Deletion of Superoxide Dismutase 1 Blunted Inflammatory Aortic Remodeling in Hypertensive Mice under Angiotensin II Infusion



Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
- WT 5′-TGAACCAGTTGTGTTGTCAGG-3′
- WT 5′- TCCATCACTGGTCACTAGCC-3′
- Mutant 5′- TGTTCTCCTCTTCCTCATCTCC-3′
- Mutant 5′- ACCCTTTCCAAATCCTCAGC-3′
2.2. AngII Infusion
2.3. VSMC Cell Culture
2.4. Measurement of SOD Activity
- Abs BL:
- Absorbance of the blind test (water)
- Abs BLBL:
- Absorbance of reagent blind test (water without XO)
- Abs S:
- Absorbance of sample test
- Abs SBL:
- Absorbance of sample blind test (sample without XO)
2.5. Measurement of Blood Pressure
2.6. Tissue Preparation and Histology
2.7. Measurement of Aorta Medial Area
2.8. Measurement of Extracellular and Intracellular H2O2 Levels
2.9. Western Blotting
2.10. Quantitative Real-Time PCR
3. Results
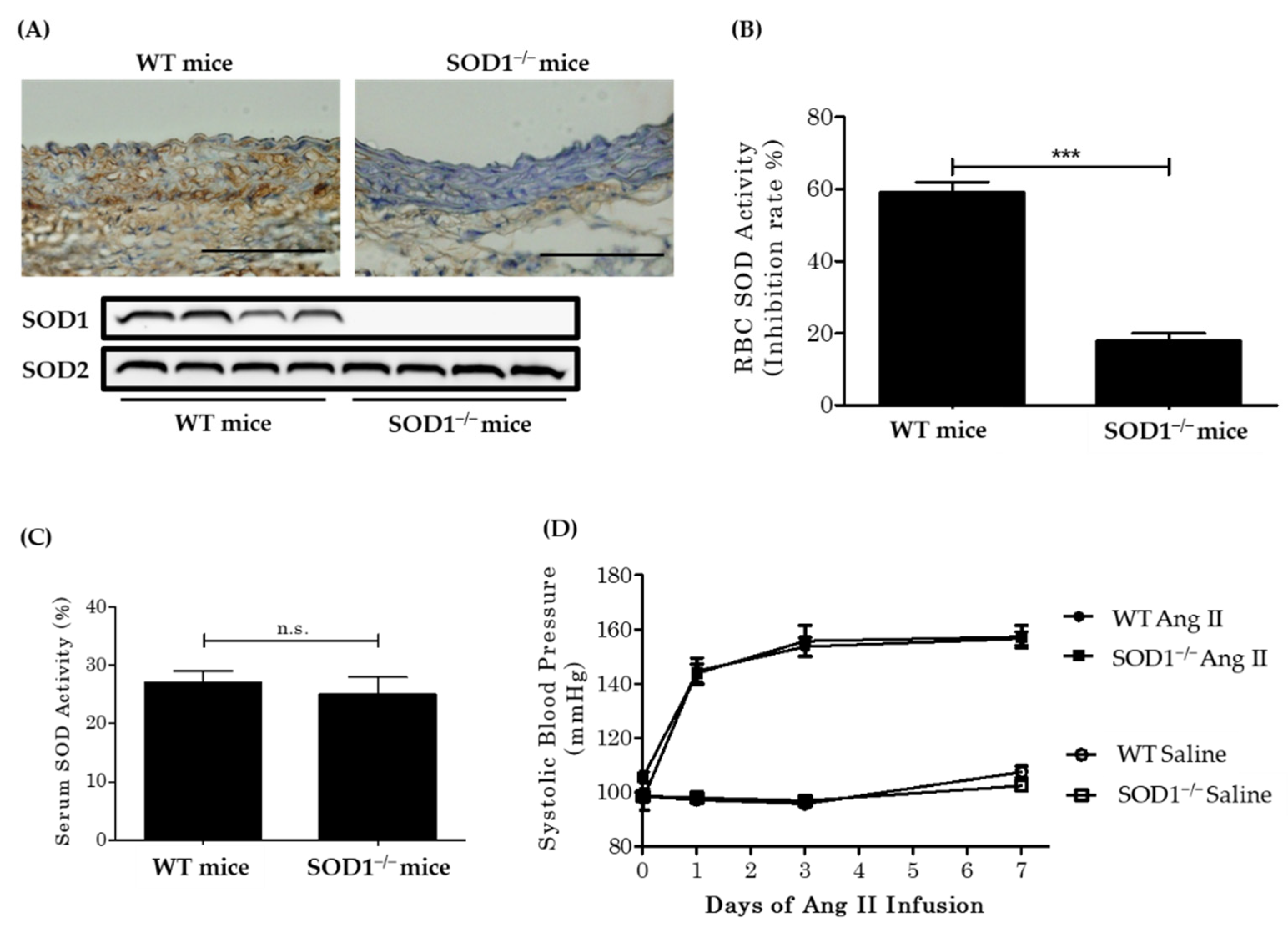
3.1. Baseline Characteristics and Blood Pressure
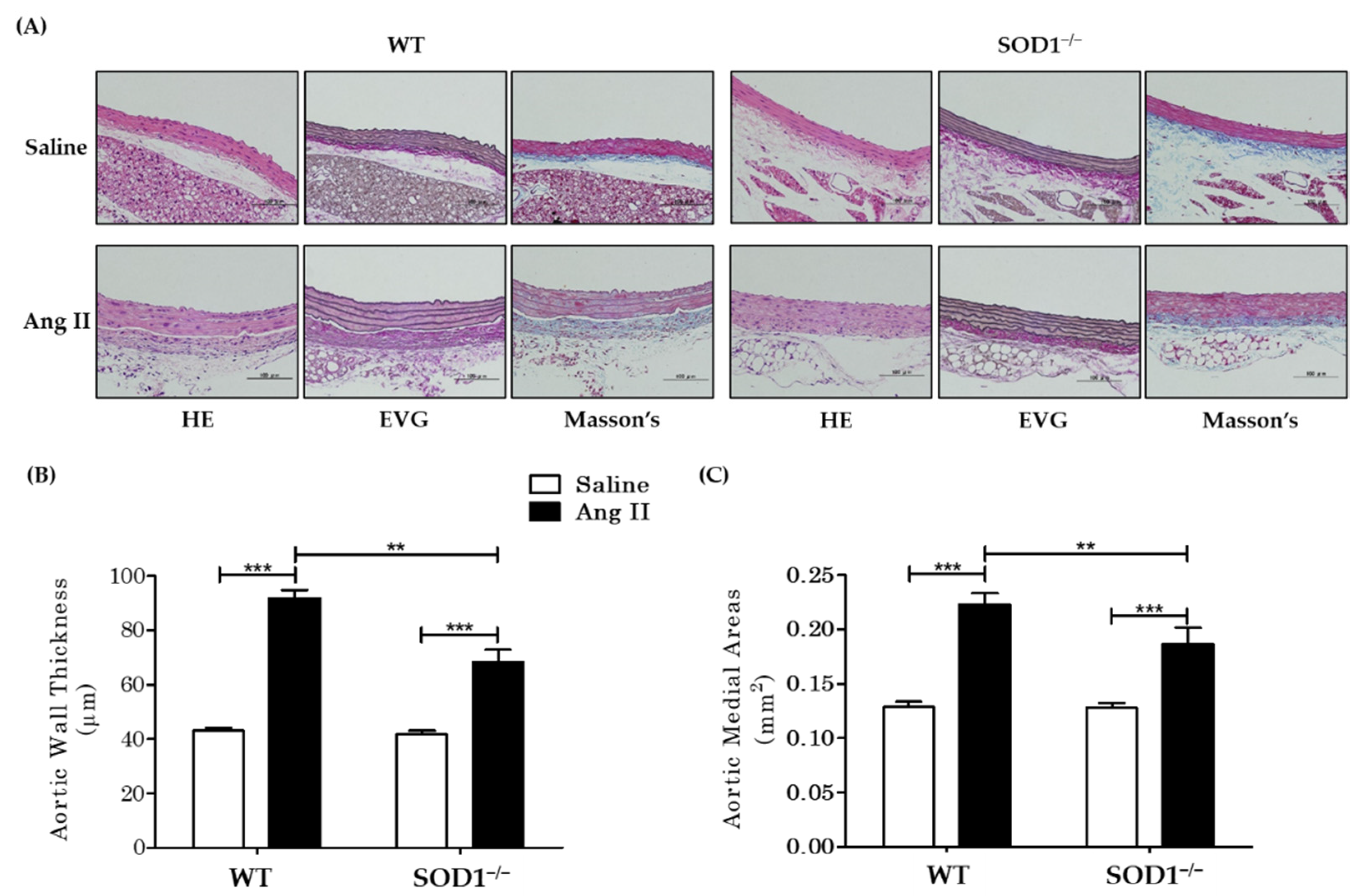
3.2. Histological and Morphometric Analysis
3.3. Extracellular and Intracellular H2O2 Levels In Vivo
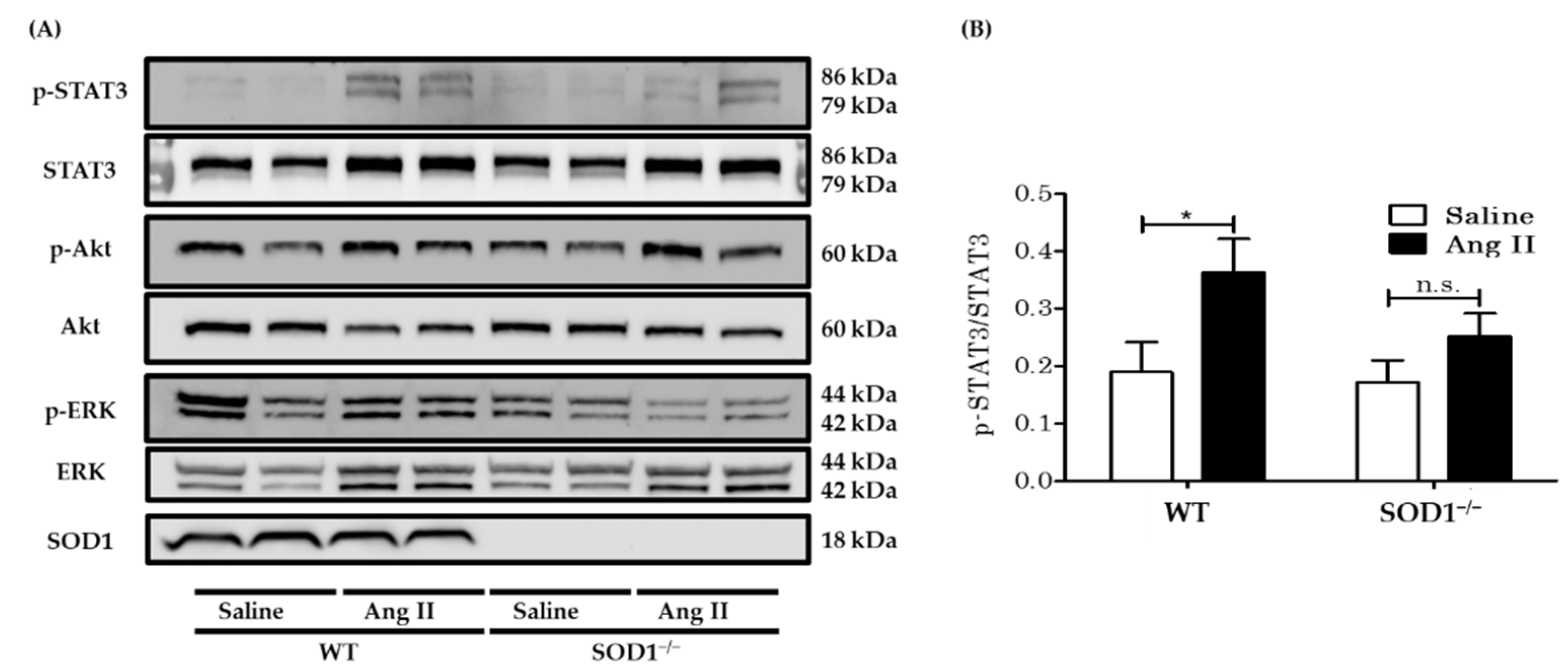
3.4. Western Blot Analysis for Downstream Signaling of AngII in Aortas In Vivo
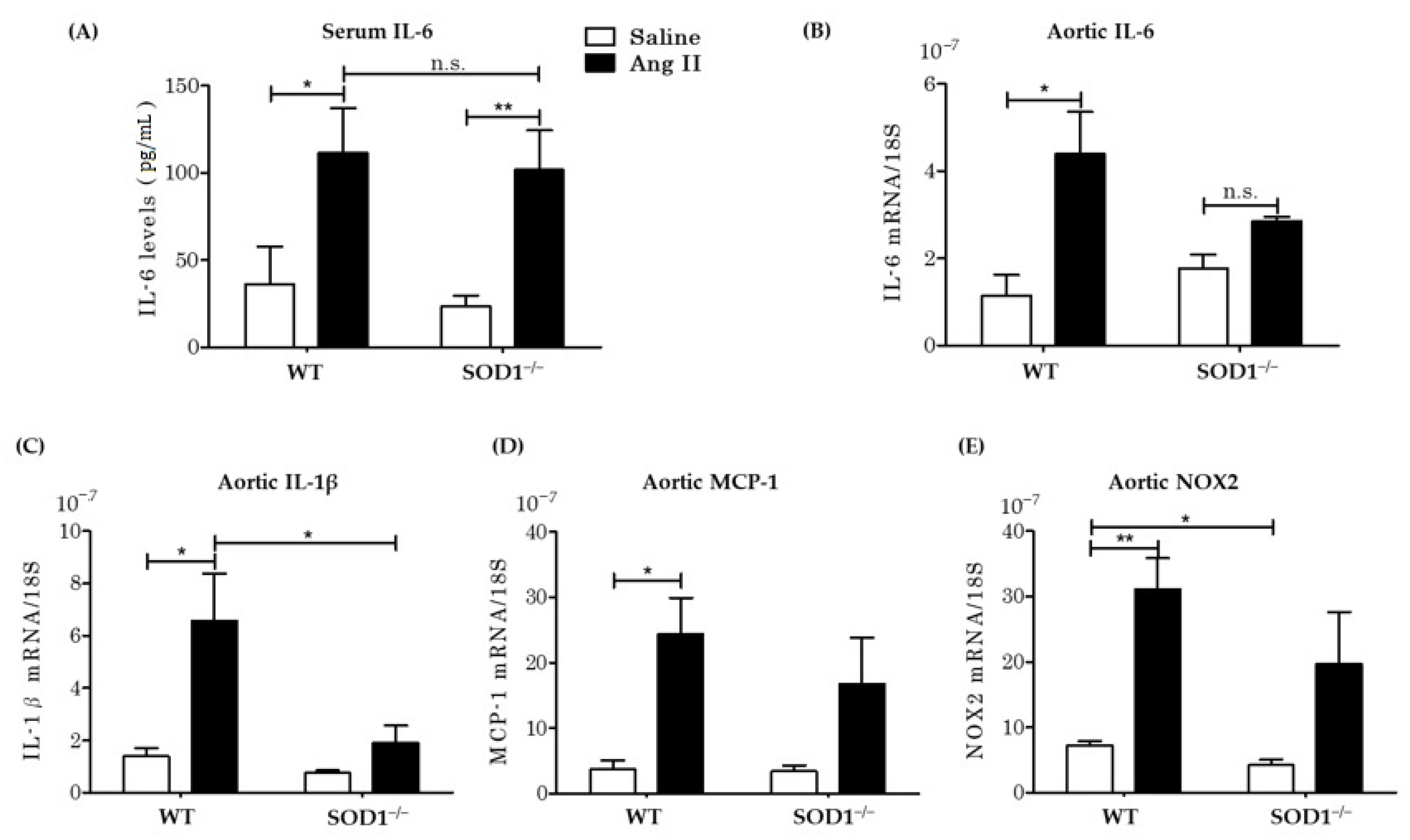
3.5. Serum Inflammatory Cytokines Levels and Aortic mRNA Expressions In Vivo
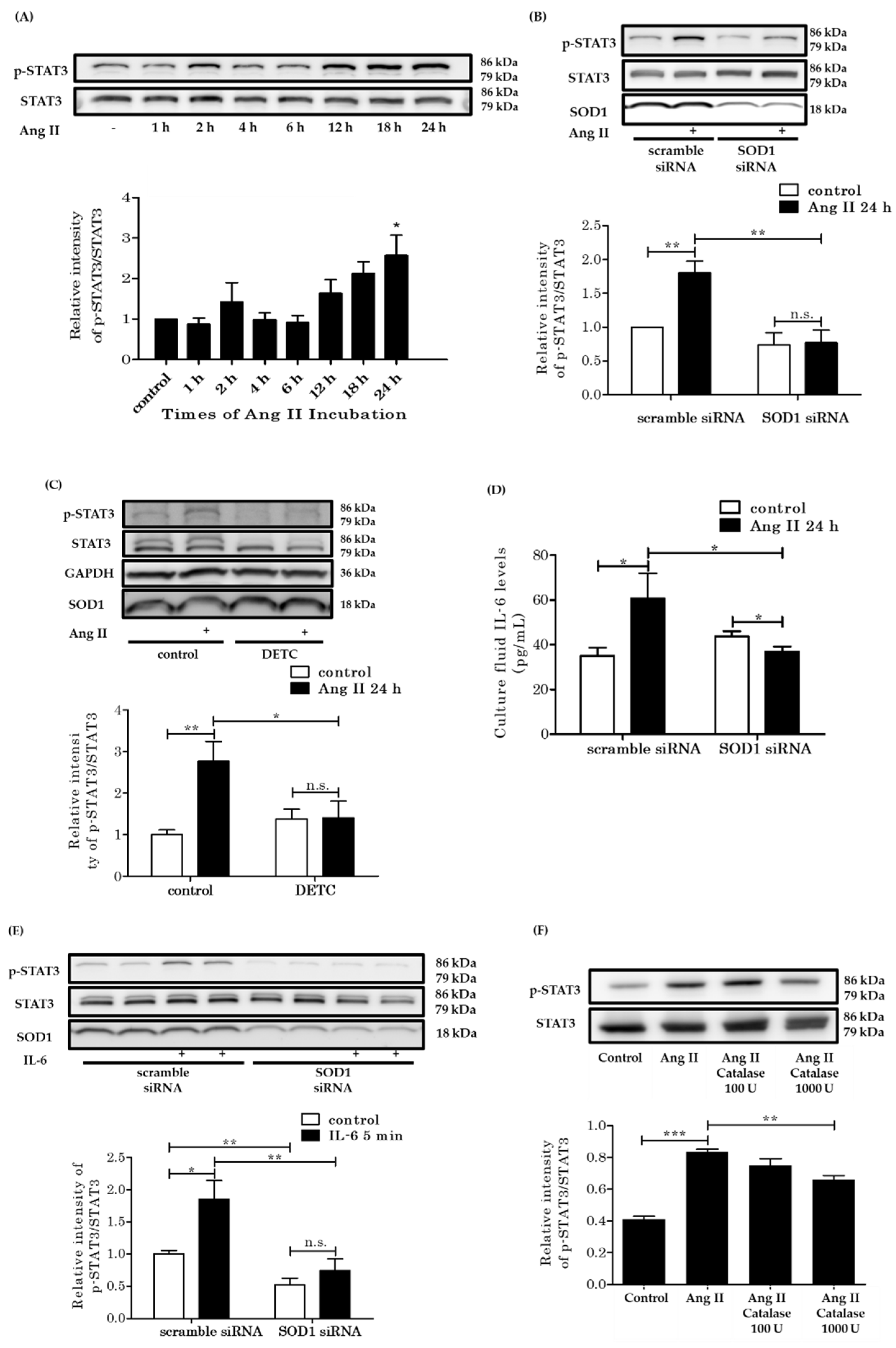
3.6. Rat VSMCs Incubated with AngII
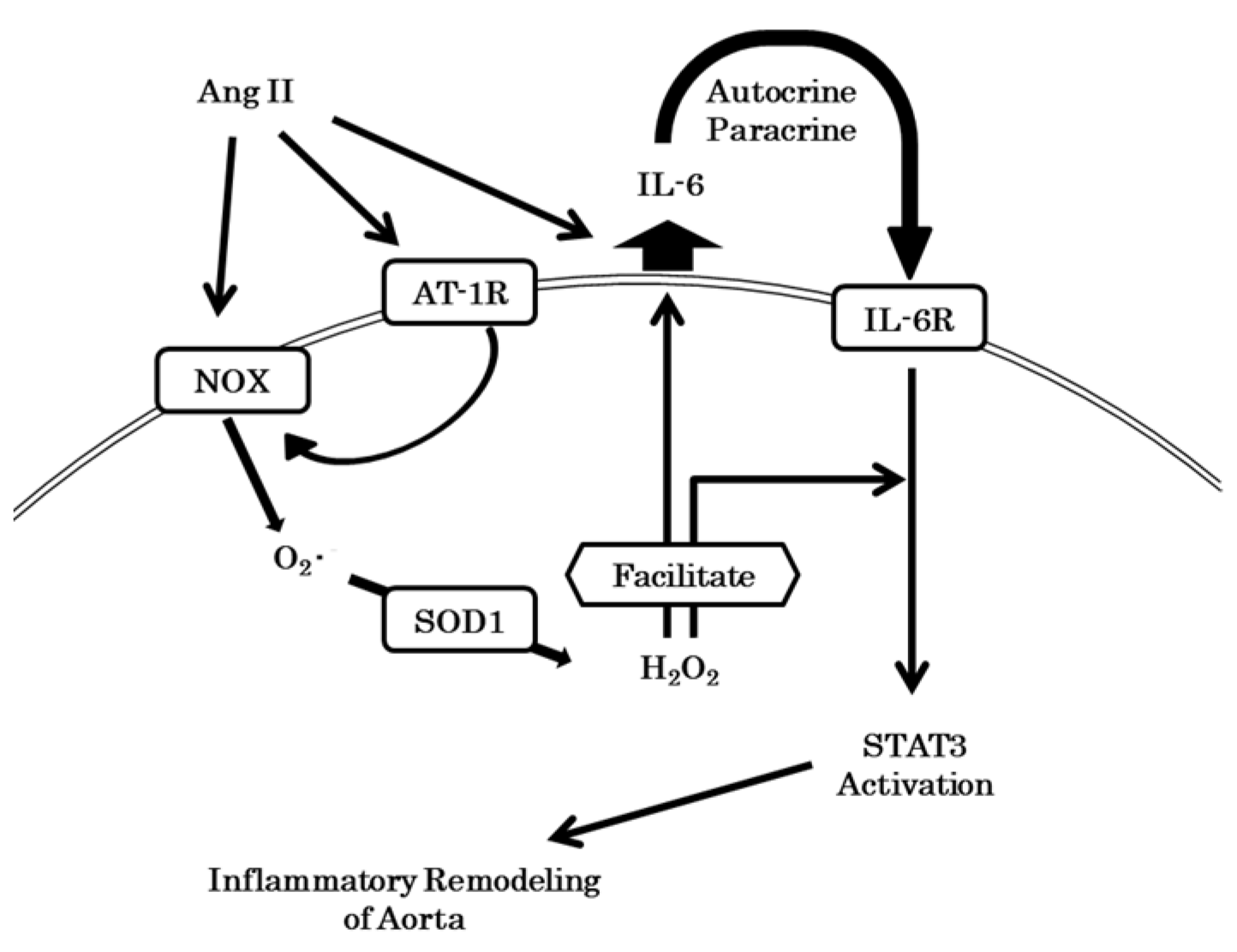
4. Discussion
4.1. SOD1 Changed ROS Levels
4.2. Hypertensive Response
4.3. Aortic Remodeling
4.4. Inflammation and IL-6 Production
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Z.; Xu, X.; Leng, X.; He, M.; Wang, J.; Cheng, S.; Wu, H. Roles of reactive oxygen species in cell signaling pathways and immune responses to viral infections. Arch. Virol. 2017, 162, 603–610. [Google Scholar] [CrossRef]
