
The Interplay between S-Glutathionylation and Phosphorylation of Cardiac Troponin I and Myosin Binding Protein C in End-Stage Human Failing Hearts

 , ,
, , 
 , ,
, ,  ,
,
Abstract
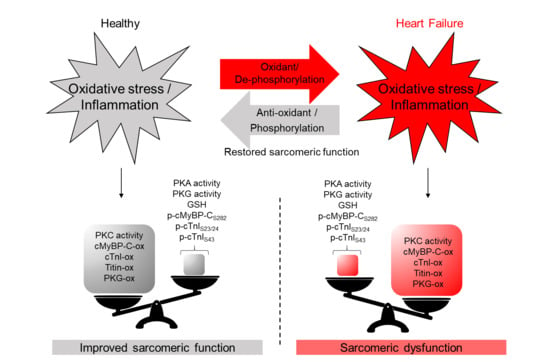
1. Introduction
2. Materials and Methods
2.1. Human Heart Tissues
2.2. Force Measurements in Single, Skinned Cardiomyocytes
2.3. Myocardial Protein Kinase a (PKA) and Myocardial Protein Kinase C (PKC) Activity
2.4. Myocardial Protein Kinase G (PKG) Activity
2.5. Protein Analysis by Western Blot
2.6. Oxidation Detection (OxICAT)
2.7. Mass Spectrometry Data Analysis
2.8. Duolink In Situ PLA (Proximity Ligation Assay (PLA)) Technology
2.9. Immunofluorescence Imaging
2.10. Quantification of Oxidative Stress and Inflammation Parameters of End-Stage Human Heart Failure Tissue
2.11. Quantification of Nitric Oxide (NO) Level of End-Stage Human Failing Hearts
2.12. Statistical Analysis
3. Results
3.1. Altered Maximum Ca2+-Activated Tension and Ca2+ Sensitivity of HF Demembranated Cardiomyocytes
3.2. Alterations in Activity and Oxidation of Major Protein Kinases and the Effect on Phosphorylation of Ctni and Cmybp-C in End-Stage Human Failing Hearts
3.3. cTnI and cMyBP-C Glutathionylation in End-Stage Human Failing Hearts
3.4. Identification of Oxidized Cysteins in Cmybp-C and Ctni Using Oxicat Together with Mass Spectrometry
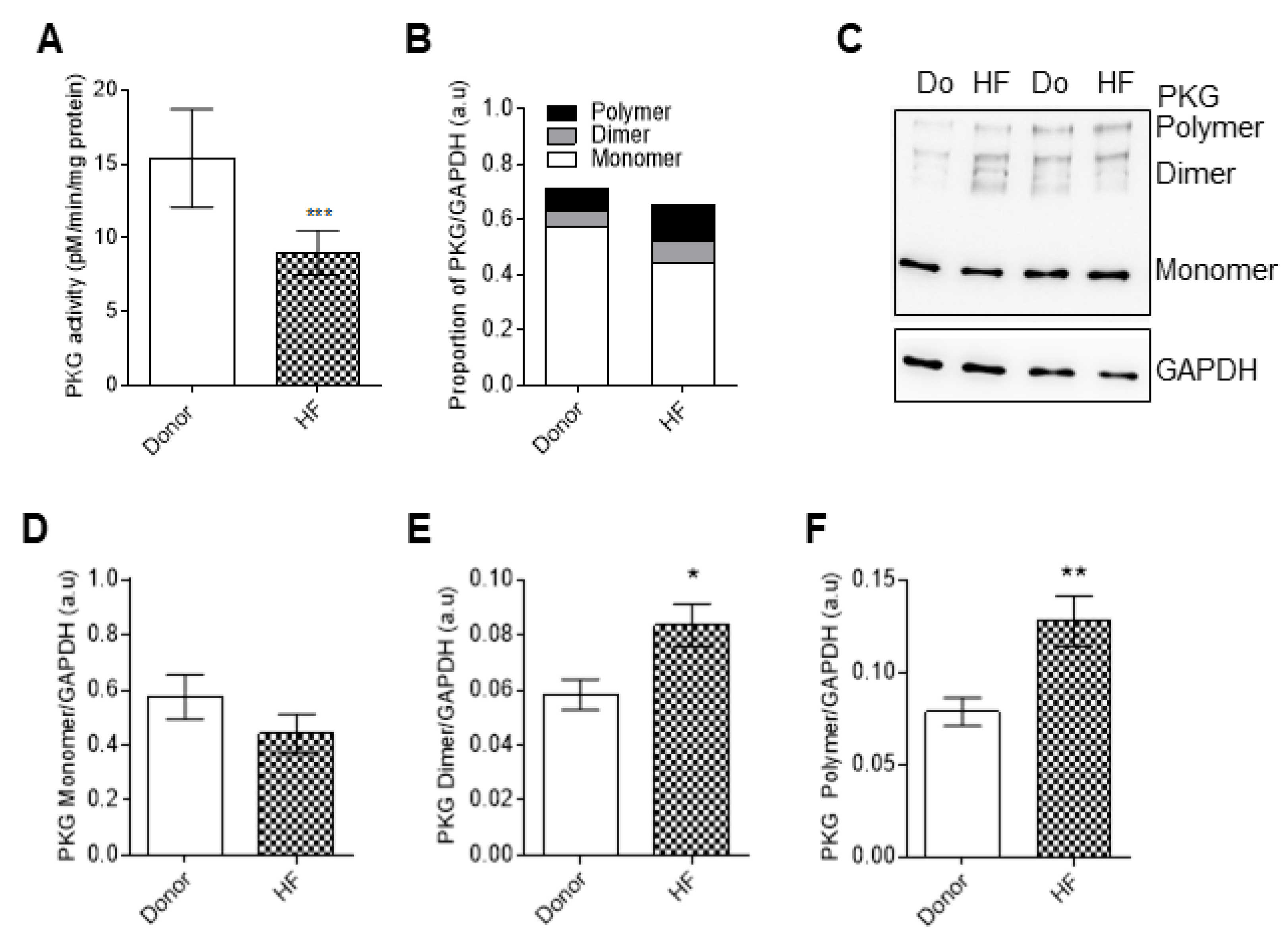
3.5. PKG Activity and Oxidation in End-Stage Human Failing Hearts
3.6. The Functional Interplay between Ctni Oxidation and Phosphorylation in End-Stage Heart Failure
3.7. Pro-Inflammatory Cytokines and Oxidative Stress Parameters in End Stage Heart Failure Human Samples before and after Reduced Glutathione (GSH) Treatment
3.8. Increased Myocardial Stiffness in HF Myocytes Is Reversed by PKA and GSH Treatment
3.9. cTnI and cMyBP-C S-Glutathionylation in End Stage of Failing Heart Samples
4. Discussion
4.1. Oxidation and Inflammation Parameters in End-Stage Heart Failure
4.2. Myofilament Proteins and Protein Kinases, Their Modification and Function under Oxidative Stress
4.3. Interplay between Myofilament Proteins Oxidation and Phosphorylation under Oxidative Stress
5. Conclusions and Therapeutic Implications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breitkreuz, M.; Hamdani, N. A Change of Heart: Oxidative Stress in Governing Muscle Function? Biophys. Rev. 2015, 7, 321–341. [Google Scholar] [CrossRef]
- Terrill, J.R.; Radley-Crabb, H.G.; Iwasaki, T.; Lemckert, F.A.; Arthur, P.G.; Grounds, M.D. Oxidative Stress and Pathology in Muscular Dystrophies: Focus on Protein Thiol Oxidation and Dysferlinopathies. FEBS J. 2013, 280, 4149–4164. [Google Scholar] [CrossRef]
- Janué, A.; Olivé, M.; Ferrer, I. Oxidative Stress in Desminopathies and Myotilinopathies: A Link between Oxidative Damage and Abnormal Protein Aggregation. Brain Pathol. 2007, 17, 377–388. [Google Scholar] [CrossRef]
