
Oxidative Stress and Vascular Damage in the Context of Obesity: The Hidden Guest

 ,
, 
Abstract
1. Introduction
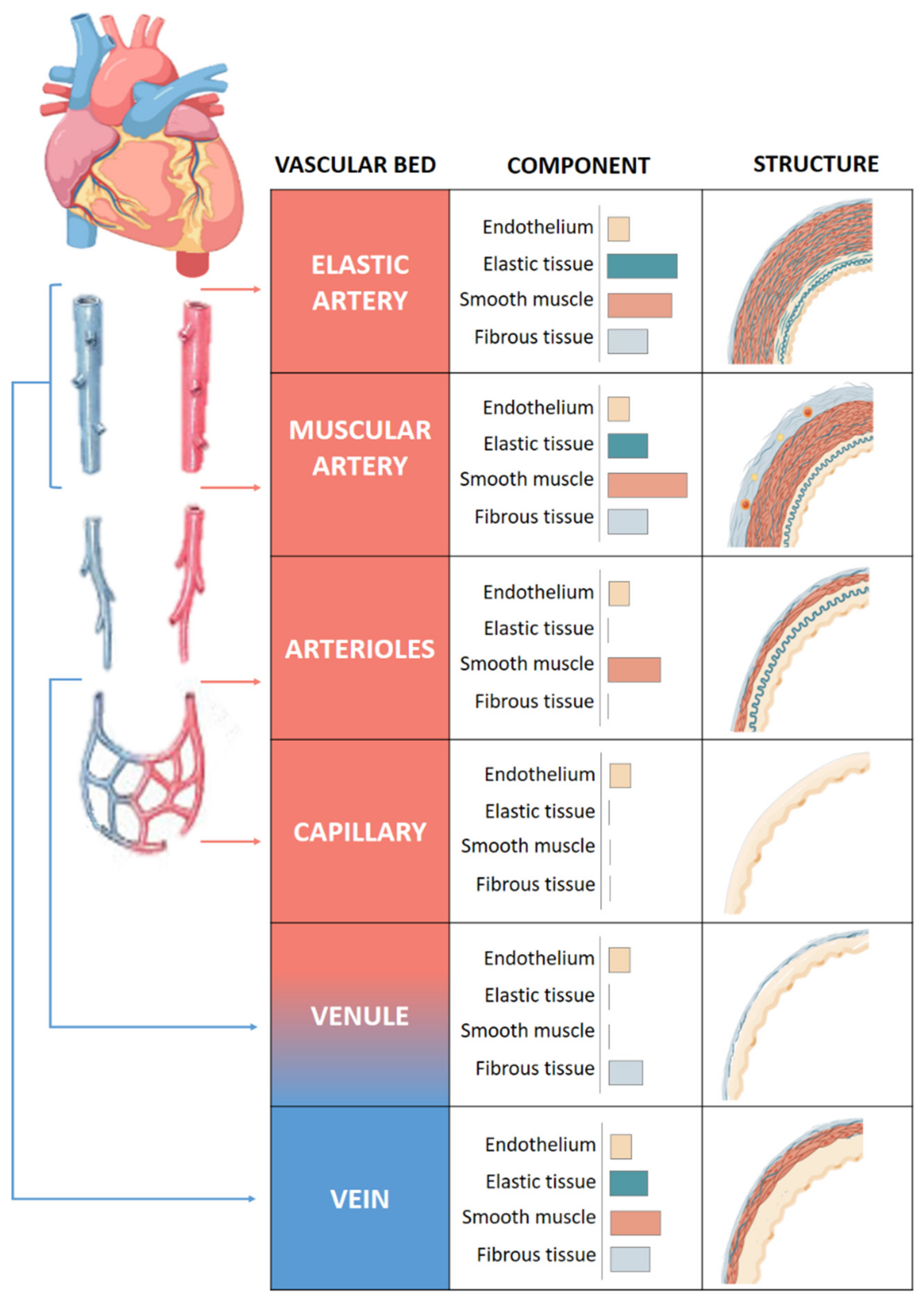
- The middle layer or media composed of elastic and muscular tissue which modulates the internal lumen of the vessel. This layer is mainly composed of vascular smooth muscle cells [5].
- The inner layer or intima, composed of endothelial cells that surrounds the interior of the vessel and provides an interface between the blood and vessel wall. These act as sensors for different stimuli, including mechanical (flow and pressure) and/or circulating humoral and inflammatory factors [6].
2. Vascular Remodeling in Obesity
- Hypertrophic involves thickening of the vascular wall due to cellular hyperplasia and/or hypertrophy or deposition of ECM, which determines an increase in wall-to-lumen ratio. This thickening can be inward or outward.
- Eutrophic involves changes in the diameter of the vessel without changes in the wall-to-lumen ratio.
- Hypotrophic involves thinning of wall and a reduction in wall-to-lumen ratio.
3. Endothelial Dysfunction in Obesity
4. Mechanisms Involved in Vascular Alterations Associated with Obesity
4.1. Perivascular Adipose Tissue
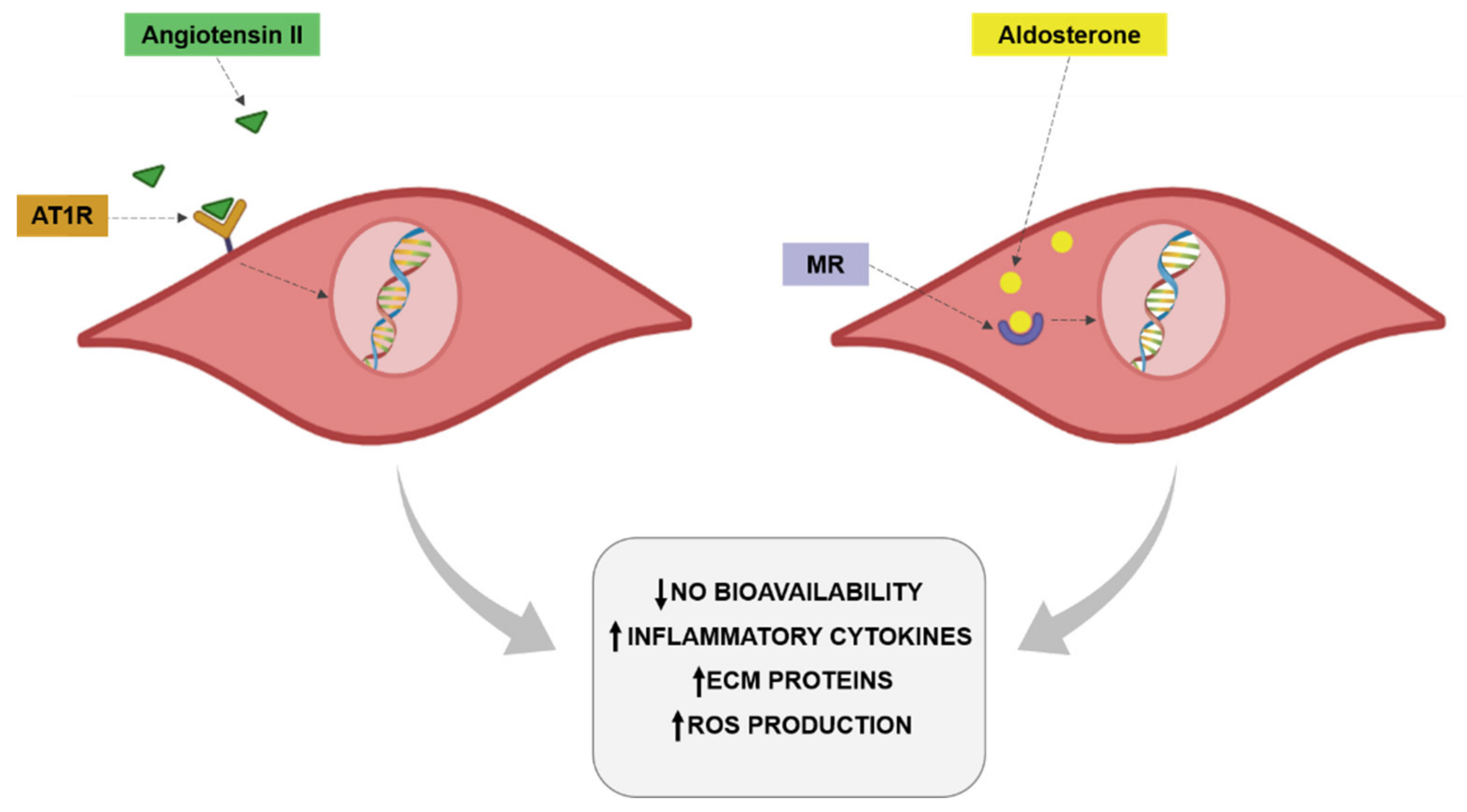
4.2. Renin-Angiotenisn-Aldosterone System
4.3. Endoplasmic Reticulum Stress
4.4. Central Role of Oxidative Stress in Vascular Alterations Associated with Obesity
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Witzleb, E. Functions of the Vascular System. In Human Phsysiology; Human Physiology; Springer: Berlin/Heidelberg, Germany, 1989; pp. 480–542. [Google Scholar] [CrossRef]
- Pugsley, M.K.; Tabrizchi, R. The vascular system. An overview of structure and function. J. Pharmacol. Toxicol. Methods 2000, 44, 333–340. [Google Scholar] [CrossRef]
- Gutterman, D.D. Adventitia-dependent influences on vascular function. Am. J. Physiol. 1999, 277, H1265–H1272. [Google Scholar] [CrossRef]
- Haurani, M.J.; Pagano, P.J. Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: Bellwether for vascular disease? Cardiovasc. Res. 2007, 75, 679–689. [Google Scholar] [CrossRef]
- Bacakova, L.; Travnickova, M.; Filova, E.; Matějka, R.; Stepanovska, J.; Musilkova, J.; Zarubova, J.; Molitor, M. The role of vascular smooth muscle cells in the physiology andpathophysiology of blood vessels. In Muscle Cell and Tissue—Current Status of Research Field; InTech Open: Rijeka, Croatia, 2018; pp. 229–257. [Google Scholar] [CrossRef]
- Feletou, M. The Endothelium: Part 1: Multiple Functions of the Endothelial Cells-Focus on Endothelium-Derived Vasoactive Mediators; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011. [Google Scholar]
- Rahman, M.; Siddik, A.B. Anatomy, Arterioles; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Caggiati, A.; Phillips, M.; Lametschwandtner, A.; Allegra, C. Valves in small veins and venules. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 2006, 32, 447–452. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Deighan, C.; Briones, A.M.; Shafaroudi, M.M.; McBride, M.; Adler, J.; Arribas, S.M.; Vila, E.; Daly, C.J. New aspects of vascular remodelling: The involvement of all vascular cell types. Exp. Physiol. 2005, 90, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.H.; Dzau, V.J. The emerging concept of vascular remodeling. N. Engl. J. Med. 1994, 330, 1431–1438. [Google Scholar] [CrossRef]
- Intengan, H.D.; Schiffrin, E.L. Vascular remodeling in hypertension: Roles of apoptosis, inflammation, and fibrosis. Hypertension 2001, 38, 581–587. [Google Scholar] [CrossRef]
- Knock, G.A. NADPH oxidase in the vasculature: Expression, regulation and signalling pathways; role in normal cardiovascular physiology and its dysregulation in hypertension. Free Radic. Biol. Med. 2019, 145, 385–427. [Google Scholar] [CrossRef]
- Labazi, H.; Trask, A.J. Coronary microvascular disease as an early culprit in the pathophysiology of diabetes and metabolic syndrome. Pharmacol. Res. 2017, 123, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Van Varik, B.J.; Rennenberg, R.J.; Reutelingsperger, C.P.; Kroon, A.A.; de Leeuw, P.W.; Schurgers, L.J. Mechanisms of arterial remodeling: Lessons from genetic diseases. Front. Genet. 2012, 3, 290. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Miana, M.; Jurado-Lopez, R.; Bartolome, M.V.; Souza Neto, F.V.; Salaices, M.; Lopez-Andres, N.; Cachofeiro, V. The potential role of leptin in the vascular remodeling associated with obesity. Int. J. Obes. 2014, 38, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Favero, G.; Lonati, C.; Giugno, L.; Castrezzati, S.; Rodella, L.F.; Rezzani, R. Obesity-related dysfunction of the aorta and prevention by melatonin treatment in ob/ob mice. Acta Histochem. 2013, 115, 783–788. [Google Scholar] [CrossRef]
- Briones, A.M.; Aras-Lopez, R.; Alonso, M.J.; Salaices, M. Small artery remodeling in obesity and insulin resistance. Curr. Vasc. Pharmacol. 2014, 12, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Gil-Ortega, M.; Martin-Ramos, M.; Arribas, S.M.; Gonzalez, M.C.; Aranguez, I.; Ruiz-Gayo, M.; Somoza, B.; Fernandez-Alfonso, M.S. Arterial stiffness is associated with adipokine dysregulation in non-hypertensive obese mice. Vasc. Pharmacol. 2016, 77, 38–47. [Google Scholar] [CrossRef]
- Aubert, C.E.; Floriani, C.; Bauer, D.C.; da Costa, B.R.; Segna, D.; Blum, M.R.; Collet, T.H.; Fink, H.A.; Cappola, A.R.; Syrogiannouli, L.; et al. Thyroid Function Tests in the Reference Range and Fracture: Individual Participant Analysis of Prospective Cohorts. J. Clin. Endocrinol. Metab. 2017, 102, 2719–2728. [Google Scholar] [CrossRef]
- Candela, J.; Velmurugan, G.V.; White, C. Hydrogen sulfide depletion contributes to microvascular remodeling in obesity. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1071–H1080. [Google Scholar] [CrossRef]
- Carroll, J.F.; Tyagi, S.C. Extracellular matrix remodeling in the heart of the homocysteinemic obese rabbit. Am. J. Hypertens. 2005, 18, 692–698. [Google Scholar] [CrossRef]
- Vinet, A.; Karpoff, L.; Walther, G.; Startun, A.; Obert, P.; Goret, L.; Dauzat, M.; Perez-Martin, A. Vascular reactivity at rest and during exercise in middle-aged obese men: Effects of short-term, low-intensity, exercise training. Int. J. Obes. 2011, 35, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Rizzoni, D.; Porteri, E.; De Ciuceis, C.; Sleiman, I.; Rodella, L.; Rezzani, R.; Paiardi, S.; Bianchi, R.; Ruggeri, G.; Boari, G.E.; et al. Effect of treatment with candesartan or enalapril on subcutaneous small artery structure in hypertensive patients with noninsulin-dependent diabetes mellitus. Hypertension 2005, 45, 659–665. [Google Scholar] [CrossRef]
- Elfimova, E.M.; Litvin, A.Y.; Chazova, I.E. The effectiveness of combination antihypertensive therapy in patients with arterial hypertension and additional risk factors: Obesity and obstructive sleep apnea syndrome. Ter. Arkhiv 2018, 90, 28–33. [Google Scholar] [CrossRef]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef]
- Soares, A.G.; de Carvalho, M.H.C.; Akamine, E. Obesity Induces Artery-Specific Alterations: Evaluation of Vascular Function and Inflammatory and Smooth Muscle Phenotypic Markers. Biomed Res. Int. 2017, 2017, 5038602. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Shi, Y.N.; Zhu, N.; Zhao, T.J.; Yi-Jie, G.; Liao, D.F.; Dai, A.G.; Qin, L. PVAT targets VSMCs to regulate vascular remodeling: Angel or demon. J. Drug Target. 2020, 1–38. [Google Scholar] [CrossRef]
- Zhang, M.J.; Zhou, Y.; Chen, L.; Wang, Y.Q.; Wang, X.; Pi, Y.; Gao, C.Y.; Li, J.C.; Zhang, L.L. An overview of potential molecular mechanisms involved in VSMC phenotypic modulation. Histochem. Cell Biol. 2016, 145, 119–130. [Google Scholar] [CrossRef]
