

Introduction of a More Glutaredoxin-like Active Site to PDI Results in Competition between Protein Substrate and Glutathione Binding

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Production and Purification
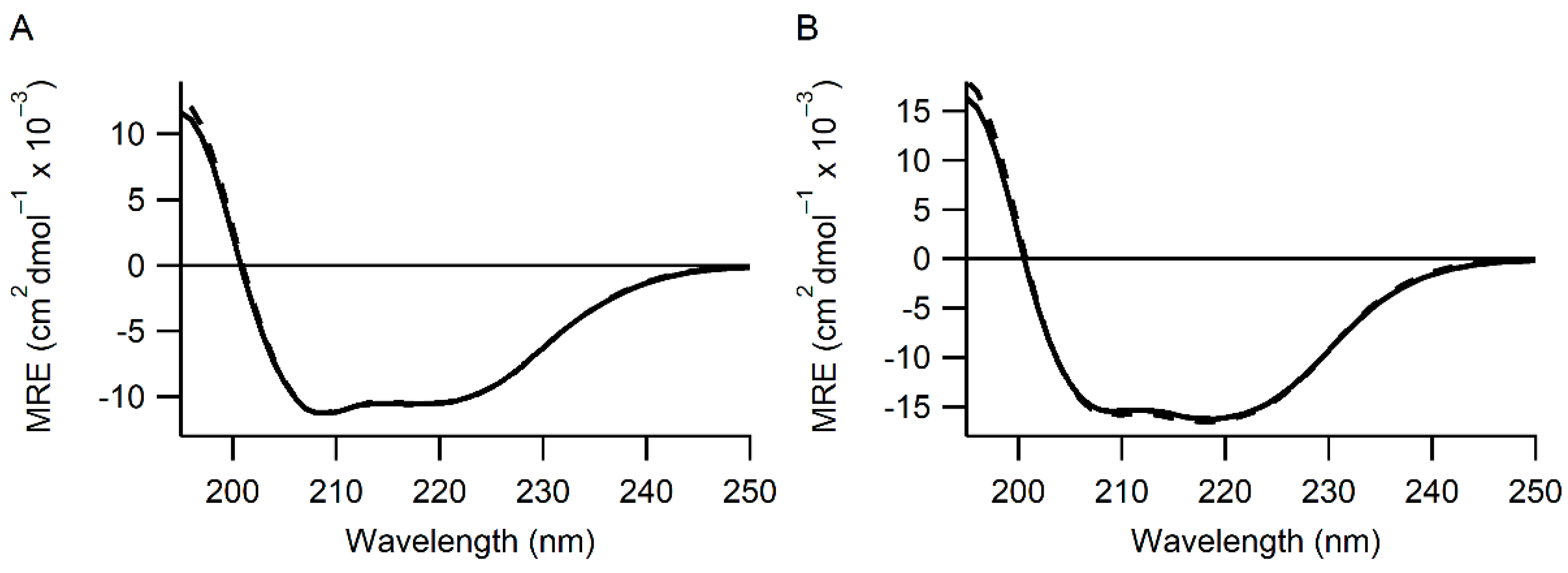
2.2. Circular Dichroism
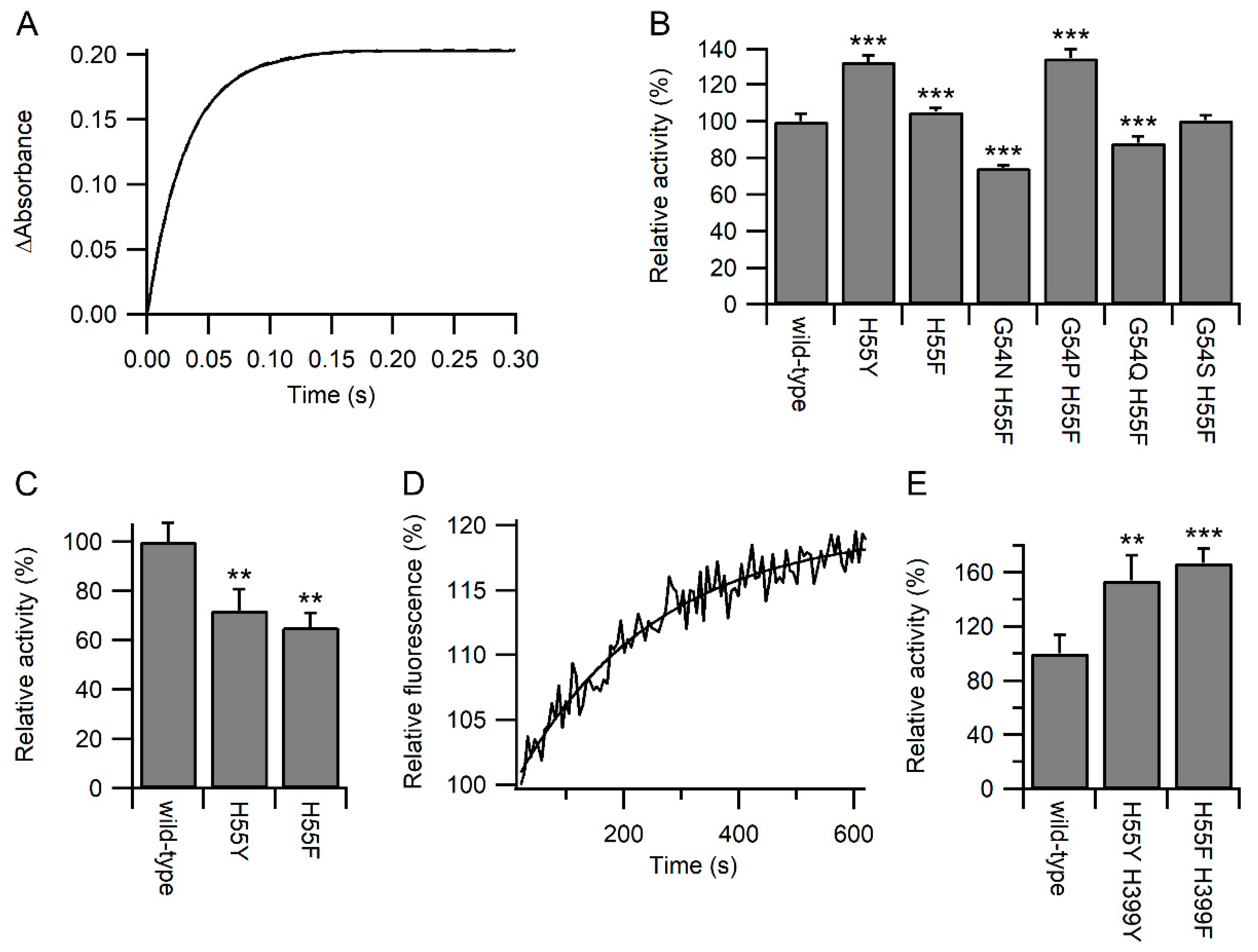
2.3. BPTI Refolding
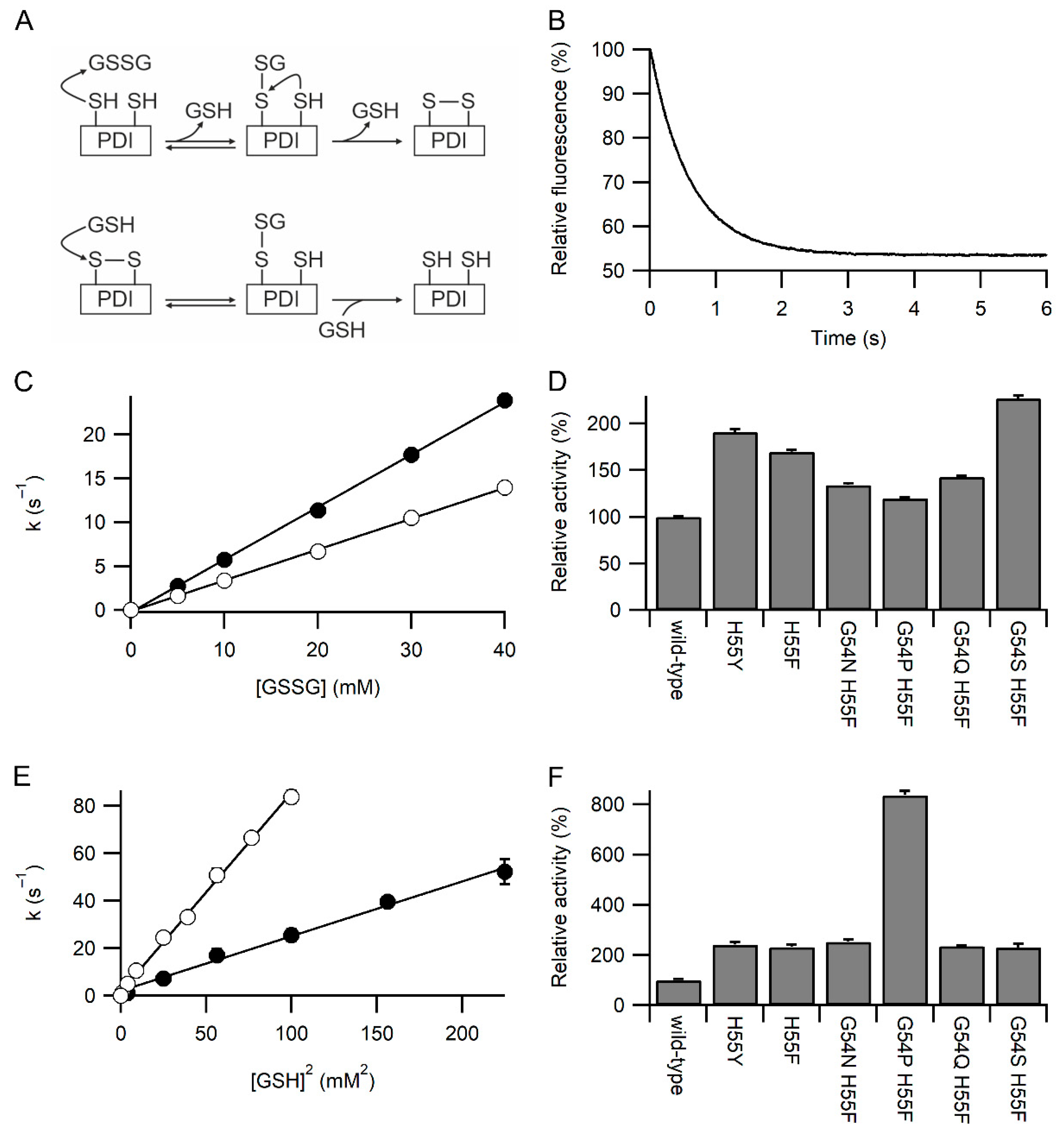
2.4. Kinetics of Oxidation and Reduction
2.5. Ellman’s Assay
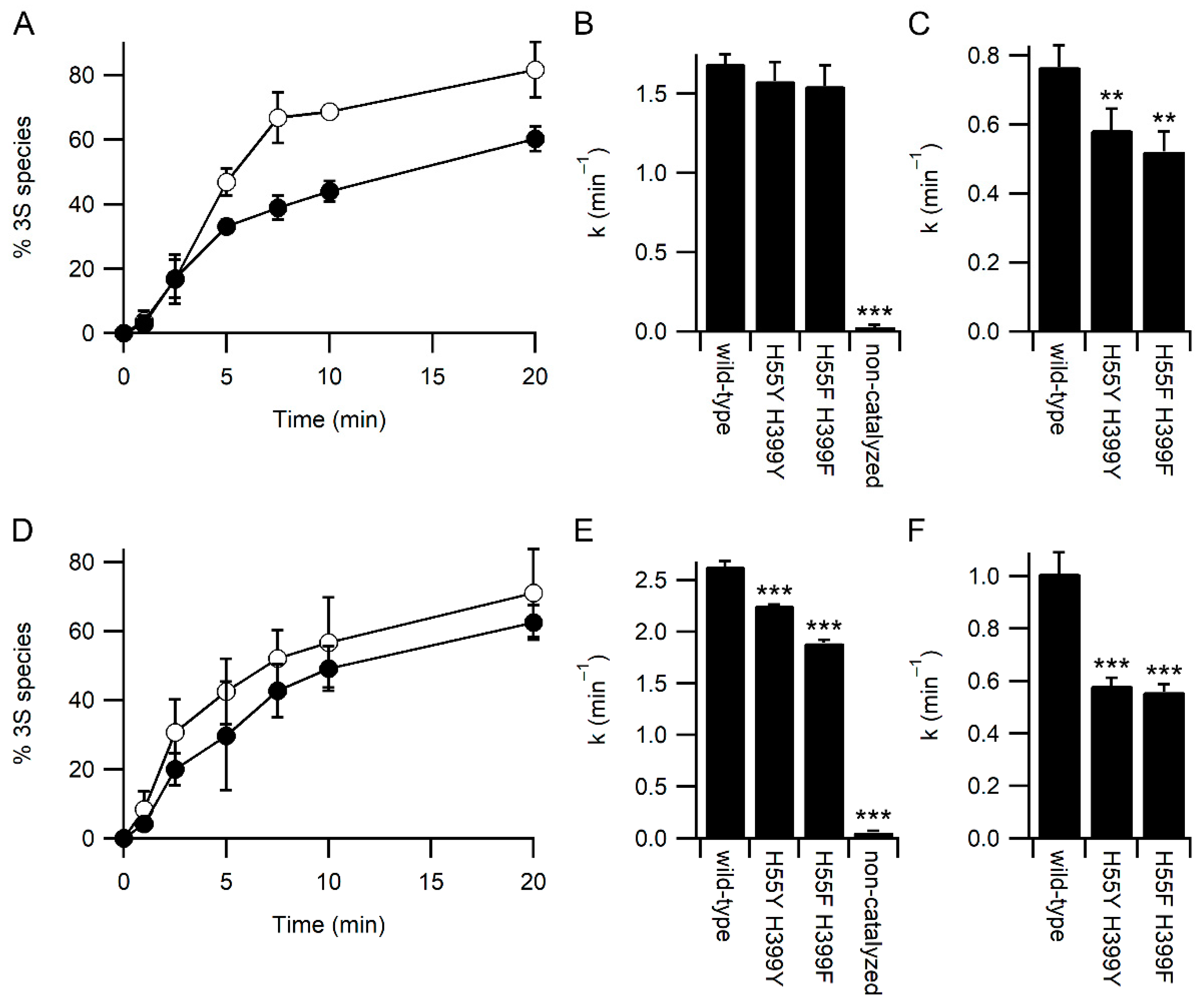
2.6. Deglutathionylation Assay
3. Results
3.1. Identification of Variants with Increased Reactivity towards Glutathione
3.2. Specificity of the Variants towards Glutathione
3.3. Glutathione Acts As a Competitor for Binding Protein Substrates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sousa, S.F.; Neves, R.P.P.; Waheed, S.O.; Fernandes, P.A.; Ramos, M.J. Structural and Mechanistic Aspects of S-S Bonds in the Thioredoxin-like Family of Proteins. Biol. Chem. 2019, 400, 575–587. [Google Scholar] [CrossRef]
- Hatahet, F.; Ruddock, L.W. Protein Disulfide Isomerase: A Critical Evaluation of Its Function in Disulfide Bond Formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef]
- Robinson, P.J.; Bulleid, N.J. Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway. Cells 2020, 9, 1994. [Google Scholar] [CrossRef]
- Bulleid, N.J.; Ellgaard, L. Multiple Ways to Make Disulfides. Trends Biochem. Sci. 2011, 36, 485–492. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized Redox State of Glutathione in the Endoplasmic Reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Gravina, S.A.; Mieyal, J.J. Thioltransferase Is a Specific Glutathionyl Mixed Disulfide Oxidoreductase. Biochemistry 1993, 32, 3368–3376. [Google Scholar] [CrossRef]
