef]
- Archambault, A.S.; Zaid, Y.; Rakotoarivelo, V.; Turcotte, C.; Doré, É.; Dubuc, I.; Martin, C.; Flamand, O.; Amar, Y.; Cheikh, A.; et al. High levels of eicosanoids and docosanoids in the lungs of intubated COVID-19 patients. FASEB J. 2021, 35, e21666. [Google Scholar] [CrossRef]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Łos, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S.; et al. Endoplasmic reticulum as a potential therapeutic target for COVID-19 infection management? Eur. J. Pharmacol. 2020, 882, 173288. [Google Scholar] [CrossRef]
- Asleh, R.; Asher, E.; Yagel, O.; Samuel, T.; Elbaz-Greener, G.; Wolak, A.; Durst, R.; Ben-Chetrit, E.; Nir-Paz, R.; Helviz, Y.; et al. Predictors of Hypoxemia and Related Adverse Outcomes in Patients Hospitalized with COVID-19: A Double-Center Retrospective Study. J. Clin. Med. 2021, 10, 3581. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J. All about Albumin; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Roche, M.; Philippe, R.; Singh, N.R.; Tarnus, E.; Bourdon, E. The antioxidant properties of serum albumin. FEBS Lett. 2008, 582, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Stocker, R. Bilirubin attenuates radical-mediated damage to serum albumin. FEBS Lett. 1993, 331, 281–284. [Google Scholar] [CrossRef]
- Cohen, M.P. Intervention strategies to prevent pathogenetic effects of glycated albumin. Arch. Biochem. Biophys. 2003, 419, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cheng, A.; Kumar, R.; Fang, Y.; Chen, G.; Zhu, Y.; Lin, S. Hypoalbuminemia predicts the outcome of COVID-19 independent of age and co-morbidity. J. Med. Virol. 2020, 92, 2152–2158. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Garcia-Rodriguez, V.; Yu, A.; Dutra, B.; Larson, S.; Cash, B.; DuPont, A.; Farooq, A. Elevated transaminases and hypoalbuminemia in COVID-19 are prognostic factors for disease severity. Sci. Rep. 2021, 11, 10308. [Google Scholar] [CrossRef]
- Schwarz, K.B. Oxidative stress during viral infection: A review. Free Radic. biol Med. 1996, 21, 641–649. [Google Scholar] [CrossRef]
- Qadri, I.; Iwahashi, M.; Capasso, J.M.; Hopken, M.W.; Flores, S.; Schaack, J.; Simon, F.R. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: Role of JNK, p38 MAPK and AP-1. Biochem. J. 2004, 378, 919–928. [Google Scholar] [CrossRef]
- Fillebeen, C.; Muckenthaler, M.; Andriopoulos, B.; Bisaillon, M.; Mounir, Z.; Hentze, M.W.; Koromilas, A.E.; Pantopoulos, K. Expression of the subgenomic hepatitis C virus replicon alters iron homeostasis in Huh7 cells. J. Hepatol. 2007, 47, 12–22. [Google Scholar] [CrossRef]
- Mehri, F.; Rahbar, A.H.; Ghane, E.T.; Souri, B.; Esfahani, M. Changes in oxidative markers in COVID-19 patients. Arch. Med. Res. 2021, 52, 843–849. [Google Scholar] [CrossRef]
- Zendelovska, D.; Atanasovska, E.; Petrushevska, M.; Spasovska, K.; Stevanovikj, M.; Demiri, I.; Labachevski, N. Evaluation of oxidative stress markers in hospitalized patients with moderate and severe COVID-19. Rom. J. Intern. Med. 2021, 59, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Lincet, H.; Wu, Z.; Coquerel, A.; Forgez, P.; Alifano, M.; Fournel, L. The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie 2021, 180, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Szarpak, L.; Ruetzler, K.; Safiejko, K.; Hampel, M.; Pruc, M.; Kanczuga-Koda, L.; Filipiak, K.J.; Jaguszewski, M.J. Lactate dehydrogenase level as a COVID-19 severity marker. Am. J. Emerg. Med. 2021, 45, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Jagt, D.L.V.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol. Cell Physiol. 2021, 320, C57–C65. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Liu, Z.; Zhu, Y.; Lu, J.; Ge, C.; Zhang, C.; Li, N.; Jin, N.; Li, Y.; Tian, M.; et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front. Microbiol. 2022, 12, 780768. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus Infection and Death Receptor-Mediated Apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef]