- Beamish, J.A.; He, P.; Kottke-Marchant, K.; Marchant, R.E. Molecular regulation of contractile smooth muscle cell phenotype: Implications for vascular tissue engineering. Tissue Eng. Part B Rev. 2010, 16, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Bunbupha, S.; Apaijit, K.; Maneesai, P.; Prasarttong, P.; Pakdeechote, P. Nobiletin ameliorates high-fat diet-induced vascular and renal changes by reducing inflammation with modulating AdipoR1 and TGF-beta1 expression in rats. Life Sci. 2020, 260, 118398. [Google Scholar] [CrossRef]
- Sowers, J.R.; Habibi, J.; Aroor, A.R.; Yang, Y.; Lastra, G.; Hill, M.A.; Whaley-Connell, A.; Jaisser, F.; Jia, G. Epithelial sodium channels in endothelial cells mediate diet-induced endothelium stiffness and impaired vascular relaxation in obese female mice. Metab. Clin. Exp. 2019, 99, 57–66. [Google Scholar] [CrossRef]
- Bhatta, A.; Yao, L.; Xu, Z.; Toque, H.A.; Chen, J.; Atawia, R.T.; Fouda, A.Y.; Bagi, Z.; Lucas, R.; Caldwell, R.B.; et al. Obesity-induced vascular dysfunction and arterial stiffening requires endothelial cell arginase 1. Cardiovasc. Res. 2017, 113, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Leite, S.; Cerqueira, R.J.; Ibarrola, J.; Fontoura, D.; Fernandez-Celis, A.; Zannad, F.; Falcao-Pires, I.; Paulus, W.J.; Leite-Moreira, A.F.; Rossignol, P.; et al. Arterial Remodeling and Dysfunction in the ZSF1 Rat Model of Heart Failure with Preserved Ejection Fraction. Circ. Heart Fail. 2019, 12, e005596. [Google Scholar] [CrossRef]
- Parkin, J.D.; San Antonio, J.D.; Persikov, A.V.; Dagher, H.; Dalgleish, R.; Jensen, S.T.; Jeunemaitre, X.; Savige, J. The collalphagen III fibril has a “flexi-rod” structure of flexible sequences interspersed with rigid bioactive domains including two with hemostatic roles. PLoS ONE 2017, 12, e0175582. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Rodriguez, C.; Galan, M.; Miana, M.; Jurado-Lopez, R.; Bartolome, M.V.; Luaces, M.; Islas, F.; Martinez-Gonzalez, J.; Lopez-Andres, N.; et al. The lysyl oxidase inhibitor (beta-aminopropionitrile) reduces leptin profibrotic effects and ameliorates cardiovascular remodeling in diet-induced obesity in rats. J. Mol. Cell. Cardiol. 2016, 92, 96–104. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, J.; Varona, S.; Canes, L.; Galan, M.; Briones, A.M.; Cachofeiro, V.; Rodriguez, C. Emerging Roles of Lysyl Oxidases in the Cardiovascular System: New Concepts and Therapeutic Challenges. Biomolecules 2019, 9, 610. [Google Scholar] [CrossRef]
- Briones, A.M.; Gonzalez, J.M.; Somoza, B.; Giraldo, J.; Daly, C.J.; Vila, E.; Gonzalez, M.C.; McGrath, J.C.; Arribas, S.M. Role of elastin in spontaneously hypertensive rat small mesenteric artery remodelling. J. Physiol. 2003, 552, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.M.; Briones, A.M.; Somoza, B.; Daly, C.J.; Vila, E.; Starcher, B.; McGrath, J.C.; Gonzalez, M.C.; Arribas, S.M. Postnatal alterations in elastic fiber organization precede resistance artery narrowing in SHR. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H804–H812. [Google Scholar] [CrossRef][Green Version]
- Martinez-Martinez, E.; Miana, M.; Jurado-Lopez, R.; Rousseau, E.; Rossignol, P.; Zannad, F.; Cachofeiro, V.; Lopez-Andres, N. A role for soluble ST2 in vascular remodeling associated with obesity in rats. PLoS ONE 2013, 8, e79176. [Google Scholar] [CrossRef]
- Sista, A.K.; O’Connell, M.K.; Hinohara, T.; Oommen, S.S.; Fenster, B.E.; Glassford, A.J.; Schwartz, E.A.; Taylor, C.A.; Reaven, G.M.; Tsao, P.S. Increased aortic stiffness in the insulin-resistant Zucker fa/fa rat. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H845–H851. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bouissou, C.; Lacolley, P.; Dabire, H.; Safar, M.E.; Gabella, G.; Duchatelle, V.; Challande, P.; Bezie, Y. Increased stiffness and cell-matrix interactions of abdominal aorta in two experimental nonhypertensive models: Long-term chemically sympathectomized and sinoaortic denervated rats. J. Hypertens. 2014, 32, 652–658. [Google Scholar] [CrossRef]
- Sloboda, N.; Feve, B.; Thornton, S.N.; Nzietchueng, R.; Regnault, V.; Simon, G.; Labat, C.; Louis, H.; Max, J.P.; Muscat, A.; et al. Fatty acids impair endothelium-dependent vasorelaxation: A link between obesity and arterial stiffness in very old Zucker rats. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2012, 67, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.D.; DeVallance, E.; d’Audiffret, A.C.; Frisbee, S.J.; Tabone, L.E.; Shrader, C.D.; Frisbee, J.C.; Chantler, P.D. Metabolic syndrome impairs reactivity and wall mechanics of cerebral resistance arteries in obese Zucker rats. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1846–H1859. [Google Scholar] [CrossRef] [PubMed]
- Czippelova, B.; Turianikova, Z.; Krohova, J.; Wiszt, R.; Lazarova, Z.; Pozorciakova, K.; Ciljakova, M.; Javorka, M. Arterial Stiffness and Endothelial Function in Young Obese Patients—Vascular Resistance Matters. J. Atheroscler. Thromb. 2019, 26, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Galerneau, L.M.; Bailly, S.; Borel, J.C.; Jullian-Desayes, I.; Joyeux-Faure, M.; Benmerad, M.; Bonsignore, M.R.; Tamisier, R.; Pepin, J.L. Long-term variations of arterial stiffness in patients with obesity and obstructive sleep apnea treated with continuous positive airway pressure. PLoS ONE 2020, 15, e0236667. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; DeMarco, V.G.; Martinez-Lemus, L.A.; Meininger, G.A.; Sowers, J.R. Vascular stiffness in insulin resistance and obesity. Front. Physiol. 2015, 6, 231. [Google Scholar] [CrossRef] [PubMed]
- Schutten, J.C.; Joris, P.J.; Mensink, R.P.; Danel, R.M.; Goorman, F.; Heiner-Fokkema, M.R.; Weersma, R.K.; Keyzer, C.A.; de Borst, M.H.; Bakker, S.J.L. Effects of magnesium citrate, magnesium oxide and magnesium sulfate supplementation on arterial stiffness in healthy overweight individuals: A study protocol for a randomized controlled trial. Trials 2019, 20, 295. [Google Scholar] [CrossRef]
- Inoue, K.; Fujie, S.; Hasegawa, N.; Horii, N.; Uchida, M.; Iemitsu, K.; Sanada, K.; Hamaoka, T.; Iemitsu, M. Aerobic exercise training-induced irisin secretion is associated with the reduction of arterial stiffness via nitric oxide production in adults with obesity. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Metab. 2020, 45, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Bonarjee, V.V.S. Arterial Stiffness: A Prognostic Marker in Coronary Heart Disease. Available Methods and Clinical Application. Front. Cardiovasc. Med. 2018, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, H.; Matsumoto, C.; Shiina, K.; Yamashina, A. Brachial-Ankle PWV: Current Status and Future Directions as a Useful Marker in the Management of Cardiovascular Disease and/or Cardiovascular Risk Factors. J. Atheroscler. Thromb. 2016, 23, 128–146. [Google Scholar] [CrossRef] [PubMed]
- Speer, M.Y.; Giachelli, C.M. Regulation of cardiovascular calcification. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2004, 13, 63–70. [Google Scholar] [CrossRef]
- Schinzari, F.; Tesauro, M.; Bertoli, A.; Valentini, A.; Veneziani, A.; Campia, U.; Cardillo, C. Calcification biomarkers and vascular dysfunction in obesity and type 2 diabetes: Influence of oral hypoglycemic agents. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E658–E666. [Google Scholar] [CrossRef] [PubMed]
- Lins, D.D.C.; Gadelha, P.S.; Santa-Cruz, F.; Siqueira, L.T.; Campos, J.M.; Ferraz, A.A.B. Bariatric surgery and the coronary artery calcium score. Rev. Do Col. Bras. De Cir. 2019, 46, e20192170. [Google Scholar] [CrossRef]