- Finkel, T. Oxygen radicals and signaling. Curr. Opin. Cell Biol. 1998, 10, 248–253. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Nguyen, D.C.A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Seshiah, P.N.; Weber, D.S.; Rocic, P.; Valppu, L.; Taniyama, Y.; Griendling, K.K. Angiotensin II stimulation of NAD(P)H oxidase activity: Upstream mediators. Circ. Res. 2002, 91, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Yao, G.; Schiffrin, E.L. c-Src induces phosphorylation and translocation of p47phox: Role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 981–987. [Google Scholar] [CrossRef]
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ. Res. 2002, 90, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Zafari, A.M.; Fukui, T.; Ishizaka, N.; Griendling, K.K. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J. Biol. Chem. 1996, 271, 23317–23321. [Google Scholar] [CrossRef]
- Ushio-Fukai, M. Localizing NADPH oxidase-derived ROS. Sci. STKE 2006, 2006, re8. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.J., Jr.; Filali, M.; Huss, G.J.; Stanic, B.; Chamseddine, A.; Barna, T.J.; Lamb, F.S. Cytokine activation of nuclear factor kappa B in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ. Res. 2007, 101, 663–671. [Google Scholar] [CrossRef]
- Chovanová, K.; Böhmer, M.; Poljovka, A.; Budiš, J.; Harichová, J.; Szemeš, T.; Zámocký, M. Parallel molecular evolution of catalases and superoxide dismutases—Focus on thermophilic fungal genomes. Antioxidants 2020, 9, 1047. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Zafari, A.M.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Shah, A.; Harrison, D.G.; Taylor, W.R.; Griendling, K.K. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension 1998, 32, 488–495. [Google Scholar] [CrossRef]
- Rao, G.N.; Berk, B.C. Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ. Res. 1992, 70, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Vekshtein, V.; Gordon, H.M.; Tsuda, T. Angiotensin II-stimulated protein synthesis in cultured vascular smooth muscle cells. Hypertension 1989, 13, 305–314. [Google Scholar] [CrossRef]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar] [CrossRef]
- Miller, A.F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef]
- Wakisaka, Y.; Chu, Y.; Miller, J.D.; Rosenberg, G.A.; Heistad, D.D. Critical role for copper/zinc-superoxide dismutase in preventing spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. Stroke 2010, 41, 790–797. [Google Scholar] [CrossRef]