- Wang, S.; Guo, H.; Zhu-Salzman, K.; Ge, F.; Sun, Y. PEBP balances apoptosis and autophagy in whitefly upon arbovirus infection. Nat. Commun. 2022, 13, 846. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 8, 881–883. [Google Scholar] [CrossRef]
- Kültz, D. Evolution of cellular stress response mechanisms. J. Exp. Zool. A Ecol. Integr. Physiol. 2020, 333, 359–378. [Google Scholar] [CrossRef]
- Taylor, E.W.; Radding, W. Understanding selenium and glutathione as antiviral factors in COVID-19: Does the viral Mpro protease target host selenoproteins and glutathione synthesis? Front. Nutr. 2020, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.F.; Lung, K.C.; Tso, E.Y.; Liu, R.; Chung, T.W.; Chu, M.Y.; Ng, Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef] [PubMed]
- Jamison, D.A., Jr.; Anand Narayanan, S.; Trovão, N.S.; Guarnieri, J.W.; Topper, M.J.; Moraes-Vieira, P.M.; Zaksas, V.; Singh, K.K.; Wurtele, E.S.; Beheshti, A.; et al. A comprehensive SARS-CoV-2 and COVID-19 review, Part 1: Intracellular overdrive for SARS-CoV-2 infection. Eur. J. Hum. Genet. 2022, 30, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Chiang, C.C.; Korinek, M.; Cheng, W.J.; Hwang, T.L. Targeting Neutrophils to Treat Acute Respiratory Distress Syndrome in Coronavirus Disease. Front. Pharmacol. 2020, 11, 572009. [Google Scholar] [CrossRef]
- Wang, S.; Guo, F.; Liu, K.; Wang, H.; Rao, S.; Yang, P.; Jiang, C. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008, 136, 8–15. [Google Scholar] [CrossRef]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef]
- Kumar, P.; Osahon, O.; Vides, D.B.; Hanania, N.; Minard, C.G.; Sekhar, R.V. Severe Glutathione Deficiency, Oxidative Stress and Oxidant Damage in Adults Hospitalized with COVID-19: Implications for GlyNAC (Glycine and N-Acetylcysteine) Supplementation. Antioxidants 2021, 11, 50. [Google Scholar] [CrossRef]
- Santolini, J.; Wootton, S.A.; Jackson, A.A.; Feelisch, M. The Redox architecture of physiological function. Curr. Opin. Physiol. 2019, 9, 34–47. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Shi, Z.; Puyo, C.A. N-Acetylcysteine to Combat COVID-19: An Evidence Review. Ther. Clin. Risk Manag. 2020, 16, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; DiNicolantonio, J.J. Nutraceuticals have potential for boosting the type 1 interferon response to RNA viruses including influenza and coronavirus. Prog. Cardiovasc. Dis. 2020, 63, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Jorge-Aarón, R.M.; Rosa-Ester, M.P. N-acetylcysteine as a potential treatment for COVID-19. Future Microbiol. 2020, 15, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Fraternale, A.; Zara, C.; De Angelis, M.; Nencioni, L.; Palamara, A.T.; Retini, M.; Mambro, T.D.; Magnani, M.; Crinelli, R. Intracellular Redox-Modulated Pathways as Targets for Effective Approaches in the Treatment of Viral Infection. Int. J. Mol. Sci. 2021, 22, 3603. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Geicu, O.I.; Bilteanu, L.; Serban, A.I. Antioxidant, anti-inflammatory and immunomodulatory roles of vitamins in COVID-19 therapy. Eur. J. Med. Chem. 2022, 232, 114175. [Google Scholar] [CrossRef]
- Soto, M.; di Zerega, G.; Rodgers, K.E. Countermeasure and therapeutic: A(1-7) to treat acute respiratory distress syndrome due to COVID-19 infection. J. Renin-Angiotensin-Aldosterone Syst. 2020, 21, 1470320320972018. [Google Scholar] [CrossRef]
- Qin, M.; Cao, Z.; Wen, J.; Yu, Q.; Liu, C.; Wang, F.; Zhang, J.; Yang, F.; Li, Y.; Fishbein, G.; et al. An Antioxidant Enzyme Therapeutic for COVID-19. Adv. Mater. 2020, 32, e2004901. [Google Scholar] [CrossRef]
- Peng, M.Y.; Liu, W.C.; Zheng, J.Q.; Lu, C.L.; Hou, Y.C.; Zheng, C.M.; Song, J.Y.; Lu, K.C.; Chao, Y.C. Immunological Aspects of SARS-CoV-2 Infection and the Putative Beneficial Role of Vitamin-D. Int. J. Mol. Sci. 2021, 22, 5251. [Google Scholar] [CrossRef]
- Mercola, J.; Grant, W.B.; Wagner, C.L. Evidence Regarding Vitamin D and Risk of COVID-19 and Its Severity. Nutrients 2020, 12, 3361. [Google Scholar] [CrossRef] [PubMed]