- Kemmink, J.; Darby, N.J.; Dijkstra, K.; Nilges, M.; Creighton, T.E. Structure Determination of the N-Terminal Thioredoxin-like Domain of Protein Disulfide Isomerase Using Multidimensional Heteronuclear 13C/15N NMR Spectroscopy. Biochemistry 1996, 35, 7684–7691. [Google Scholar] [CrossRef]
- Xia, T.-H.; Bushweller, J.H.; Sodano, P.; Billeter, M.; Björnberg, O.; Holmgren, A.; Wüthrich, K. NMR Structure of Oxidized Escherichia coli Glutaredoxin: Comparison with Reduced E. Coli Glutaredoxin and Functionally Related Proteins. Protein Sci. 1992, 1, 310–321. [Google Scholar] [CrossRef]
- Yang, Y.; Jao, S.; Nanduri, S.; Starke, D.W.; Mieyal, J.J.; Qin, J. Reactivity of the Human Thioltransferase (Glutaredoxin) C7S, C25S, C78S, C82S Mutant and NMR Solution Structure of Its Glutathionyl Mixed Disulfide Intermediate Reflect Catalytic Specificity. Biochemistry 1998, 37, 17145–17156. [Google Scholar] [CrossRef]
- Bushweller, J.H.; Billeter, M.; Holmgren, A.; Wüthrich, K. The Nuclear Magnetic Resonance Solution Structure of the Mixed Disulfide between Escherichia Coli Glutaredoxin(C14S) and Glutathione. J. Mol. Biol. 1994, 235, 1585–1597. [Google Scholar] [CrossRef]
- Lappi, A.K.; Ruddock, L.W. Reexamination of the Role of Interplay between Glutathione and Protein Disulfide Isomerase. J. Mol. Biol. 2011, 409, 238–249. [Google Scholar] [CrossRef]