- Baumbach, G.L.; Didion, S.P.; Faraci, F.M. Hypertrophy of cerebral arterioles in mice deficient in expression of the gene for CuZn superoxide dismutase. Stroke 2006, 37, 1850–1855. [Google Scholar] [CrossRef]
- Wang, H.D.; Johns, D.G.; Xu, S.; Cohen, R.A. Role of superoxide anion in regulating pressor and vascular hypertrophic response to angiotensin II. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1697–H1702. [Google Scholar] [CrossRef] [PubMed]
- Carlstrom, M.; Lai, E.Y.; Ma, Z.; Steege, A.; Patzak, A.; Eriksson, U.J.; Lundberg, J.O.; Wilcox, C.S.; Persson, A.E. Superoxide dismutase 1 limits renal microvascular remodeling and attenuates arteriole and blood pressure responses to angiotensin II via modulation of nitric oxide bioavailability. Hypertension 2010, 56, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Ryan, M.J.; Didion, L.A.; Fegan, P.E.; Sigmund, C.D.; Faraci, F.M. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ. Res. 2002, 91, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, N.; Isoda, K.; Adachi, T.; Niida, T.; Kujiraoka, T.; Hakuno, D.; Kondo, H.; Kusuhara, M.; Ohsuzu, F. Deficiency of CuZn superoxide dismutase promotes inflammation and alters medial structure following vascular injury. J. Atheroscler. Thromb. 2018, 18, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.D.; Xu, S.; Johns, D.G.; Du, Y.; Quinn, M.T.; Cayatte, A.J.; Cohen, R.A. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ. Res. 2001, 88, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.A.; Tzanov, T.; Filipa Carneiro, A.; Paar, A.; Gübitz, G.M.; Cavaco-Paulo, A. Studies of stabilization of native catalase using additives. Enzym. Microb. Technol. 2002, 30, 387–391. [Google Scholar] [CrossRef]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Endo, J.; Sano, M.; Katayama, T.; Hishiki, T.; Shinmura, K.; Morizane, S.; Matsuhashi, T.; Katsumata, Y.; Zhang, Y.; Ito, H.; et al. Metabolic remodeling induced by mitochondrial aldehyde stress stimulates tolerance to oxidative stress in the heart. Circ. Res. 2009, 105, 1118–1127. [Google Scholar] [CrossRef]