- Rando, T.A.; Disatnik, M.H.; Yu, Y.; Franco, A. Muscle Cells from Mdx Mice Have an Increased Susceptibility to Oxidative Stress. Neuromuscul. Disord. 1998, 8, 14–21. [Google Scholar] [CrossRef]
- Canton, M.; Menazza, S.; Di Lisa, F. Oxidative Stress in Muscular Dystrophy: From Generic Evidence to Specific Sources and Targets. J. Muscle Res. Cell Motil. 2014, 35, 23–36. [Google Scholar] [CrossRef]
- Haycock, J.W.; Mac Neil, S.; Jones, P.; Harris, J.B.; Mantle, D. Oxidative Damage to Muscle Protein in Duchenne Muscular Dystrophy. Neuroreport 1996, 8, 357–361. [Google Scholar] [CrossRef]
- Zhazykbayeva, S.; Pabel, S.; Mügge, A.; Sossalla, S.; Hamdani, N. The Molecular Mechanisms Associated with the Physiological Responses to Inflammation and Oxidative Stress in Cardiovascular Diseases. Biophys. Rev. 2020, 12, 947–968. [Google Scholar] [CrossRef]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative Stress and Redox Signalling in Cardiac Hypertrophy and Heart Failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef]
- Cline, M.J.; Lehrer, R.I. D-Amino Acid Oxidase in Leukocytes: A Possible D-Amino-Acid-Linked Antimicrobial System. Proc. Natl. Acad. Sci. USA 1969, 62, 756–763. [Google Scholar] [CrossRef]
- Nishino, T. The Conversion of Xanthine Dehydrogenase to Xanthine Oxidase and the Role of the Enzyme in Reperfusion Injury. J. Biochem. 1994, 116, 1–6. [Google Scholar] [CrossRef]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms That Regulate Production of Reactive Oxygen Species by Cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. [Google Scholar] [CrossRef]
- Rahman, K. Studies on Free Radicals, Antioxidants, and Co-Factors. Clin. Interv. Aging 2007, 2, 219. [Google Scholar]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, Á.; Fülöp, G.Á.; Falcão-Pires, I.; et al. Empagliflozin Improves Endothelial and Cardiomyocyte Function in Human Heart Failure with Preserved Ejection Fraction via Reduced Pro-Inflammatory-Oxidative Pathways and Protein Kinase Gα Oxidation. Cardiovasc. Res. 2021, 117, 495–507. [Google Scholar] [CrossRef]
- Kolijn, D.; Kovács, Á.; Herwig, M.; Lódi, M.; Sieme, M.; Alhaj, A.; Sandner, P.; Papp, Z.; Reusch, P.H.; Haldenwang, P.; et al. Enhanced Cardiomyocyte Function in Hypertensive Rats With Diastolic Dysfunction and Human Heart Failure Patients After Acute Treatment With Soluble Guanylyl Cyclase (SGC) Activator. Front. Physiol. 2020, 11, 345. [Google Scholar] [CrossRef]
- Shishehbor, M.H.; Aviles, R.J.; Brennan, M.-L.; Fu, X.; Goormastic, M.; Pearce, G.L.; Gokce, N.; Keaney, J.F., Jr.; Penn, M.S.; Sprecher, D.L.; et al. Association of Nitrotyrosine Levels With Cardiovascular Disease and Modulation by Statin Therapy. JAMA 2003, 289, 1675–1680. [Google Scholar] [CrossRef]