- Carmo, L.S.; Burdmann, E.A.; Fessel, M.R.; Almeida, Y.E.; Pescatore, L.A.; Farias-Silva, E.; Gamarra, L.F.; Lopes, G.H.; Aloia, T.P.A.; Liberman, M. Expansive Vascular Remodeling and Increased Vascular Calcification Response to Cholecalciferol in a Murine Model of Obesity and Insulin Resistance. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 200–211. [Google Scholar] [CrossRef]
- Rios, R.; Raya, A.I.; Pineda, C.; Rodriguez, M.; Lopez, I.; Aguilera-Tejero, E. Vitamin E protects against extraskeletal calcification in uremic rats fed high fat diets. BMC Nephrol. 2017, 18, 374. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Ramirez, A.; Montes de Oca, A.; Raya, A.I.; Pineda, C.; Lopez, I.; Guerrero, F.; Diez, E.; Munoz-Castaneda, J.R.; Martinez, J.; Almaden, Y.; et al. Vitamin E protection of obesity-enhanced vascular calcification in uremic rats. Am. J. Physiol. Ren. Physiol. 2014, 306, F422–F429. [Google Scholar] [CrossRef]
- Sorop, O.; Olver, T.D.; van de Wouw, J.; Heinonen, I.; van Duin, R.W.; Duncker, D.J.; Merkus, D. The microcirculation: A key player in obesity-associated cardiovascular disease. Cardiovasc. Res. 2017, 113, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Paavonsalo, S.; Hariharan, S.; Lackman, M.H.; Karaman, S. Capillary Rarefaction in Obesity and Metabolic Diseases-Organ-Specificity and Possible Mechanisms. Cells 2020, 9, 2683. [Google Scholar] [CrossRef]
- Bagi, Z.; Feher, A.; Cassuto, J. Microvascular responsiveness in obesity: Implications for therapeutic intervention. Br. J. Pharmacol. 2012, 165, 544–560. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hao, H.; Leeper, N.J.; Zhu, L.; Early Career, C. Thrombotic Regulation From the Endothelial Cell Perspectives. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e90–e95. [Google Scholar] [CrossRef]
- Su, J.B. Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 2015, 7, 719–741. [Google Scholar] [CrossRef]
- Park, K.H.; Park, W.J. Endothelial Dysfunction: Clinical Implications in Cardiovascular Disease and Therapeutic Approaches. J. Korean Med. Sci. 2015, 30, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardiovasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef]
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and its alterations in cardiovascular diseases: Life style intervention. Biomed Res. Int. 2014, 2014, 801896. [Google Scholar] [CrossRef]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Luscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef]
- Virdis, A.; Ghiadoni, L.; Versari, D.; Giannarelli, C.; Salvetti, A.; Taddei, S. Endothelial function assessment in complicated hypertension. Curr. Pharm. Des. 2008, 14, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, M.; Fleming, I. The eNOS signalosome and its link to endothelial dysfunction. Pflug. Arch. Eur. J. Physiol. 2016, 468, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2018, 67, 391–405. [Google Scholar] [CrossRef]
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef]
- Virdis, A.; Masi, S.; Colucci, R.; Chiriaco, M.; Uliana, M.; Puxeddu, I.; Bernardini, N.; Blandizzi, C.; Taddei, S. Microvascular Endothelial Dysfunction in Patients with Obesity. Curr. Hypertens. Rep. 2019, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Onat, A. Metabolic syndrome: Nature, therapeutic solutions and options. Expert Opin. Pharmacother. 2011, 12, 1887–1900. [Google Scholar] [CrossRef]
- Sanchez, A.; Contreras, C.; Villalba, N.; Martinez, P.; Martinez, A.C.; Briones, A.; Salaices, M.; Garcia-Sacristan, A.; Hernandez, M.; Prieto, D. Altered arachidonic acid metabolism via COX-1 and COX-2 contributes to the endothelial dysfunction of penile arteries from obese Zucker rats. Br. J. Pharmacol. 2010, 159, 604–616. [Google Scholar] [CrossRef]
- Nishimatsu, H.; Suzuki, E.; Satonaka, H.; Takeda, R.; Omata, M.; Fujita, T.; Nagai, R.; Kitamura, T.; Hirata, Y. Endothelial dysfunction and hypercontractility of vascular myocytes are ameliorated by fluvastatin in obese Zucker rats. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1770–H1776. [Google Scholar] [CrossRef]
- Winters, B.; Mo, Z.; Brooks-Asplund, E.; Kim, S.; Shoukas, A.; Li, D.; Nyhan, D.; Berkowitz, D.E. Reduction of obesity, as induced by leptin, reverses endothelial dysfunction in obese (Lep(ob)) mice. J. Appl. Physiol. 2000, 89, 2382–2390. [Google Scholar] [CrossRef][Green Version]
- Munoz, M.; Lopez-Oliva, M.E.; Rodriguez, C.; Martinez, M.P.; Saenz-Medina, J.; Sanchez, A.; Climent, B.; Benedito, S.; Garcia-Sacristan, A.; Rivera, L.; et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2020, 28, 101330. [Google Scholar] [CrossRef]
- Oishi, J.C.; Castro, C.A.; Silva, K.A.; Fabricio, V.; Carnio, E.C.; Phillips, S.A.; Duarte, A.; Rodrigues, G.J. Endothelial Dysfunction and Inflammation Precedes Elevations in Blood Pressure Induced by a High-Fat Diet. Arq. Bras. Cardiol. 2018, 110, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Kobayasi, R.; Akamine, E.H.; Davel, A.P.; Rodrigues, M.A.; Carvalho, C.R.; Rossoni, L.V. Oxidative stress and inflammatory mediators contribute to endothelial dysfunction in high-fat diet-induced obesity in mice. J. Hypertens. 2010, 28, 2111–2119. [Google Scholar] [CrossRef]
- Majewski, M.; Jurgonski, A.; Fotschki, B.; Juskiewicz, J. The toxic effects of monosodium glutamate (MSG)—The involvement of nitric oxide, prostanoids and potassium channels in the reactivity of thoracic arteries in MSG-obese rats. Toxicol. Appl. Pharmacol. 2018, 359, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Leao, V.F.; Ferreira, L.; Melo, C.M.; Bonfleur, M.L.; da Silva, L.L.; Carneiro, E.M.; Raimundo, J.M.; Ribeiro, R.A. Taurine supplementation prevents endothelial dysfunction and attenuates structural changes in aortas from hypothalamic obese rats. Eur. J. Nutr. 2019, 58, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Dimassi, S.; Chahed, K.; Boumiza, S.; Canault, M.; Tabka, Z.; Laurant, P.; Riva, C. Role of eNOS- and NOX-containing microparticles in endothelial dysfunction in patients with obesity. Obesity 2016, 24, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Apovian, C.M.; Bigornia, S.; Mott, M.; Meyers, M.R.; Ulloor, J.; Gagua, M.; McDonnell, M.; Hess, D.; Joseph, L.; Gokce, N. Adipose macrophage infiltration is associated with insulin resistance and vascular endothelial dysfunction in obese subjects. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1654–1659. [Google Scholar] [CrossRef]
- Winkler, G.; Lakatos, P.; Salamon, F.; Nagy, Z.; Speer, G.; Kovacs, M.; Harmos, G.; Dworak, O.; Cseh, K. Elevated serum TNF-alpha level as a link between endothelial dysfunction and insulin resistance in normotensive obese patients. Diabet. Med. A J. Br. Diabet. Assoc. 1999, 16, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Ciotola, M.; Schisano, B.; Gualdiero, R.; Sardelli, L.; Misso, L.; Giannetti, G.; Giugliano, D. Endothelial microparticles correlate with endothelial dysfunction in obese women. J. Clin. Endocrinol. Metab. 2006, 91, 3676–3679. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Feher, A.; Davila, A.C.; Romero, M.J.; Patel, V.S.; Kamath, V.M.; Gooz, M.B.; Rudic, R.D.; Lucas, R.; Fulton, D.J.; et al. Role of Adipose Tissue Endothelial ADAM17 in Age-Related Coronary Microvascular Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Ortiz Segura, M.D.C.; Del Rio Navarro, B.E.; Rodriguez Espino, B.A.; Marchat, L.A.; Sanchez Munoz, F.; Villafana, S.; Hong, E.; Meza-Cuenca, F.; Mailloux Salinas, P.; Bolanos-Jimenez, F.; et al. Abnormality of adipokines and endothelial dysfunction in Mexican obese adolescents with insulin resistance. Endocr. Res. 2017, 42, 252–259. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Alotaibi, W.H.; Kheirandish-Gozal, L.; Capdevila, O.S.; Gozal, D. Endothelial dysfunction in obese non-hypertensive children without evidence of sleep disordered breathing. BMC Pediatrics 2010, 10, 8. [Google Scholar] [CrossRef]