- Vassalle, C.; Boni, C.; Di Cecco, P.; Ndreu, R.; Zucchelli, G.C. Automation and validation of a fast method for the assessment of in vivo oxidative stress levels. Clin. Chem. Lab. Med. 2006, 44, 1372–1375. [Google Scholar] [CrossRef]
- Cornelli, U.; Terranova, R.; Luca, S.; Cornelli, M.; Alberti, A. Bioavailability and antioxidant activity of some food supplements in men and women using the D-Roms test as a marker of oxidative stress. J. Nutr. 2001, 131, 3208–3211. [Google Scholar] [CrossRef] [PubMed]
- Tavender, T.J.; Bulleid, N.J. Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J. Cell Sci. 2010, 123, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Ushio-Fukai, M.; Lassegue, B.; Alexander, R.W. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension 1997, 29, 366–373. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Yin, Q.; Fujio, Y.; Walsh, K.; Griendling, K.K. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 22699–22704. [Google Scholar] [CrossRef]
- Marrero, M.B.; Schieffer, B.; Paxton, W.G.; Heerdt, L.; Berk, B.C.; Delafontaine, P.; Bernstein, K.E. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 1995, 375, 247–250. [Google Scholar] [CrossRef]
- Eguchi, S.; Matsumoto, T.; Motley, E.D.; Utsunomiya, H.; Inagami, T. Identification of an essential signaling cascade for mitogen-activated protein kinase activation by angiotensin II in cultured rat vascular smooth muscle cells. Possible requirement of Gq-mediated p21ras activation coupled to a Ca2+/calmodulin-sensitive tyrosine kinase. J. Biol. Chem. 1996, 271, 14169–14175. [Google Scholar]
- Taniyama, Y.; Ushio-Fukai, M.; Hitomi, H.; Rocic, P.; Kingsley, M.J.; Pfahnl, C.; Weber, D.S.; Alexander, R.W.; Griendling, K.K. Role of p38 MAPK and MAPKAPK-2 in angiotensin II-induced Akt activation in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2004, 287, C494–C499. [Google Scholar] [CrossRef]