- Loescher, C.M.; Breitkreuz, M.; Li, Y.; Nickel, A.; Unger, A.; Dietl, A.; Schmidt, A.; Mohamed, B.A.; Kötter, S.; Schmitt, J.P.; et al. Regulation of Titin-Based Cardiac Stiffness by Unfolded Domain Oxidation (UnDOx). Proc. Natl. Acad. Sci. USA 2020, 117, 24545–24556. [Google Scholar] [CrossRef]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernández, J.M. S-Glutathionylation of Cryptic Cysteines Enhances Titin Elasticity by Blocking Protein Folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.M.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Taube, A.; Schlich, R.; Sell, H.; Eckardt, K.; Eckel, J. Inflammation and Metabolic Dysfunction: Links to Cardiovascular Diseases. Am. J. Physiol. Circ. Physiol. 2012, 302, H2148–H2165. [Google Scholar] [CrossRef]
- Tiago, T.; Simão, S.; Aureliano, M.; Martín-Romero, F.J.; Gutiérrez-Merino, C. Inhibition of Skeletal Muscle S1-Myosin ATPase by Peroxynitrite. Biochemistry 2006, 45, 3794–3804. [Google Scholar] [CrossRef]
- Solaro, R.J. Nitroxyl Effects on Myocardium Provide New Insights into the Significance of Altered Myofilament Response to Calcium in the Regulation of Contractility. J. Physiol. 2007, 580, 697. [Google Scholar] [CrossRef]
- Gao, W.D.; Murray, C.I.; Tian, Y.; Zhong, X.; DuMond, J.F.; Shen, X.; Stanley, B.A.; Foster, D.B.; Wink, D.A.; King, S.B.; et al. Nitroxyl-Mediated Disulfide Bond Formation between Cardiac Myofilament Cysteines Enhances Contractile Function. Circ. Res. 2012, 111, 1002–1011. [Google Scholar] [CrossRef]
- Grützner, A.; Garcia-Manyes, S.; Kötter, S.; Badilla, C.L.; Fernandez, J.M.; Linke, W.A. Modulation of Titin-Based Stiffness by Disulfide Bonding in the Cardiac Titin N2-B Unique Sequence. Biophys. J. 2009, 97, 825–834. [Google Scholar] [CrossRef]
- Lovelock, J.D.; Monasky, M.M.; Jeong, E.-M.; Lardin, H.A.; Liu, H.; Patel, B.G.; Taglieri, D.M.; Gu, L.; Kumar, P.; Pokhrel, N.; et al. Ranolazine Improves Cardiac Diastolic Dysfunction Through Modulation of Myofilament Calcium Sensitivity. Circ. Res. 2012, 110, 841–850. [Google Scholar] [CrossRef]
- Kobayashi, T.; Solaro, R.J. Calcium, Thin Filaments, and the Integrative Biology of Cardiac Contractility. Annu. Rev. Physiol. 2005, 67, 39–67. [Google Scholar] [CrossRef]
- Metzger, J.M.; Westfall, M. V Covalent and Noncovalent Modification of Thin Filament Action. Circ. Res. 2004, 94, 146–158. [Google Scholar] [CrossRef]