- Karala, A.; Ruddock, L.W. Does S-Methyl Methanethiosulfonate Trap the Thiol–Disulfide State of Proteins? Antioxid. Redox Signal. 2007, 9, 527–531. [Google Scholar] [CrossRef]
- Gąciarz, A.; Ruddock, L.W. Complementarity Determining Regions and Frameworks Contribute to the Disulfide Bond Independent Folding of Intrinsically Stable ScFv. PLoS ONE 2017, 12, e0189964. [Google Scholar] [CrossRef]
- Lappi, A.K.; Lensink, M.F.; Alanen, H.I.; Salo, K.E.H.; Lobell, M.; Juffer, A.H.; Ruddock, L.W. A Conserved Arginine Plays a Role in the Catalytic Cycle of the Protein Disulphide Isomerases. J. Mol. Biol. 2004, 335, 283–295. [Google Scholar] [CrossRef]
- Alanen, H.I.; Salo, K.E.H.; Pekkala, M.; Siekkinen, H.M.; Pirneskoski, A.; Ruddock, L.W. Defining the Domain Boundaries of the Human Protein Disulfide Isomerases. Antioxid. Redox Signal. 2003, 5, 367–374. [Google Scholar] [CrossRef]
- Woehlbier, U.; Colombo, A.; Saaranen, M.J.; Pérez, V.; Ojeda, J.; Bustos, F.J.; Andreu, C.I.; Torres, M.; Valenzuela, V.; Medinas, D.B.; et al. ALS-Linked Protein Disulfide Isomerase Variants Cause Motor Dysfunction. EMBO J. 2016, 35, 845–865. [Google Scholar] [CrossRef]
- Saaranen, M.J.; Karala, A.-R.; Lappi, A.-K.; Ruddock, L.W. The Role of Dehydroascorbate in Disulfide Bond Formation. Antioxid. Redox Signal. 2009, 12, 15–25. [Google Scholar] [CrossRef]
- Karala, A.R.; Lappi, A.K.; Ruddock, L.W. Modulation of an Active-Site Cysteine PKa Allows PDI to Act as a Catalyst of Both Disulfide Bond Formation and Isomerization. J. Mol. Biol. 2010, 396, 883–892. [Google Scholar] [CrossRef]