- Schieffer, B.; Luchtefeld, M.; Braun, S.; Hilfiker, A.; Hilfiker-Kleiner, D.; Drexler, H. Role of NAD(P)H oxidase in angiotensin II-induced JAK/STAT signaling and cytokine induction. Circ. Res. 2000, 87, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, B.; Schieffer, E.; Hilfiker-Kleiner, D.; Hilfiker, A.; Kovanen, P.T.; Kaartinen, M.; Nussberger, J.; Harringer, W.; Drexler, H. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: Potential implications for inflammation and plaque instability. Circulation 2000, 101, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Kranzhofer, R.; Schmidt, J.; Pfeiffer, C.A.; Hagl, S.; Libby, P.; Kubler, W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1623–1629. [Google Scholar] [CrossRef]
- Park, S.Y.; Ives, S.J.; Gifford, J.R.; Andtbacka, R.H.; Hyngstrom, J.R.; Reese, V.; Layec, G.; Bharath, L.P.; Symons, J.D.; Richardson, R.S. Impact of age on the vasodilatory function of human skeletal muscle feed arteries. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H217–H225. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Zhou, B.; Cox, A.D.; Campbell, S.L. Rho GTPases, oxidation, and cell redox control. Small GTPases 2014, 5, e28579. [Google Scholar] [CrossRef]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar] [CrossRef]
- Rao, R.K.; Clayton, L.W. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem. Biophys. Res. Commun. 2002, 293, 610–616. [Google Scholar] [CrossRef]
- Manea, S.-A.; Constantin, A.; Manda, G.; Sasson, S.; Manea, A. Regulation of Nox enzymes expression in vascular pathophysiology: Focusing on transcription factors and epigenetic mechanisms. Redox Biol. 2015, 5, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.M.; Griendling, K.K. NADPH oxidases and angiotensin II receptor signaling. Mol. Cell Endocrinol. 2009, 302, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, K.; Shimokawa, H.; Matoba, T.; Kubota, H.; Akaike, T.; Talukder, M.A.; Hatanaka, M.; Fujiki, T.; Maeda, H.; Takahashi, S.; et al. Pivotal role of Cu,Zn-superoxide dismutase in endothelium-dependent hyperpolarization. J. Clin. Investig. 2003, 112, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Gongora, M.C.; Qin, Z.; Laude, K.; Kim, H.W.; McCann, L.; Folz, J.R.; Dikalov, S.; Fukai, T.; Harrison, D.G. Role of extracellular superoxide dismutase in hypertension. Hypertension 2006, 48, 473–481. [Google Scholar] [CrossRef]