- Hamdani, N.; Kooij, V.; van Dijk, S.; Merkus, D.; Paulus, W.J.; dos Remedios, C.; Duncker, D.J.; Stienen, G.J.M.; van der Velden, J. Sarcomeric Dysfunction in Heart Failure. Cardiovasc. Res. 2008, 77, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Linke, W.A.; Hamdani, N. Gigantic Business: Titin Properties and Function through Thick and Thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-Glutathionylation in Protein Redox Regulation. Free Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Krysiak, J.; Kreusser, M.M.; Neef, S.; Dos Remedios, C.G.; Maier, L.S.; Krüger, M.; Backs, J.; Linke, W.A. Crucial Role for Ca2(+)/Calmodulin-Dependent Protein Kinase-II in Regulating Diastolic Stress of Normal and Failing Hearts via Titin Phosphorylation. Circ. Res. 2013, 112, 664–674. [Google Scholar] [CrossRef]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged Myofilament Phosphorylation and Function in Experimental Heart Failure with Preserved Ejection Fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Franssen, C.; Lourenço, A.; Falcão-Pires, I.; Fontoura, D.; Leite, S.; Plettig, L.; López, B.; Ottenheijm, C.A.; Becher, P.M.; et al. Myocardial Titin Hypophosphorylation Importantly Contributes to Heart Failure with Preserved Ejection Fraction in a Rat Metabolic Risk Model. Circ. Heart Fail. 2013, 6, 1239–1249. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.V.; Bronzwaer, J.G.F.; van der Velden, J.; Stienen, G.J.M.; Laarman, G.J.; Somsen, A.; et al. Low Myocardial Protein Kinase G Activity in Heart Failure With Preserved Ejection Fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef]
- Marston, S.; Zamora, J.E. Troponin Structure and Function: A View of Recent Progress. J. Muscle Res. Cell Motil. 2020, 41, 71–89. [Google Scholar] [CrossRef]
- Kötter, S.; Gout, L.; Von Frieling-Salewsky, M.; Müller, A.E.; Helling, S.; Marcus, K.; Dos Remedios, C.; Linke, W.A.; Krüger, M. Differential Changes in Titin Domain Phosphorylation Increase Myofilament Stiffness in Failing Human Hearts. Cardiovasc. Res. 2013, 99, 648–656. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of Oxidative Stress in Cardiovascular Diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Lang, D. Cardiac Hypertrophy and Oxidative Stress: A Leap of Faith or Stark Reality? Heart 2002, 87, 316–317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hopps, E.; Noto, D.; Caimi, G.; Averna, M.R. A Novel Component of the Metabolic Syndrome: The Oxidative Stress. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 72–77. [Google Scholar] [CrossRef]
- Torre-Amione, G. Immune Activation in Chronic Heart Failure. Am. J. Cardiol. 2005, 95, 3C–8C, discussion 38C–40C. [Google Scholar] [CrossRef]
- Anderson, E.J.; Katunga, L.A.; Willis, M.S. Mitochondria as a Source and Target of Lipid Peroxidation Products in Healthy and Diseased Heart. Clin. Exp. Pharmacol. Physiol. 2012, 39, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Davies, S.S.; Nakajima, T.; Ong, B.-H.; Kupershmidt, S.; Fessel, J.; Amarnath, V.; Anderson, M.E.; Boyden, P.A.; Viswanathan, P.C.; et al. Oxidative Mediated Lipid Peroxidation Recapitulates Proarrhythmic Effects on Cardiac Sodium Channels. Circ. Res. 2005, 97, 1262–1269. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Lassègue, B.; Ushio-Fukai, M. Modulation of Protein Kinase Activity and Gene Expression by Reactive Oxygen Species and Their Role in Vascular Physiology and Pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2175–2183. [Google Scholar] [CrossRef]