- Valle Jimenez, M.; Estepa, R.M.; Camacho, R.M.; Estrada, R.C.; Luna, F.G.; Guitarte, F.B. Endothelial dysfunction is related to insulin resistance and inflammatory biomarker levels in obese prepubertal children. Eur. J. Endocrinol. 2007, 156, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Rosenstand, K.; Andersen, K.; Terp, R.; Gennemark, P.; Ellman, D.G.; Reznichenko, A.; Lambertsen, K.L.; Vanhoutte, P.M.; Hansen, P.B.L.; Svenningsen, P. Deficiency of T-type voltage-gated calcium channels results in attenuated weight gain and improved endothelium-dependent dilatation of resistance vessels induced by a high-fat diet in mice. J. Physiol. Biochem. 2020, 76, 135–145. [Google Scholar] [CrossRef]
- Villalba, N.; Martinez, P.; Briones, A.M.; Sanchez, A.; Salaices, M.; Garcia-Sacristan, A.; Hernandez, M.; Benedito, S.; Prieto, D. Differential structural and functional changes in penile and coronary arteries from obese Zucker rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H696–H707. [Google Scholar] [CrossRef]
- Shahidi, M.; Hashemi, S.R.; Fattahi, N.; Roshani, D.; Vahedi, S.; Sharifi, P.; Moradveisi, B. The Effects of L-Carnitine on Echocardiographic Changes in Patients With beta-Thalassemia Major and Intermedia. J. Pediatric Hematol. Oncol. 2020, 42, 386–390. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Y.; Zhang, R.; Pan, J.; Qi, D.; Wang, J.; Yang, X. The protective effects of walnut green husk polysaccharide on liver injury, vascular endothelial dysfunction and disorder of gut microbiota in high fructose-induced mice. Int. J. Biol. Macromol. 2020, 162, 92–106. [Google Scholar] [CrossRef]
- Roque, F.R.; Hernanz, R.; Salaices, M.; Briones, A.M. Exercise training and cardiometabolic diseases: Focus on the vascular system. Curr. Hypertens. Rep. 2013, 15, 204–214. [Google Scholar] [CrossRef]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef]
- Montani, J.P.; Carroll, J.F.; Dwyer, T.M.; Antic, V.; Yang, Z.; Dulloo, A.G. Ectopic fat storage in heart, blood vessels and kidneys in the pathogenesis of cardiovascular diseases. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 2004, 28 (Suppl. S4), S58–S65. [Google Scholar] [CrossRef]
- Brown, N.K.; Zhou, Z.; Zhang, J.; Zeng, R.; Wu, J.; Eitzman, D.T.; Chen, Y.E.; Chang, L. Perivascular adipose tissue in vascular function and disease: A review of current research and animal models. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1621–1630. [Google Scholar] [CrossRef]
- Soltis, E.E.; Cassis, L.A. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin. Exp. Hypertens. Part A Theory Pract. 1991, 13, 277–296. [Google Scholar] [CrossRef]
- Szasz, T.; Bomfim, G.F.; Webb, R.C. The influence of perivascular adipose tissue on vascular homeostasis. Vasc. Health Risk Manag. 2013, 9, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Gollasch, M.; Dubrovska, G. Paracrine role for periadventitial adipose tissue in the regulation of arterial tone. Trends Pharmacol. Sci. 2004, 25, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, S.; Meier, C.A. Local adipose tissue depots as cardiovascular risk factors. Cardiovasc. Res. 2007, 75, 690–701. [Google Scholar] [CrossRef]
- Omar, A.; Chatterjee, T.K.; Tang, Y.; Hui, D.Y.; Weintraub, N.L. Proinflammatory phenotype of perivascular adipocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1631–1636. [Google Scholar] [CrossRef]
- Gil-Ortega, M.; Somoza, B.; Huang, Y.; Gollasch, M.; Fernandez-Alfonso, M.S. Regional differences in perivascular adipose tissue impacting vascular homeostasis. Trends Endocrinol. Metab. Tem 2015, 26, 367–375. [Google Scholar] [CrossRef]
- Akoumianakis, I.; Antoniades, C. The interplay between adipose tissue and the cardiovascular system: Is fat always bad? Cardiovasc. Res. 2017, 113, 999–1008. [Google Scholar] [CrossRef]
- Fitzgibbons, T.P.; Czech, M.P. Epicardial and perivascular adipose tissues and their influence on cardiovascular disease: Basic mechanisms and clinical associations. J. Am. Heart Assoc. 2014, 3, e000582. [Google Scholar] [CrossRef]
- Agabiti-Rosei, C.; Paini, A.; De Ciuceis, C.; Withers, S.; Greenstein, A.; Heagerty, A.M.; Rizzoni, D. Modulation of Vascular Reactivity by Perivascular Adipose Tissue (PVAT). Curr. Hypertens. Rep. 2018, 20, 44. [Google Scholar] [CrossRef]
- Lohn, M.; Dubrovska, G.; Lauterbach, B.; Luft, F.C.; Gollasch, M.; Sharma, A.M. Periadventitial fat releases a vascular relaxing factor. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, G.; Verlohren, S.; Luft, F.C.; Gollasch, M. Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1107–H1113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fesus, G.; Dubrovska, G.; Gorzelniak, K.; Kluge, R.; Huang, Y.; Luft, F.C.; Gollasch, M. Adiponectin is a novel humoral vasodilator. Cardiovasc. Res. 2007, 75, 719–727. [Google Scholar] [CrossRef]
- Lu, C.; Su, L.Y.; Lee, R.M.; Gao, Y.J. Mechanisms for perivascular adipose tissue-mediated potentiation of vascular contraction to perivascular neuronal stimulation: The role of adipocyte-derived angiotensin II. Eur. J. Pharmacol. 2010, 634, 107–112. [Google Scholar] [CrossRef]
- Lu, C.; Zhao, A.X.; Gao, Y.J.; Lee, R.M. Modulation of vein function by perivascular adipose tissue. Eur. J. Pharmacol. 2011, 657, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.J.; Lu, C.; Su, L.Y.; Sharma, A.M.; Lee, R.M. Modulation of vascular function by perivascular adipose tissue: The role of endothelium and hydrogen peroxide. Br. J. Pharmacol. 2007, 151, 323–331. [Google Scholar] [CrossRef]
- Galvez-Prieto, B.; Somoza, B.; Gil-Ortega, M.; Garcia-Prieto, C.F.; de Las Heras, A.I.; Gonzalez, M.C.; Arribas, S.; Aranguez, I.; Bolbrinker, J.; Kreutz, R.; et al. Anticontractile Effect of Perivascular Adipose Tissue and Leptin are Reduced in Hypertension. Front Pharm. 2012, 3, 103. [Google Scholar] [CrossRef]
- Gil-Ortega, M.; Stucchi, P.; Guzman-Ruiz, R.; Cano, V.; Arribas, S.; Gonzalez, M.C.; Ruiz-Gayo, M.; Fernandez-Alfonso, M.S.; Somoza, B. Adaptative nitric oxide overproduction in perivascular adipose tissue during early diet-induced obesity. Endocrinology 2010, 151, 3299–3306. [Google Scholar] [CrossRef] [PubMed]
- Margaritis, M.; Antonopoulos, A.S.; Digby, J.; Lee, R.; Reilly, S.; Coutinho, P.; Shirodaria, C.; Sayeed, R.; Petrou, M.; De Silva, R.; et al. Interactions between vascular wall and perivascular adipose tissue reveal novel roles for adiponectin in the regulation of endothelial nitric oxide synthase function in human vessels. Circulation 2013, 127, 2209–2221. [Google Scholar] [CrossRef]
- Almabrouk, T.A.M.; White, A.D.; Ugusman, A.B.; Skiba, D.S.; Katwan, O.J.; Alganga, H.; Guzik, T.J.; Touyz, R.M.; Salt, I.P.; Kennedy, S. High Fat Diet Attenuates the Anticontractile Activity of Aortic PVAT via a Mechanism Involving AMPK and Reduced Adiponectin Secretion. Front. Physiol. 2018, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Lehman, S.J.; Massaro, J.M.; Schlett, C.L.; O’Donnell, C.J.; Hoffmann, U.; Fox, C.S. Peri-aortic fat, cardiovascular disease risk factors, and aortic calcification: The Framingham Heart Study. Atherosclerosis 2010, 210, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, A.S.; Khavandi, K.; Withers, S.B.; Sonoyama, K.; Clancy, O.; Jeziorska, M.; Laing, I.; Yates, A.P.; Pemberton, P.W.; Malik, R.A.; et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 2009, 119, 1661–1670. [Google Scholar] [CrossRef]