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: Matched cohort study. BMJ 2021, 372, n579. [Google Scholar] [CrossRef] [PubMed]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 And Novel Therapeutics Against Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022; Volume 2022. [Google Scholar]
- Childs, A.; Jacobs, C.; Kaminski, T.; Halliwell, B.; Leeuwenburgh, C. Supplementation with vitamin C and N-acetyl-cysteine increases oxidative stress in humans after an acute muscle injury induced by eccentric exercise. Free Radic. Biol. Med. 2001, 31, 745–753. [Google Scholar] [CrossRef]
- Fukuda, S.; Nojima, J.; Kajimoto, O.; Yamaguti, K.; Nakatomi, Y.; Kuratsune, H.; Watanabe, Y. Ubiquinol-10 supplementation improves autonomic nervous function and cognitive function in chronic fatigue syndrome. Biofactors 2016, 42, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Castro-Marrero, J.; Cordero, M.D.; Segundo, M.J.; Sáez-Francàs, N.; Calvo, N.; Román-Malo, L.; Aliste, L.; Sevilla, T.F.D.; Alegre, J. Does oral coenzyme Q10 plus NADH supplementation improve fatigue and biochemical parameters in chronic fatigue syndrome? Antioxid. Redox Signal. 2015, 22, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Faller, S.; Seiler, R.; Donus, R.; Engelstaedter, H.; Hoetzel, A.; Spassov, S.G. Pre- and posttreatment with hydrogen sulfide prevents ventilator-induced lung injury by limiting inflammation and oxidation. PLoS ONE 2017, 12, e0176649. [Google Scholar]
- Chavarría, A.P.; Vázquez, R.R.V.; Cherit, J.G.D.; Bello, H.H.; Suastegui, H.C.; Moreno-Castañeda, L.; Estrada, G.A.; Hernández, F.; González-Marcos, O.; Saucedo-Orozco, H.; et al. Antioxidants and pentoxifylline as coadjuvant measures to standard therapy to improve prognosis of patients with pneumonia by COVID-19. Comput. Struct. Biotechnol. J. 2021, 19, 1379–1390. [Google Scholar] [CrossRef]
- Wise, J. COVID-19: Critically ill patients treated with arthritis drug tocilizumab show improved outcomes, researchers report. BMJ 2020, 371, m4530. [Google Scholar] [CrossRef]
- Hu, Y.P.; Peng, Y.B.; Zhang, Y.F.; Wang, Y.; Yu, W.R.; Yao, M.; Fu, X.J. Reactive Oxygen Species Mediated Prostaglandin E2 Contributes to Acute Response of Epithelial Injury. Oxidative Med. Cell. Longev. 2017, 2017, 4123854. [Google Scholar] [CrossRef]
- Biagiotti, S.; Menotta, M.; Orazi, S.; Spapperi, C.; Brundu, S.; Fraternale, A.; Bianchi, M.; Rossi, L.; Chessa, L.; Magnani, M.. Dexamethasone improves redox state in ataxia telangiectasia cells by promoting an NRF2-mediated antioxidant response. FEBS J. 2016, 283, 3962–3978. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. After 62 years of regulating immunity, dexamethasone meets COVID-19. Nat. Rev. Immunol. 2020, 20, 587–588. [Google Scholar]
- Beltran Gonzalez, J.L.; González Gámez, M.; Mendoza Enciso, E.A.; Esparza Maldonado, R.J.; Hernández Palacios, D. Efficacy and Safety of Ivermectin and Hydroxychloroquine in Patients with Severe COVID-19: A Randomized Controlled Trial. Infect. Dis. Rep. 2022, 14, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Mu, C.; Kwok, H.F.; Xu, J.; Wu, Y.; Liu, W.; Sabatier, J.M.; Annweiler, C.; Li, X.; Cao, Z.; et al. Capivasertib restricts SARS-CoV-2 cellular entry: A potential clinical application for COVID-19. Int. J. Biol. Sci. 2021, 17, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Patrì, A.; Fabbrocini, G. Hydroxychloroquine and ivermectin: A synergistic combination for COVID-19 chemoprophylaxis and treatment? J. Am. Acad. Dermatol. 2020, 82, e221. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Choi, K.; Jeong, E.; Kwon, D.; Benveniste, E.N.; Choi, C. Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia 2004, 47, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Devrim-Lanpir, A.; Hill, L.; Knechtle, B. How N-Acetylcysteine Supplementation Affects Redox Regulation, Especially at Mitohormesis and Sarcohormesis Level: Current Perspective. Antioxidants 2021, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).