- Herwig, M.; Kolijn, D.; Lódi, M.; Hölper, S.; Kovács, Á.; Papp, Z.; Jaquet, K.; Haldenwang, P.; Dos Remedios, C.; Reusch, P.H.; et al. Modulation of Titin-Based Stiffness in Hypertrophic Cardiomyopathy via Protein Kinase D. Front. Physiol. 2020, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Böhm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; et al. ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in Collaboration with the Hear. Eur. Heart J. 2012, 33, 1787–1847. [Google Scholar] [CrossRef] [PubMed]
- Cowie, M.R.; Wood, D.A.; Coats, A.J.; Thompson, S.G.; Suresh, V.; Poole-Wilson, P.A.; Sutton, G.C. Survival of Patients with a New Diagnosis of Heart Failure: A Population Based Study. Heart 2000, 83, 505–510. [Google Scholar] [CrossRef]
- Kain, D.; Amit, U.; Yagil, C.; Landa, N.; Naftali-Shani, N.; Molotski, N.; Aviv, V.; Feinberg, M.S.; Goitein, O.; Kushnir, T.; et al. Macrophages Dictate the Progression and Manifestation of Hypertensive Heart Disease. Int. J. Cardiol. 2016, 203, 381–395. [Google Scholar] [CrossRef]
- Hwang, S.-J.; Melenovsky, V.; Borlaug, B.A. Implications of Coronary Artery Disease in Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2014, 63, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, J.; Bullard, B.; Mercola, D. Interaction of Troponin Subunits. The Interaction between the Inhibitory and Tropomyosin-Binding Subunits. J. Biol. Chem. 1979, 254, 350–355. [Google Scholar] [CrossRef]
- Chong, P.C.; Hodges, R.S. Proximity of Sulfhydryl Groups to the Sites of Interaction between Components of the Troponin Complex from Rabbit Skeletal Muscle. J. Biol. Chem. 1982, 257, 2549–2555. [Google Scholar] [CrossRef]
- Aihara, T.; Nakamura, M.; Ueki, S.; Hara, H.; Miki, M.; Arata, T. Switch Action of Troponin on Muscle Thin Filament as Revealed by Spin Labeling and Pulsed EPR. J. Biol. Chem. 2010, 285, 10671–10677. [Google Scholar] [CrossRef]
- Chong, P.C.; Hodges, R.S. A New Heterobifunctional Cross-Linking Reagent for the Study of Biological Interactions between Proteins. I. Design, Synthesis, and Characterization. J. Biol. Chem. 1981, 256, 5064–5070. [Google Scholar] [CrossRef]
- Jeong, E.-M.; Monasky, M.M.; Gu, L.; Taglieri, D.M.; Patel, B.G.; Liu, H.; Wang, Q.; Greener, I.; Dudley, S.C.J.; Solaro, R.J. Tetrahydrobiopterin Improves Diastolic Dysfunction by Reversing Changes in Myofilament Properties. J. Mol. Cell. Cardiol. 2013, 56, 44–54. [Google Scholar] [CrossRef]