- Ketonen, J.; Shi, J.; Martonen, E.; Mervaala, E. Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese C57Bl/6 mice. Circ. J. Off. J. Jpn. Circ. Soc. 2010, 74, 1479–1487. [Google Scholar] [CrossRef]
- Ma, L.; Ma, S.; He, H.; Yang, D.; Chen, X.; Luo, Z.; Liu, D.; Zhu, Z. Perivascular fat-mediated vascular dysfunction and remodeling through the AMPK/mTOR pathway in high-fat diet-induced obese rats. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2010, 33, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Huang Cao, Z.F.; Stoffel, E.; Cohen, P. Role of Perivascular Adipose Tissue in Vascular Physiology and Pathology. Hypertension 2017, 69, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Sanna, F.; Sabharwal, N.; Thomas, S.; Oikonomou, E.K.; Herdman, L.; Margaritis, M.; Shirodaria, C.; Kampoli, A.M.; Akoumianakis, I.; et al. Detecting human coronary inflammation by imaging perivascular fat. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Payne, G.A.; Borbouse, L.; Kumar, S.; Neeb, Z.; Alloosh, M.; Sturek, M.; Tune, J.D. Epicardial perivascular adipose-derived leptin exacerbates coronary endothelial dysfunction in metabolic syndrome via a protein kinase C-beta pathway. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1711–1717. [Google Scholar] [CrossRef]
- Gil-Ortega, M.; Condezo-Hoyos, L.; Garcia-Prieto, C.F.; Arribas, S.M.; Gonzalez, M.C.; Aranguez, I.; Ruiz-Gayo, M.; Somoza, B.; Fernandez-Alfonso, M.S. Imbalance between pro and anti-oxidant mechanisms in perivascular adipose tissue aggravates long-term high-fat diet-derived endothelial dysfunction. PLoS ONE 2014, 9, e95312. [Google Scholar] [CrossRef]
- Briones, A.M.; Nguyen Dinh Cat, A.; Callera, G.E.; Yogi, A.; Burger, D.; He, Y.; Correa, J.W.; Gagnon, A.M.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: Implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension 2012, 59, 1069–1078. [Google Scholar] [CrossRef]
- Landgraf, K.; Friebe, D.; Ullrich, T.; Kratzsch, J.; Dittrich, K.; Herberth, G.; Adams, V.; Kiess, W.; Erbs, S.; Korner, A. Chemerin as a mediator between obesity and vascular inflammation in children. J. Clin. Endocrinol. Metab. 2012, 97, E556–E564. [Google Scholar] [CrossRef]
- Chatterjee, T.K.; Stoll, L.L.; Denning, G.M.; Harrelson, A.; Blomkalns, A.L.; Idelman, G.; Rothenberg, F.G.; Neltner, B.; Romig-Martin, S.A.; Dickson, E.W.; et al. Proinflammatory phenotype of perivascular adipocytes: Influence of high-fat feeding. Circ. Res. 2009, 104, 541–549. [Google Scholar] [CrossRef]
- Da Costa, R.M.; Fais, R.S.; Dechandt, C.R.P.; Louzada-Junior, P.; Alberici, L.C.; Lobato, N.S.; Tostes, R.C. Increased mitochondrial ROS generation mediates the loss of the anti-contractile effects of perivascular adipose tissue in high-fat diet obese mice. Br. J. Pharmacol. 2017, 174, 3527–3541. [Google Scholar] [CrossRef]
- Theccanat, T.; Philip, J.L.; Razzaque, A.M.; Ludmer, N.; Li, J.; Xu, X.; Akhter, S.A. Regulation of cellular oxidative stress and apoptosis by G protein-coupled receptor kinase-2; The role of NADPH oxidase 4. Cell Signal 2016, 28, 190–203. [Google Scholar] [CrossRef]
- Gonzalez-Amor, M.; Vila-Bedmar, R.; Rodrigues-Diez, R.; Moreno-Carriles, R.; Arcones, A.C.; Cruces-Sande, M.; Salaices, M.; Mayor, F., Jr.; Briones, A.M.; Murga, C. Myeloid GRK2 Regulates Obesity-Induced Endothelial Dysfunction by Modulating Inflammatory Responses in Perivascular Adipose Tissue. Antioxidants 2020, 9, 953. [Google Scholar] [CrossRef] [PubMed]
- Ozen, G.; Daci, A.; Norel, X.; Topal, G. Human perivascular adipose tissue dysfunction as a cause of vascular disease: Focus on vascular tone and wall remodeling. Eur. J. Pharmacol. 2015, 766, 16–24. [Google Scholar] [CrossRef]
- Verhagen, S.N.; Buijsrogge, M.P.; Vink, A.; van Herwerden, L.A.; van der Graaf, Y.; Visseren, F.L. Secretion of adipocytokines by perivascular adipose tissue near stenotic and non-stenotic coronary artery segments in patients undergoing CABG. Atherosclerosis 2014, 233, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Manrique, C.; Lastra, G.; Gardner, M.; Sowers, J.R. The renin angiotensin aldosterone system in hypertension: Roles of insulin resistance and oxidative stress. Med. Clin. North Am. 2009, 93, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E. The kidney, hypertension, and obesity. Hypertension 2003, 41, 625–633. [Google Scholar] [CrossRef]
- Cabandugama, P.K.; Gardner, M.J.; Sowers, J.R. The Renin Angiotensin Aldosterone System in Obesity and Hypertension: Roles in the Cardiorenal Metabolic Syndrome. Med. Clin. North Am. 2017, 101, 129–137. [Google Scholar] [CrossRef]
- Toyama, K.; Nakamura, T.; Kataoka, K.; Yasuda, O.; Fukuda, M.; Tokutomi, Y.; Dong, Y.F.; Ogawa, H.; Kim-Mitsuyama, S. Telmisartan protects against diabetic vascular complications in a mouse model of obesity and type 2 diabetes, partially through peroxisome proliferator activated receptor-gamma-dependent activity. Biochem. Biophys. Res. Commun. 2011, 410, 508–513. [Google Scholar] [CrossRef]
- Kagota, S.; Tada, Y.; Kubota, Y.; Nejime, N.; Yamaguchi, Y.; Nakamura, K.; Kunitomo, M.; Shinozuka, K. Peroxynitrite is Involved in the dysfunction of vasorelaxation in SHR/NDmcr-cp rats, spontaneously hypertensive obese rats. J. Cardiovasc. Pharmacol. 2007, 50, 677–685. [Google Scholar] [CrossRef]
- Kagota, S.; Fukushima, K.; Umetani, K.; Tada, Y.; Nejime, N.; Nakamura, K.; Mori, H.; Sugimura, K.; Kunitomo, M.; Shinozuka, K. Coronary vascular dysfunction promoted by oxidative-nitrative stress in SHRSP.Z-Lepr(fa) /IzmDmcr rats with metabolic syndrome. Clin. Exp. Pharmacol. Physiol. 2010, 37, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Ashcheulova, T.; Gerasimchuk, N.; Kovalyova, O.; Honchar, O. Beneficial effects of combined therapy with lacidipine and candesartan in obese hypertensive patients. Rom. J. Intern. Med. Rev. Roum. De Med. Interne 2018, 56, 257–264. [Google Scholar] [CrossRef]
- Duarte, J.; Martinez, A.; Bermejo, A.; Vera, B.; Gamez, M.J.; Cabo, P.; Zarzuelo, A. Cardiovascular effects of captopril and enalapril in obese Zucker rats. Eur. J. Pharmacol. 1999, 365, 225–232. [Google Scholar] [CrossRef]
- Russell, J.C.; Kelly, S.E.; Schafer, S. Vasopeptidase inhibition improves insulin sensitivity and endothelial function in the JCR:LA-cp rat. J. Cardiovasc. Pharmacol. 2004, 44, 258–265. [Google Scholar] [CrossRef]
- Oltman, C.L.; Davidson, E.P.; Coppey, L.J.; Kleinschmidt, T.L.; Lund, D.D.; Yorek, M.A. Attenuation of vascular/neural dysfunction in Zucker rats treated with enalapril or rosuvastatin. Obesity 2008, 16, 82–89. [Google Scholar] [CrossRef]
- Feher, A.; Cassuto, J.; Szabo, A.; Patel, V.; Vinayak Kamath, M.; Bagi, Z. Increased tissue angiotensin-converting enzyme activity impairs bradykinin-induced dilation of coronary arterioles in obesity. Circ. J. Off. J. Jpn. Circ. Soc. 2013, 77, 1867–1876. [Google Scholar] [CrossRef]
- Ashcheulova, T.; Gerasimchuk, N.; Rezunenko, Y.; Demydenko, G.; Kochubiei, O. Pathogenetic Advances of Fosinopril Sodium with Hydrochlorothiazide in Obese Hypertensive Patients. Georgian Med. News 2017, 271, 55–61. [Google Scholar]
- Zaman, A.K.; Fujii, S.; Goto, D.; Furumoto, T.; Mishima, T.; Nakai, Y.; Dong, J.; Imagawa, S.; Sobel, B.E.; Kitabatake, A. Salutary effects of attenuation of angiotensin II on coronary perivascular fibrosis associated with insulin resistance and obesity. J. Mol. Cell. Cardiol. 2004, 37, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Kappert, K.; Foryst-Ludwig, A.; Kramer, F.; Clemenz, M.; Grzesiak, A.; Sommerfeld, M.; Paul Frese, J.; Greiner, A.; Kintscher, U.; et al. AT1-receptor blockade attenuates outward aortic remodeling associated with diet-induced obesity in mice. Clin. Sci. 2017, 131, 1989–2005. [Google Scholar] [CrossRef]