- Peltoniemi, M.J.; Karala, A.R.; Jurvansuu, J.K.; Kinnula, V.L.; Ruddock, L.W. Insights into Deglutathionylation Reactions: Different Intermediates in the Glutaredoxin and Protein Disulfide Isomerase Catalyzed Reactions Are Defined by the Gamma-Linkage Present in Glutathione. J. Biol. Chem. 2006, 281, 33107–33114. [Google Scholar] [CrossRef]
- Ruddock, L.W.; Hirst, T.R.; Freedman, R.B. PH-Dependence of the Dithiol-Oxidizing Activity of DsbA (a Periplasmic Protein Thiol:Disulphide Oxidoreductase) and Protein Disulphide-Isomerase: Studies with a Novel Simple Peptide Substrate. Biochem. J. 1996, 315, 1001–1005. [Google Scholar] [CrossRef]
- Weissman, J.S.; Kim, P.S. A Kinetic Explanation for the Rearrangement Pathway of BPTI Folding. Nat. Struct. Mol. Biol. 1995, 2, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Weissman, J.S.; Kim, P.S. Reexamination of the Folding of BPTI: Predominance of Native Intermediates. Science 1991, 253, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Mezghrani, A.; Fassio, A.; Benham, A.; Simmen, T.; Braakman, I.; Sitia, R. Manipulation of Oxidative Protein Folding and PDI Redox State in Mammalian Cells. EMBO J. 2001, 20, 6288–6296. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Riemer, J.; Zito, E.; Chin, K.-T.; Ron, D.; Spiess, M.; Ellgaard, L. Disulphide Production by Ero1α–PDI Relay Is Rapid and Effectively Regulated. EMBO J. 2010, 29, 3318–3329. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Wang, B.; Li, W.; Rapoport, T.A. Vitamin K Epoxide Reductase Prefers ER Membrane-Anchored Thioredoxin-like Redox Partners. Proc. Natl. Acad. Sci. USA 2010, 107, 15027–15032. [Google Scholar] [CrossRef] [PubMed]