- Lob, H.E.; Vinh, A.; Li, L.; Blinder, Y.; Offermanns, S.; Harrison, D.G. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension 2011, 58, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Lob, H.E.; Marvar, P.J.; Guzik, T.J.; Sharma, S.; McCann, L.A.; Weyand, C.; Gordon, F.J.; Harrison, D.G. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension 2009, 55, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Iida, S.; Lund, D.D.; Weiss, R.M.; DiBona, G.F.; Watanabe, Y.; Faraci, F.M.; Heistad, D.D. Gene transfer of extracellular superoxide dismutase reduces arterial pressure in spontaneously hypertensive rats: Role of heparin-binding domain. Circ. Res. 2003, 92, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Zhang, W.; Lee, D.L.; Romero, M.J.; Nguyen, D.T.; Al-Shabrawey, M.; Tsai, N.T.; Liou, G.I.; Brands, M.W.; Caldwell, R.W.; et al. Role of IL-6 in angiotensin II-induced retinal vascular inflammation. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1709–1718. [Google Scholar] [CrossRef]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. 1998, 275, C1640–C1652. [Google Scholar] [CrossRef]
- Tieu, B.C.; Ju, X.; Lee, C.; Sun, H.; Lejeune, W.; Recinos, A., 3rd; Brasier, A.R.; Tilton, R.G. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J. Vasc. Res. 2011, 8, 261–272. [Google Scholar] [CrossRef]
- Tieu, B.C.; Lee, C.; Sun, H.; Lejeune, W.; Recinos, A., 3rd; Ju, X.; Spratt, H.; Guo, D.C.; Milewicz, D.; Tilton, R.G.; et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Investig. 2009, 119, 3637–3651. [Google Scholar] [CrossRef]
- Johns, C.; Gavras, I.; Handy, D.E.; Salomao, A.; Gavras, H. Models of experimental hypertension in mice. Hypertension 1996, 28, 1064–1069. [Google Scholar] [CrossRef]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef]
- Zhang, Y.; Griendling, K.K.; Dikalova, A.; Owens, G.K.; Taylor, W.R. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension 2005, 46, 732–737. [Google Scholar] [CrossRef]