- Patel, B.; Wilder, T.; Solaro, R. Novel Control of Cardiac Myofilament Response to Calcium by S-Glutathionylation at Specific Sites of Myosin Binding Protein C. Front. Physiol. 2013, 4, 336. [Google Scholar] [CrossRef]
- Linke, W.A. Sense and Stretchability: The Role of Titin and Titin-Associated Proteins in Myocardial Stress-Sensing and Mechanical Dysfunction. Cardiovasc. Res. 2008, 77, 637–648. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. Lysine Acetylation: Codified Crosstalk with Other Posttranslational Modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef]
- Meng, T.-C.; Fukada, T.; Tonks, N.K. Reversible Oxidation and Inactivation of Protein Tyrosine Phosphatases in Vivo. Mol. Cell 2002, 9, 387–399. [Google Scholar] [CrossRef]
- Kensler, R.W.; Craig, R.; Moss, R.L. Phosphorylation of Cardiac Myosin Binding Protein C Releases Myosin Heads from the Surface of Cardiac Thick Filaments. Proc. Natl. Acad. Sci. USA 2017, 114, E1355–E1364. [Google Scholar] [CrossRef]
- Krüger, M.; Kötter, S.; Grützner, A.; Lang, P.; Andresen, C.; Redfield, M.M.; Butt, E.; dos Remedios, C.G.; Linke, W.A. Protein Kinase G Modulates Human Myocardial Passive Stiffness by Phosphorylation of the Titin Springs. Circ. Res. 2009, 104, 87–94. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Oka, S.; Ale-Agha, N.; Eaton, P. Hydrogen Peroxide Sensing and Signaling by Protein Kinases in the Cardiovascular System. Antioxid. Redox Signal. 2013, 18, 1042–1052. [Google Scholar] [CrossRef]
- Kuster, D.W.D.; Bawazeer, A.C.; Zaremba, R.; Goebel, M.; Boontje, N.M.; van der Velden, J. Cardiac Myosin Binding Protein C Phosphorylation in Cardiac Disease. J. Muscle Res. Cell Motil. 2012, 33, 43–52. [Google Scholar] [CrossRef]
- Aslam, N. Increase in PKCα Activity during Heart Failure Despite the Stimulation of PKCα Braking Mechanism. Int. J. Mol. Sci. 2020, 21, 2561. [Google Scholar] [CrossRef]
- Cimiotti, D.; Fujita-Becker, S.; Möhner, D.; Smolina, N.; Budde, H.; Wies, A.; Morgenstern, L.; Gudkova, A.; Sejersen, T.; Sjöberg, G.; et al. Infantile Restrictive Cardiomyopathy: CTnI-R170G/W Impair the Interplay of Sarcomeric Proteins and the Integrity of Thin Filaments. PLoS ONE 2020, 15, e0229227. [Google Scholar] [CrossRef]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zhao, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; et al. PKC-α Regulates Cardiac Contractility and Propensity toward Heart Failure. Nat. Med. 2004, 10, 248–254. [Google Scholar] [CrossRef]
- Bilchick, K.C.; Duncan, J.G.; Ravi, R.; Takimoto, E.; Champion, H.C.; Gao, W.D.; Stull, L.B.; Kass, D.A.; Murphy, A.M. Heart Failure-Associated Alterations in Troponin I Phosphorylation Impair Ventricular Relaxation-Afterload and Force-Frequency Responses and Systolic Function. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H318–H325. [Google Scholar] [CrossRef][Green Version]