- Karala, A.; Lappi, A.; Saaranen, M.J.; Ruddock, L.W. Efficient Peroxide-Mediated Oxidative Refolding of a Protein at Physiological PH and Implications for Oxidative Folding in the Endoplasmic Reticulum. Antioxid. Redox Signal. 2009, 11, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.H.; Wang, C.C.; Ruddock, L.W. Two Endoplasmic Reticulum PDI Peroxidases Increase the Efficiency of the Use of Peroxide during Disulfide Bond Formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, Å.; Neubert, T.A.; Ron, D. Oxidative Protein Folding by an Endoplasmic Reticulum-Localized Peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef]
- Poet, G.J.; Oka, O.B.; Lith, M.; Cao, Z.; Robinson, P.J.; Pringle, M.A.; Arnér, E.S.; Bulleid, N.J. Cytosolic Thioredoxin Reductase 1 Is Required for Correct Disulfide Formation in the ER. EMBO J. 2017, 36, 693–702. [Google Scholar] [CrossRef]
- Cao, X.; Lilla, S.; Cao, Z.; Pringle, M.A.; Oka, O.B.; Robinson, P.J.; Szmaja, T.; van Lith, M.; Zanivan, S.; Bulleid, N.J. The Mammalian Cytosolic Thioredoxin Reductase Pathway Acts via a Membrane Protein to Reduce ER-Localised Proteins. J. Cell Sci. 2020, 133, jcs241976. [Google Scholar] [CrossRef]
- Tsunoda, S.; Avezov, E.; Zyryanova, A.; Konno, T.; Mendes-Silva, L.; Pinho Melo, E.; Harding, H.P.; Ron, D. Intact Protein Folding in the Glutathione-Depleted Endoplasmic Reticulum Implicates Alternative Protein Thiol Reductants. eLife 2014, 3, e03421. [Google Scholar] [CrossRef]