- Higashi, M.; Shimokawa, H.; Hattori, T.; Hiroki, J.; Mukai, Y.; Morikawa, K.; Ichiki, T.; Takahashi, S.; Takeshita, A. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: Effect on endothelial NAD(P)H oxidase system. Circ. Res. 2003, 93, 767–775. [Google Scholar] [CrossRef]
- Adachi, T.; Pimentel, D.R.; Heibeck, T.; Hou, X.; Lee, Y.J.; Jiang, B.; Ido, Y.; Cohen, R.A. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 2004, 279, 29857–29862. [Google Scholar] [CrossRef]
- Frank, G.D.; Saito, S.; Motley, E.D.; Sasaki, T.; Ohba, M.; Kuroki, T.; Inagami, T.; Eguchi, S. Requirement of Ca2+ and PKCdelta for Janus kinase 2 activation by angiotensin II: Involvement of PYK2. Mol. Endocrinol. 2002, 16, 367–377. [Google Scholar] [PubMed]
- Madamanchi, N.R.; Li, S.; Patterson, C.; Runge, M.S. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 321–326. [Google Scholar] [CrossRef]
- Cheng, J.F.; Ni, G.H.; Chen, M.F.; Li, Y.J.; Wang, Y.J.; Wang, C.L.; Yuan, Q.; Shi, R.Z.; Hu, C.P.; Yang, T.L. Involvement of profilin-1 in angiotensin II-induced vascular smooth muscle cell proliferation. Vasc. Pharmacol. 2011, 55, 34–41. [Google Scholar] [CrossRef]
- Schrader, L.I.; Kinzenbaw, D.A.; Johnson, A.W.; Faraci, F.M.; Didion, S.P. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2576–2581. [Google Scholar] [CrossRef]
- Sano, M.; Fukuda, K.; Kodama, H.; Pan, J.; Saito, M.; Matsuzaki, J.; Takahashi, T.; Makino, S.; Kato, T.; Ogawa, S. Interleukin-6 family of cytokines mediate angiotensin II-induced cardiac hypertrophy in rodent cardiomyocytes. J. Biol. Chem. 2000, 275, 29717–29723. [Google Scholar] [CrossRef]
- Norlander, A.E.; Madhur, M.S.; Harrison, D.G. The immunology of hypertension. J. Exp. Med. 2018, 215, 21–33. [Google Scholar] [CrossRef]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef]
- Libermann, T.A.; Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell. Biol. 1990, 10, 2327–2334. [Google Scholar] [CrossRef]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-kappaB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef]
- Wormald, S.; Hilton, D.J. Inhibitors of cytokine signal transduction. J. Biol. Chem. 2004, 279, 821–824. [Google Scholar] [CrossRef]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Donate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence | Product Suze (bp) | |