- Kooij, V.; Holewinski, R.J.; Murphy, A.M.; Van Eyk, J.E. Characterization of the Cardiac Myosin Binding Protein-C Phosphoproteome in Healthy and Failing Human Hearts. J. Mol. Cell. Cardiol. 2013, 60, 116–120. [Google Scholar] [CrossRef]
- Mohamed, A.S.; Dignam, J.D.; Schlender, K.K. Cardiac Myosin-Binding Protein C (MyBP-C): Identification of Protein Kinase A and Protein Kinase C Phosphorylation Sites. Arch. Biochem. Biophys. 1998, 358, 313–319. [Google Scholar] [CrossRef]
- Lang, S.E.; Stevenson, T.K.; Schatz, T.M.; Biesiadecki, B.J.; Westfall, M.V. Functional Communication between PKC-Targeted Cardiac Troponin I Phosphorylation Sites. Arch. Biochem. Biophys. 2017, 627, 1–9. [Google Scholar] [CrossRef]
- Nixon, B.R.; Thawornkaiwong, A.; Jin, J.; Brundage, E.A.; Little, S.C.; Davis, J.P.; Solaro, R.J.; Biesiadecki, B.J. AMP-Activated Protein Kinase Phosphorylates Cardiac Troponin I at Ser-150 to Increase Myofilament Calcium Sensitivity and Blunt PKA-Dependent Function *. J. Biol. Chem. 2012, 287, 19136–19147. [Google Scholar] [CrossRef]
- Farías, J.G.; Molina, V.M.; Carrasco, R.A.; Zepeda, A.B.; Figueroa, E.; Letelier, P.; Castillo, R.L. Antioxidant Therapeutic Strategies for Cardiovascular Conditions Associated with Oxidative Stress. Nutrients 2017, 9, 966. [Google Scholar] [CrossRef]
- Pashkow, F.J.; Watumull, D.G.; Campbell, C.L. Astaxanthin: A Novel Potential Treatment for Oxidative Stress and Inflammation in Cardiovascular Disease. Am. J. Cardiol. 2008, 101, S58–S68. [Google Scholar] [CrossRef]
- Fassett, R.G.; Coombes, J.S. Astaxanthin, Oxidative Stress, Inflammation and Cardiovascular Disease. Future Cardiol. 2009, 5, 333–342. [Google Scholar] [CrossRef]
- Luo, M.; Sun, Q.; Zhao, H.; Tao, J.; Yan, D. The Effects of Dimethyl Fumarate on Atherosclerosis in the Apolipoprotein E-Deficient Mouse Model with Streptozotocin-Induced Hyperglycemia Mediated By the Nuclear Factor Erythroid 2-Related Factor 2/Antioxidant Response Element (Nrf2/ARE) Signaling Pathw. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 7966. [Google Scholar] [CrossRef] [PubMed]
- Tita, C.; Gilbert, E.M.; Van Bakel, A.B.; Grzybowski, J.; Haas, G.J.; Jarrah, M.; Dunlap, S.H.; Gottlieb, S.S.; Klapholz, M.; Patel, P.C. A Phase 2a Dose-escalation Study of the Safety, Tolerability, Pharmacokinetics and Haemodynamic Effects of BMS-986231 in Hospitalized Patients with Heart Failure with Reduced Ejection Fraction. Eur. J. Heart Fail. 2017, 19, 1321–1332. [Google Scholar] [CrossRef]
- Pabel, S.; Wagner, S.; Bollenberg, H.; Bengel, P.; Kovács, Á.; Schach, C.; Tirilomis, P.; Mustroph, J.; Renner, A.; Gummert, J.; et al. Empagliflozin Directly Improves Diastolic Function in Human Heart Failure. Eur. J. Heart Fail. 2018, 20, 1690–1700. [Google Scholar] [CrossRef]