- Montero, D.; Tachibana, C.; Rahr Winther, J.; Appenzeller-Herzog, C. Intracellular Glutathione Pools Are Heterogeneously Concentrated. Redox Biol. 2013, 1, 508–513. [Google Scholar] [CrossRef]
- Dixon, B.M.; Heath, S.-H.D.; Kim, R.; Suh, J.H.; Hagen, T.M. Assessment of Endoplasmic Reticulum Glutathione Redox Status Is Confounded by Extensive Ex Vivo Oxidation. Antioxid. Redox Signal. 2008, 10, 963–972. [Google Scholar] [CrossRef]
- Bass, R.; Ruddock, L.W.; Klappa, P.; Freedman, R.B. A Major Fraction of Endoplasmic Reticulum-Located Glutathione Is Present as Mixed Disulfides with Protein. J. Biol. Chem. 2004, 279, 5257–5262. [Google Scholar] [CrossRef]
- Ponsero, A.J.; Igbaria, A.; Darch, M.A.; Miled, S.; Outten, C.E.; Winther, J.R.; Palais, G.; D’Autréaux, B.; Delaunay-Moisan, A.; Toledano, M.B. Endoplasmic Reticulum Transport of Glutathione by Sec61 Is Regulated by Ero1 and Bip. Mol. Cell 2017, 67, 962–973.e5. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid Name | Construct | References |
|---|---|---|
| pAKL2 | human PDI a domain (MH6M-D18-A137) W128F | [14] |
| pHIA441 | human PDI a domain H55Y W128F | This study |
| pHIA440 | human PDI a domain H55F W128F | This study |
| pKEHS961 | human PDI a domain G54N H55F W128F | This study |
| pPK2 | human PDI a domain G54Q H55F W128F | This study |
| pEDN3 | human PDI a domain G54S H55F W128F | This study |