|---|---|---|---|
| Il6 (IL-6) | Forward: | 5′-ACAACCACGGCCTTCCCTACTT-3′ | 129 |
| Reverse: | 5′-CACGATTTCCCAGAGAACATGTG-3′ | ||
| Ccl2 (MCP-1) | Forward: | 5′-CCACTCACCTGCTGCTACTCAT-3′ | 76 |
| Reverse: | 5′-TGGTGATCCTCTTGTAGCTCTCC-3′ | ||
| Il1b (IL-1beta) | Forward: | 5′-GCTGCTTCCAAACCTTTGAC-3′ | 118 |
| Reverse: | 5′-TTCTCCACAGCCACAATGAG-3′ | ||
| Nox1 | Forward: | 5′-CTACAGAAGAAGCCAACAGGCCAT-3′ | 117 |
| Reverse: | 5′-ACTGTCATGTTTGGAGACTGGATG-3′ | ||
| Cybb (Nox2) | Forward: | 5′-CCCTTTGGTACAGCCAGTGAAGAT-3′ | 67 |
| Reverse: | 5′-CAATCCCGGCTCCCACTAACATCA-3′ | ||
| Nox4 | Forward: | 5′-GGATCACAGAAGGTCCCTAGCAG-3′ | 113 |
| Reverse: | 5′-GCGGCTACATGCACACCTGAGAA-3′ | ||
| Rn18s (18S rRNA) | Forward: | 5′-TTCCGATAACGAACGAGACTCT-3′ | 99 |
| Reverse: | 5′-TGGCTGAACGCCACTTGTC-3′ | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiraishi, Y.; Ishigami, N.; Kujiraoka, T.; Sato, A.; Fujita, M.; Ido, Y.; Adachi, T. Deletion of Superoxide Dismutase 1 Blunted Inflammatory Aortic Remodeling in Hypertensive Mice under Angiotensin II Infusion. Antioxidants 2021, 10, 471. https://doi.org/10.3390/antiox10030471
Shiraishi Y, Ishigami N, Kujiraoka T, Sato A, Fujita M, Ido Y, Adachi T. Deletion of Superoxide Dismutase 1 Blunted Inflammatory Aortic Remodeling in Hypertensive Mice under Angiotensin II Infusion. Antioxidants. 2021; 10(3):471. https://doi.org/10.3390/antiox10030471
Chicago/Turabian StyleShiraishi, Yasunaga, Norio Ishigami, Takehiko Kujiraoka, Atsushi Sato, Masanori Fujita, Yasuo Ido, and Takeshi Adachi. 2021. "Deletion of Superoxide Dismutase 1 Blunted Inflammatory Aortic Remodeling in Hypertensive Mice under Angiotensin II Infusion" Antioxidants 10, no. 3: 471. https://doi.org/10.3390/antiox10030471
APA StyleShiraishi, Y., Ishigami, N., Kujiraoka, T., Sato, A., Fujita, M., Ido, Y., & Adachi, T. (2021). Deletion of Superoxide Dismutase 1 Blunted Inflammatory Aortic Remodeling in Hypertensive Mice under Angiotensin II Infusion. Antioxidants, 10(3), 471. https://doi.org/10.3390/antiox10030471