- Shende, P.; Plaisance, I.; Morandi, C.; Pellieux, C.; Berthonneche, C.; Zorzato, F.; Krishnan, J.; Lerch, R.; Hall, M.N.; Rüegg, M.A.; et al. Cardiac Raptor Ablation Impairs Adaptive Hypertrophy, Alters Metabolic Gene Expression, and Causes Heart Failure in Mice. Circulation 2011, 123, 1073–1082. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Product Number | Buffer | Dilution |
|---|---|---|---|---|
| Cardiac Troponin I antibody | Abcam | ab 47003 | BSA-TBST | 1:1000 |
| Phospho-Troponin I (Cardiac) (S23/S24) antibody | Cell Signaling Technology | 4004S | BSA-TBST | 1:1000 |
| Cardiac Troponin I (phospho S43) antibody/mouse canonical sequence | Abcam | ab 196005 | BSA-TBST | 1:1000 |
| Cardiac Troponin I (phospho T143) antibody | Abcam | ab 58546 | BSA-TBST | 1:1000 |
| PKA C-α antibody | Cell Signaling | 4782 | BSA-TBST | 1:1000 |
| PKCα antibody | Cell Signaling | 2056 | BSA-TBST | 1:1000 |
| PKG antibody | Cell Signaling | 13511 | BSA-TBST | 1:1000 |
| Cardiac myosin binding protein C (phospho S282) antibody | Enzolife sciences | ALX-215-057-R050 | BSA-TBST | 1:500 |
| Glutathione | Abcam | ab19534 | BSA-TBST | 1:200 |
| Total cardiac myosin binding protein C antibody | Sigma-Aldrich | HPA040147 | BSA-TBST | 1:500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budde, H.; Hassoun, R.; Tangos, M.; Zhazykbayeva, S.; Herwig, M.; Varatnitskaya, M.; Sieme, M.; Delalat, S.; Sultana, I.; Kolijn, D.; et al. The Interplay between S-Glutathionylation and Phosphorylation of Cardiac Troponin I and Myosin Binding Protein C in End-Stage Human Failing Hearts. Antioxidants 2021, 10, 1134. https://doi.org/10.3390/antiox10071134
Budde H, Hassoun R, Tangos M, Zhazykbayeva S, Herwig M, Varatnitskaya M, Sieme M, Delalat S, Sultana I, Kolijn D, et al. The Interplay between S-Glutathionylation and Phosphorylation of Cardiac Troponin I and Myosin Binding Protein C in End-Stage Human Failing Hearts. Antioxidants. 2021; 10(7):1134. https://doi.org/10.3390/antiox10071134
Chicago/Turabian StyleBudde, Heidi, Roua Hassoun, Melina Tangos, Saltanat Zhazykbayeva, Melissa Herwig, Marharyta Varatnitskaya, Marcel Sieme, Simin Delalat, Innas Sultana, Detmar Kolijn, and et al. 2021. "The Interplay between S-Glutathionylation and Phosphorylation of Cardiac Troponin I and Myosin Binding Protein C in End-Stage Human Failing Hearts" Antioxidants 10, no. 7: 1134. https://doi.org/10.3390/antiox10071134
APA StyleBudde, H., Hassoun, R., Tangos, M., Zhazykbayeva, S., Herwig, M., Varatnitskaya, M., Sieme, M., Delalat, S., Sultana, I., Kolijn, D., Gömöri, K., Jarkas, M., Lódi, M., Jaquet, K., Kovács, Á., Mannherz, H. G., Sequeira, V., Mügge, A., Leichert, L. I., ... Hamdani, N. (2021). The Interplay between S-Glutathionylation and Phosphorylation of Cardiac Troponin I and Myosin Binding Protein C in End-Stage Human Failing Hearts. Antioxidants, 10(7), 1134. https://doi.org/10.3390/antiox10071134