| pCS1 | human PDI a domain G54P H55F W128F | This study |
| pLWRP64 | human PDI (MH6M-D18-L508) | [15] |
| pHIA452 | human PDI H55Y H399Y | This study |
| pHIA453 | human PDI H55F H399F | This study |
| pARK32 | mature BPTI (M-R36-A93) | [12] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saaranen, M.J.; Alanen, H.I.; Salo, K.E.H.; Nji, E.; Kärkkäinen, P.; Schmotz, C.; Ruddock, L.W. Introduction of a More Glutaredoxin-like Active Site to PDI Results in Competition between Protein Substrate and Glutathione Binding. Antioxidants 2022, 11, 1920. https://doi.org/10.3390/antiox11101920
Saaranen MJ, Alanen HI, Salo KEH, Nji E, Kärkkäinen P, Schmotz C, Ruddock LW. Introduction of a More Glutaredoxin-like Active Site to PDI Results in Competition between Protein Substrate and Glutathione Binding. Antioxidants. 2022; 11(10):1920. https://doi.org/10.3390/antiox11101920
Chicago/Turabian StyleSaaranen, Mirva J., Heli I. Alanen, Kirsi E. H. Salo, Emmanuel Nji, Pekka Kärkkäinen, Constanze Schmotz, and Lloyd W. Ruddock. 2022. "Introduction of a More Glutaredoxin-like Active Site to PDI Results in Competition between Protein Substrate and Glutathione Binding" Antioxidants 11, no. 10: 1920. https://doi.org/10.3390/antiox11101920
APA StyleSaaranen, M. J., Alanen, H. I., Salo, K. E. H., Nji, E., Kärkkäinen, P., Schmotz, C., & Ruddock, L. W. (2022). Introduction of a More Glutaredoxin-like Active Site to PDI Results in Competition between Protein Substrate and Glutathione Binding. Antioxidants, 11(10), 1920. https://doi.org/10.3390/antiox11101920