- Savoia, C.; Touyz, R.M.; Endemann, D.H.; Pu, Q.; Ko, E.A.; De Ciuceis, C.; Schiffrin, E.L. Angiotensin receptor blocker added to previous antihypertensive agents on arteries of diabetic hypertensive patients. Hypertension 2006, 48, 271–277. [Google Scholar] [CrossRef]
- Rong, X.; Li, Y.; Ebihara, K.; Zhao, M.; Naowaboot, J.; Kusakabe, T.; Kuwahara, K.; Murray, M.; Nakao, K. Angiotensin II type 1 receptor-independent beneficial effects of telmisartan on dietary-induced obesity, insulin resistance and fatty liver in mice. Diabetologia 2010, 53, 1727–1731. [Google Scholar] [CrossRef][Green Version]
- De las Heras, N.; Martin-Fernandez, B.; Miana, M.; Ballesteros, S.; Oubina, M.P.; Lopez-Farre, A.J.; Cachofeiro, V.; Lahera, V. The protective effect of irbesartan in rats fed a high fat diet is associated with modification of leptin-adiponectin imbalance. J. Hypertens. Suppl. Off. J. Int. Soc. Hypertens. 2009, 27, S37–S41. [Google Scholar] [CrossRef] [PubMed]
- Loloi, J.; Miller, A.J.; Bingaman, S.S.; Silberman, Y.; Arnold, A.C. Angiotensin-(1-7) contributes to insulin-sensitizing effects of angiotensin-converting enzyme inhibition in obese mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E1204–E1211. [Google Scholar] [CrossRef]
- Paulis, L.; Foulquier, S.; Namsolleck, P.; Recarti, C.; Steckelings, U.M.; Unger, T. Combined Angiotensin Receptor Modulation in the Management of Cardio-Metabolic Disorders. Drugs 2016, 76, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Souza-Mello, V. Hepatic structural enhancement and insulin resistance amelioration due to AT1 receptor blockade. World J. Hepatol. 2017, 9, 74–79. [Google Scholar] [CrossRef]
- Saeed, S.; Waje-Andreassen, U.; Nilsson, P.M. The association of the metabolic syndrome with target organ damage: Focus on the heart, brain, and central arteries. Expert Rev. Cardiovasc. Ther. 2020, 18, 601–614. [Google Scholar] [CrossRef]
- Sposito, A.C.; Berwanger, O.; de Carvalho, L.S.F.; Saraiva, J.F.K. GLP-1RAs in type 2 diabetes: Mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovasc. Diabetol. 2018, 17, 157. [Google Scholar] [CrossRef]
- Graus-Nunes, F.; Souza-Mello, V. The renin-angiotensin system as a target to solve the riddle of endocrine pancreas homeostasis. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 109, 639–645. [Google Scholar] [CrossRef]
- McCurley, A.; Jaffe, I.Z. Mineralocorticoid receptors in vascular function and disease. Mol. Cell Endocrinol. 2012, 350, 256–265. [Google Scholar] [CrossRef]
- Feraco, A.; Marzolla, V.; Scuteri, A.; Armani, A.; Caprio, M. Mineralocorticoid Receptors in Metabolic Syndrome: From Physiology to Disease. Trends Endocrinol. Metab. 2020, 31, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Biwer, L.A.; Wallingford, M.C.; Jaffe, I.Z. Vascular Mineralocorticoid Receptor: Evolutionary Mediator of Wound Healing Turned Harmful by Our Modern Lifestyle. Am. J. Hypertens. 2019, 32, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Gorini, S.; Kim, S.K.; Infante, M.; Mammi, C.; La Vignera, S.; Fabbri, A.; Jaffe, I.Z.; Caprio, M. Role of Aldosterone and Mineralocorticoid Receptor in Cardiovascular Aging. Front. Endocrinol. 2019, 10, 584. [Google Scholar] [CrossRef]
- Dudenbostel, T.; Li, P.; Calhoun, D.A. Paradoxical increase of 24-hour urinary aldosterone levels in obese patients with resistant hypertension on a high salt diet. Am. J. Hypertens. 2020. [Google Scholar] [CrossRef]
- Gutierrez-Tenorio, J.; Marin-Royo, G.; Martinez-Martinez, E.; Martin, R.; Miana, M.; Lopez-Andres, N.; Jurado-Lopez, R.; Gallardo, I.; Luaces, M.; San Roman, J.A.; et al. The role of oxidative stress in the crosstalk between leptin and mineralocorticoid receptor in the cardiac fibrosis associated with obesity. Sci. Rep. 2017, 7, 16802. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Dinh Cat, A.; Callera, G.E.; Friederich-Persson, M.; Sanchez, A.; Dulak-Lis, M.G.; Tsiropoulou, S.; Montezano, A.C.; He, Y.; Briones, A.M.; Jaisser, F.; et al. Vascular dysfunction in obese diabetic db/db mice involves the interplay between aldosterone/mineralocorticoid receptor and Rho kinase signaling. Sci. Rep. 2018, 8, 2952. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Briones, A.M.; Callera, G.E.; Yogi, A.; He, Y.; Montezano, A.C.; Touyz, R.M. Adipocyte-derived factors regulate vascular smooth muscle cells through mineralocorticoid and glucocorticoid receptors. Hypertension 2011, 58, 479–488. [Google Scholar] [CrossRef]
- Gorini, S.; Marzolla, V.; Mammi, C.; Armani, A.; Caprio, M. Mineralocorticoid Receptor and Aldosterone-Related Biomarkers of End-Organ Damage in Cardiometabolic Disease. Biomolecules 2018, 8, 96. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Habibi, J.; Aroor, A.R.; Martinez-Lemus, L.A.; DeMarco, V.G.; Ramirez-Perez, F.I.; Sun, Z.; Hayden, M.R.; Meininger, G.A.; Mueller, K.B.; et al. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ. Res. 2016, 118, 935–943. [Google Scholar] [CrossRef]
- De Rita, O.; Hackam, D.G.; Spence, J.D. Effects of aldosterone on human atherosclerosis: Plasma aldosterone and progression of carotid plaque. Can. J. Cardiol. 2012, 28, 706–711. [Google Scholar] [CrossRef]
- Marzolla, V.; Armani, A.; Mammi, C.; Moss, M.E.; Pagliarini, V.; Pontecorvo, L.; Antelmi, A.; Fabbri, A.; Rosano, G.; Jaffe, I.Z.; et al. Essential role of ICAM-1 in aldosterone-induced atherosclerosis. Int. J. Cardiol. 2017, 232, 233–242. [Google Scholar] [CrossRef]
- Schafer, N.; Lohmann, C.; Winnik, S.; van Tits, L.J.; Miranda, M.X.; Vergopoulos, A.; Ruschitzka, F.; Nussberger, J.; Berger, S.; Luscher, T.F.; et al. Endothelial mineralocorticoid receptor activation mediates endothelial dysfunction in diet-induced obesity. Eur. Heart J. 2013, 34, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Huang, X.; Yan, Y.; Chen, J.; Wang, Z.; Xie, M.; Li, J. Rac1 is a possible link between obesity and oxidative stress in Chinese overweight adolescents. Obesity 2012, 20, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- McCurley, A.; Pires, P.W.; Bender, S.B.; Aronovitz, M.; Zhao, M.J.; Metzger, D.; Chambon, P.; Hill, M.A.; Dorrance, A.M.; Mendelsohn, M.E.; et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat. Med. 2012, 18, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.Z.; Morgan, J.; Tesch, G.H.; Rickard, A.J.; Chrissobolis, S.; Drummond, G.R.; Fuller, P.J.; Young, M.J. Cardiac Tissue Injury and Remodeling Is Dependent Upon MR Regulation of Activation Pathways in Cardiac Tissue Macrophages. Endocrinology 2016, 157, 3213–3223. [Google Scholar] [CrossRef]
- Michea, L.; Delpiano, A.M.; Hitschfeld, C.; Lobos, L.; Lavandero, S.; Marusic, E.T. Eplerenone blocks nongenomic effects of aldosterone on the Na+/H+ exchanger, intracellular Ca2+ levels, and vasoconstriction in mesenteric resistance vessels. Endocrinology 2005, 146, 973–980. [Google Scholar] [CrossRef]
- Lacolley, P.; Labat, C.; Pujol, A.; Delcayre, C.; Benetos, A.; Safar, M. Increased carotid wall elastic modulus and fibronectin in aldosterone-salt-treated rats: Effects of eplerenone. Circulation 2002, 106, 2848–2853. [Google Scholar] [CrossRef]
- DeMarco, V.G.; Habibi, J.; Jia, G.; Aroor, A.R.; Ramirez-Perez, F.I.; Martinez-Lemus, L.A.; Bender, S.B.; Garro, M.; Hayden, M.R.; Sun, Z.; et al. Low-Dose Mineralocorticoid Receptor Blockade Prevents Western Diet-Induced Arterial Stiffening in Female Mice. Hypertension 2015, 66, 99–107. [Google Scholar] [CrossRef]
- Aroor, A.R.; Habibi, J.; Nistala, R.; Ramirez-Perez, F.I.; Martinez-Lemus, L.A.; Jaffe, I.Z.; Sowers, J.R.; Jia, G.; Whaley-Connell, A. Diet-Induced Obesity Promotes Kidney Endothelial Stiffening and Fibrosis Dependent on the Endothelial Mineralocorticoid Receptor. Hypertension 2019, 73, 849–858. [Google Scholar] [CrossRef]
- O’Neill, H.; Lebeck, J.; Collins, P.B.; Kwon, T.H.; Frokiaer, J.; Nielsen, S. Aldosterone-mediated apical targeting of ENaC subunits is blunted in rats with streptozotocin-induced diabetes mellitus. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2008, 23, 1546–1555. [Google Scholar] [CrossRef]
- Catena, C.; Lapenna, R.; Baroselli, S.; Nadalini, E.; Colussi, G.; Novello, M.; Favret, G.; Melis, A.; Cavarape, A.; Sechi, L.A. Insulin sensitivity in patients with primary aldosteronism: A follow-up study. J. Clin. Endocrinol. Metab. 2006, 91, 3457–3463. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.M.; Meuth, A.I.; Davis, J.W.; Rector, R.S.; Bender, S.B. Mineralocorticoid receptor antagonism reverses diabetes-related coronary vasodilator dysfunction: A unique vascular transcriptomic signature. Pharmacol. Res. 2018, 134, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Vecchiola, A.; Fuentes, C.A.; Solar, I.; Lagos, C.F.; Opazo, M.C.; Munoz-Durango, N.; Riedel, C.A.; Owen, G.I.; Kalergis, A.M.; Fardella, C.E. Eplerenone Implantation Improved Adipose Dysfunction Averting RAAS Activation and Cell Division. Front. Endocrinol. 2020, 11, 223. [Google Scholar] [CrossRef] [PubMed]
- Kassan, M.; Galan, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef]
- Safiedeen, Z.; Andriantsitohaina, R.; Martinez, M.C. Dialogue between endoplasmic reticulum and mitochondria as a key actor of vascular dysfunction associated to metabolic disorders. Int. J. Biochem. Cell Biol. 2016, 77, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Galan, M.; Kassan, M.; Kadowitz, P.J.; Trebak, M.; Belmadani, S.; Matrougui, K. Mechanism of endoplasmic reticulum stress-induced vascular endothelial dysfunction. Biochim. Biophys. Acta 2014, 1843, 1063–1075. [Google Scholar] [CrossRef]
- Yao, Y.; Lu, Q.; Hu, Z.; Yu, Y.; Chen, Q.; Wang, Q.K. A non-canonical pathway regulates ER stress signaling and blocks ER stress-induced apoptosis and heart failure. Nat. Commun. 2017, 8, 133. [Google Scholar] [CrossRef]
- Cimellaro, A.; Perticone, M.; Fiorentino, T.V.; Sciacqua, A.; Hribal, M.L. Role of endoplasmic reticulum stress in endothelial dysfunction. Nutr. Metab. Cardiovasc. Dis. Nmcd 2016, 26, 863–871. [Google Scholar] [CrossRef]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef]
- Read, D.E.; Gupta, A.; Ladilov, Y.; Samali, A.; Gupta, S. miRNA signature of unfolded protein response in H9c2 rat cardiomyoblasts. Cell Biosci. 2014, 4, 56. [Google Scholar] [CrossRef]
- Choi, S.K.; Lim, M.; Yeon, S.I.; Lee, Y.H. Inhibition of endoplasmic reticulum stress improves coronary artery function in type 2 diabetic mice. Exp. Physiol. 2016, 101, 768–777. [Google Scholar] [CrossRef]
- Battson, M.L.; Lee, D.M.; Gentile, C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H355–H367. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cheng, J.; Chen, L.; Li, C.; Chen, G.; Gui, L.; Shen, B.; Zhang, Q. Endoplasmic reticulum stress involved in high-fat diet and palmitic acid-induced vascular damages and fenofibrate intervention. Biochem. Biophys. Res. Commun. 2015, 458, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Montagnani, M.; Chandrasekran, S.; Quon, M.J. Role of lipotoxicity in endothelial dysfunction. Heart Fail. Clin. 2012, 8, 589–607. [Google Scholar] [CrossRef]
- Luo, J.; Huang, L.; Wang, A.; Liu, Y.; Cai, R.; Li, W.; Zhou, M.S. Resistin-Induced Endoplasmic Reticulum Stress Contributes to the Impairment of Insulin Signaling in Endothelium. Front. Pharm. 2018, 9, 1226. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Yan, Z.; Zhu, Z. Mitochondria-Associated Endoplasmic Reticulum Membranes in Cardiovascular Diseases. Front. Cell Dev. Biol. 2020, 8, 604240. [Google Scholar] [CrossRef]
- Gorlach, A.; Klappa, P.; Kietzmann, T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals, antioxidants, and human disease: Curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Forstermann, U. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 338–349. [Google Scholar] [CrossRef]
- Marques, J.; Cortes, A.; Pejenaute, A.; Zalba, G. Implications of NADPH oxidase 5 in vascular diseases. Int. J. Biochem. Cell Biol. 2020, 128, 105851. [Google Scholar] [CrossRef]
- Yu, W.; Li, S.; Wu, H.; Hu, P.; Chen, L.; Zeng, C.; Tong, X. Endothelial Nox4 dysfunction aggravates atherosclerosis by inducing endoplasmic reticulum stress and soluble epoxide hydrolase. Free Radic. Biol. Med. 2021, 164, 44–57. [Google Scholar] [CrossRef]
- San Jose, G.; Moreno, M.U.; Olivan, S.; Beloqui, O.; Fortuno, A.; Diez, J.; Zalba, G. Functional effect of the p22phox -930A/G polymorphism on p22phox expression and NADPH oxidase activity in hypertension. Hypertension 2004, 44, 163–169. [Google Scholar] [CrossRef]
- Harrison, C.B.; Trevelin, S.C.; Richards, D.A.; Santos, C.X.C.; Sawyer, G.; Markovinovic, A.; Zhang, X.; Zhang, M.; Brewer, A.C.; Yin, X.; et al. Fibroblast Nox2 (NADPH Oxidase-2) Regulates ANG II (Angiotensin II)-Induced Vascular Remodeling and Hypertension via Paracrine Signaling to Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2020. [Google Scholar] [CrossRef]
- Rezende, F.; Moll, F.; Walter, M.; Helfinger, V.; Hahner, F.; Janetzko, P.; Ringel, C.; Weigert, A.; Fleming, I.; Weissmann, N.; et al. The NADPH organizers NoxO1 and p47phox are both mediators of diabetes-induced vascular dysfunction in mice. Redox Biol. 2018, 15, 12–21. [Google Scholar] [CrossRef]
- Chen, S.; Meng, X.F.; Zhang, C. Role of NADPH oxidase-mediated reactive oxygen species in podocyte injury. Biomed Res. Int. 2013, 2013, 839761. [Google Scholar] [CrossRef]
- Brandes, R.P.; Kreuzer, J. Vascular NADPH oxidases: Molecular mechanisms of activation. Cardiovasc. Res. 2005, 65, 16–27. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef]
- Lassegue, B.; Sorescu, D.; Szocs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef]
- Chen, X.L.; Zhang, Q.; Zhao, R.; Medford, R.M. Superoxide, H2O2, and iron are required for TNF-alpha-induced MCP-1 gene expression in endothelial cells: Role of Rac1 and NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1001–H1007. [Google Scholar] [CrossRef]
- DeVallance, E.; Li, Y.; Jurczak, M.J.; Cifuentes-Pagano, E.; Pagano, P.J. The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology. Antioxid. Redox Signal. 2019, 31, 687–709. [Google Scholar] [CrossRef]
- Silver, A.E.; Beske, S.D.; Christou, D.D.; Donato, A.J.; Moreau, K.L.; Eskurza, I.; Gates, P.E.; Seals, D.R. Overweight and obese humans demonstrate increased vascular endothelial NAD(P)H oxidase-p47(phox) expression and evidence of endothelial oxidative stress. Circulation 2007, 115, 627–637. [Google Scholar] [CrossRef]
- Kim, F.; Pham, M.; Maloney, E.; Rizzo, N.O.; Morton, G.J.; Wisse, B.E.; Kirk, E.A.; Chait, A.; Schwartz, M.W. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.H.; Vendrov, A.E.; Tchivilev, I.; Niu, X.L.; Molnar, K.C.; Rojas, M.; Carter, J.D.; Tong, H.; Stouffer, G.A.; Madamanchi, N.R.; et al. Mitochondrial oxidative stress in aortic stiffening with age: The role of smooth muscle cell function. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Siu, K.L.; Lob, H.E.; Itani, H.; Harrison, D.G.; Cai, H. Role of vascular oxidative stress in obesity and metabolic syndrome. Diabetes 2014, 63, 2344–2355. [Google Scholar] [CrossRef]
- Tam, H.K.; Kelly, A.S.; Metzig, A.M.; Steinberger, J.; Johnson, L.A. Xanthine oxidase and cardiovascular risk in obese children. Child. Obes. 2014, 10, 175–180. [Google Scholar] [CrossRef]
- Erdei, N.; Toth, A.; Pasztor, E.T.; Papp, Z.; Edes, I.; Koller, A.; Bagi, Z. High-fat diet-induced reduction in nitric oxide-dependent arteriolar dilation in rats: Role of xanthine oxidase-derived superoxide anion. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2107–H2115. [Google Scholar] [CrossRef] [PubMed]
- Abate, G.; Vezzoli, M.; Sandri, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Mitochondria and cellular redox state on the route from ageing to Alzheimer’s disease. Mech. Ageing Dev. 2020, 192, 111385. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Fukuto, J.M.; Miller, T.; Zhang, H.; Rinna, A.; Levy, S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch. Biochem. Biophys. 2008, 477, 183–195. [Google Scholar] [CrossRef]
- Frohnert, B.I.; Bernlohr, D.A. Protein carbonylation, mitochondrial dysfunction, and insulin resistance. Adv. Nutr. 2013, 4, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; Grune, T.; Speckmann, B. The two faces of reactive oxygen species (ROS) in adipocyte function and dysfunction. Biol. Chem. 2016, 397, 709–724. [Google Scholar] [CrossRef]
- Matsuda, M.; Shimomura, I. Roles of adiponectin and oxidative stress in obesity-associated metabolic and cardiovascular diseases. Rev. Endocr. Metab. Disord. 2014, 15, 1–10. [Google Scholar] [CrossRef]
- Maslov, L.N.; Naryzhnaya, N.V.; Boshchenko, A.A.; Popov, S.V.; Ivanov, V.V.; Oeltgen, P.R. Is oxidative stress of adipocytes a cause or a consequence of the metabolic syndrome? J. Clin. Transl. Endocrinol. 2019, 15, 1–5. [Google Scholar] [CrossRef]
- Le Lay, S.; Simard, G.; Martinez, M.C.; Andriantsitohaina, R. Oxidative stress and metabolic pathologies: From an adipocentric point of view. Oxidative Med. Cell. Longev. 2014, 2014, 908539. [Google Scholar] [CrossRef]
- Touyz, R.M. Reactive oxygen species and angiotensin II signaling in vascular cells: Implications in cardiovascular disease. Braz. J. Med Biol. Res. Rev. Bras. Pesqui. Med. Biol. 2004, 37, 1263–1273. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Lopez-Andres, N.; Jurado-Lopez, R.; Rousseau, E.; Bartolome, M.V.; Fernandez-Celis, A.; Rossignol, P.; Islas, F.; Antequera, A.; Prieto, S.; et al. Galectin-3 Participates in Cardiovascular Remodeling Associated With Obesity. Hypertension 2015, 66, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola, J.; Arrieta, V.; Sadaba, R.; Martinez-Martinez, E.; Garcia-Pena, A.; Alvarez, V.; Fernandez-Celis, A.; Gainza, A.; Santamaria, E.; Fernandez-Irigoyen, J.; et al. Galectin-3 down-regulates antioxidant peroxiredoxin-4 in human cardiac fibroblasts: A new pathway to induce cardiac damage. Clin. Sci. 2018, 132, 1471–1485. [Google Scholar] [CrossRef]
- Galili, O.; Versari, D.; Sattler, K.J.; Olson, M.L.; Mannheim, D.; McConnell, J.P.; Chade, A.R.; Lerman, L.O.; Lerman, A. Early experimental obesity is associated with coronary endothelial dysfunction and oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H904–H911. [Google Scholar] [CrossRef]
- Dunn, S.; Hilgers, R.H.; Das, K.C. Thioredoxin deficiency exacerbates vascular dysfunction during diet-induced obesity in small mesenteric artery in mice. Microcirculation 2020, e12674. [Google Scholar] [CrossRef]
- Neves, K.B.; Nguyen Dinh Cat, A.; Alves-Lopes, R.; Harvey, K.Y.; Costa, R.M.D.; Lobato, N.S.; Montezano, A.C.; Oliveira, A.M.; Touyz, R.M.; Tostes, R.C. Chemerin receptor blockade improves vascular function in diabetic obese mice via redox-sensitive and Akt-dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1851–H1860. [Google Scholar] [CrossRef]
- Silva, M.A.; Cau, S.B.; Lopes, R.A.; Manzato, C.P.; Neves, K.B.; Bruder-Nascimento, T.; Mestriner, F.L.; Montezano, A.C.; Nguyen Dinh Cat, A.; Touyz, R.M.; et al. Mineralocorticoid receptor blockade prevents vascular remodelling in a rodent model of type 2 diabetes mellitus. Clin. Sci. 2015, 129, 533–545. [Google Scholar] [CrossRef]
- DeVallance, E.; Branyan, K.W.; Lemaster, K.; Olfert, I.M.; Smith, D.M.; Pistilli, E.E.; Frisbee, J.C.; Chantler, P.D. Aortic dysfunction in metabolic syndrome mediated by perivascular adipose tissue TNFalpha- and NOX2-dependent pathway. Exp. Physiol. 2018, 103, 590–603. [Google Scholar] [CrossRef]
- Gonzaga, N.A.; Awata, W.M.C.; Ficher, S.P.; Assis, V.O.; Alves, J.V.; Tostes, R.C.; Tirapelli, C.R. Melatonin reverses the loss of the anticontractile effect of perivascular adipose tissue in obese rats. J. Pineal Res. 2020, e12710. [Google Scholar] [CrossRef]
- Agabiti-Rosei, C.; De Ciuceis, C.; Rossini, C.; Porteri, E.; Rodella, L.F.; Withers, S.B.; Heagerty, A.M.; Favero, G.; Agabiti-Rosei, E.; Rizzoni, D.; et al. Anticontractile activity of perivascular fat in obese mice and the effect of long-term treatment with melatonin. J. Hypertens. 2014, 32, 1264–1274. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Jurado-Lopez, R.; Valero-Munoz, M.; Bartolome, M.V.; Ballesteros, S.; Luaces, M.; Briones, A.M.; Lopez-Andres, N.; Miana, M.; Cachofeiro, V. Leptin induces cardiac fibrosis through galectin-3, mTOR and oxidative stress: Potential role in obesity. J. Hypertens. 2014, 32, 1104–1114. [Google Scholar] [CrossRef]
- Wong, W.T.; Tian, X.Y.; Xu, A.; Ng, C.F.; Lee, H.K.; Chen, Z.Y.; Au, C.L.; Yao, X.; Huang, Y. Angiotensin II type 1 receptor-dependent oxidative stress mediates endothelial dysfunction in type 2 diabetic mice. Antioxid. Redox Signal. 2010, 13, 757–768. [Google Scholar] [CrossRef]
- Ionica, M.; Aburel, O.M.; Vaduva, A.; Petrus, A.; Ratiu, S.; Olariu, S.; Sturza, A.; Muntean, D.M. Vitamin D alleviates oxidative stress in adipose tissue and mesenteric vessels from obese patients with subclinical inflammation. Can. J. Physiol. Pharmacol. 2020, 98, 85–92. [Google Scholar] [CrossRef]
- Lastra, G.; Manrique, C.; Jia, G.; Aroor, A.R.; Hayden, M.R.; Barron, B.J.; Niles, B.; Padilla, J.; Sowers, J.R. Xanthine oxidase inhibition protects against Western diet-induced aortic stiffness and impaired vasorelaxation in female mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R67–R77. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Bioactive Compounds | Effect | Reference |
|---|---|---|
| Acetylcholine | Vasodilator | [63,65,66,70,71] |
| Nitric oxide | Vasodilator | [63,64,65,70,71] |
| Bradykinin | Vasodilator | [65,66] |
| Prostacyclin | Vasodilator | [63,64,65] |
| Endothelium-Derived Hyperpolarizing Factor | Vasodilator | [63,64,65,70] |
| Endothelin-1 | vasoconstrictor | [63,64,65,70] |
| Thromboxane A2 | vasoconstrictor | [63] |
| Angiotensin II | vasoconstrictor | [63,64,70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Martínez, E.; Souza-Neto, F.V.; Jiménez-González, S.; Cachofeiro, V. Oxidative Stress and Vascular Damage in the Context of Obesity: The Hidden Guest. Antioxidants 2021, 10, 406. https://doi.org/10.3390/antiox10030406
Martínez-Martínez E, Souza-Neto FV, Jiménez-González S, Cachofeiro V. Oxidative Stress and Vascular Damage in the Context of Obesity: The Hidden Guest. Antioxidants. 2021; 10(3):406. https://doi.org/10.3390/antiox10030406
Chicago/Turabian StyleMartínez-Martínez, Ernesto, Francisco V. Souza-Neto, Sara Jiménez-González, and Victoria Cachofeiro. 2021. "Oxidative Stress and Vascular Damage in the Context of Obesity: The Hidden Guest" Antioxidants 10, no. 3: 406. https://doi.org/10.3390/antiox10030406
APA StyleMartínez-Martínez, E., Souza-Neto, F. V., Jiménez-González, S., & Cachofeiro, V. (2021). Oxidative Stress and Vascular Damage in the Context of Obesity: The Hidden Guest. Antioxidants, 10(3), 406. https://doi.org/10.3390/antiox10030406