
ACE Inhibition Modulates Myeloid Hematopoiesis after Acute Myocardial Infarction and Reduces Cardiac and Vascular Inflammation in Ischemic Heart Failure

 ,
,  ,
,  , and
, and 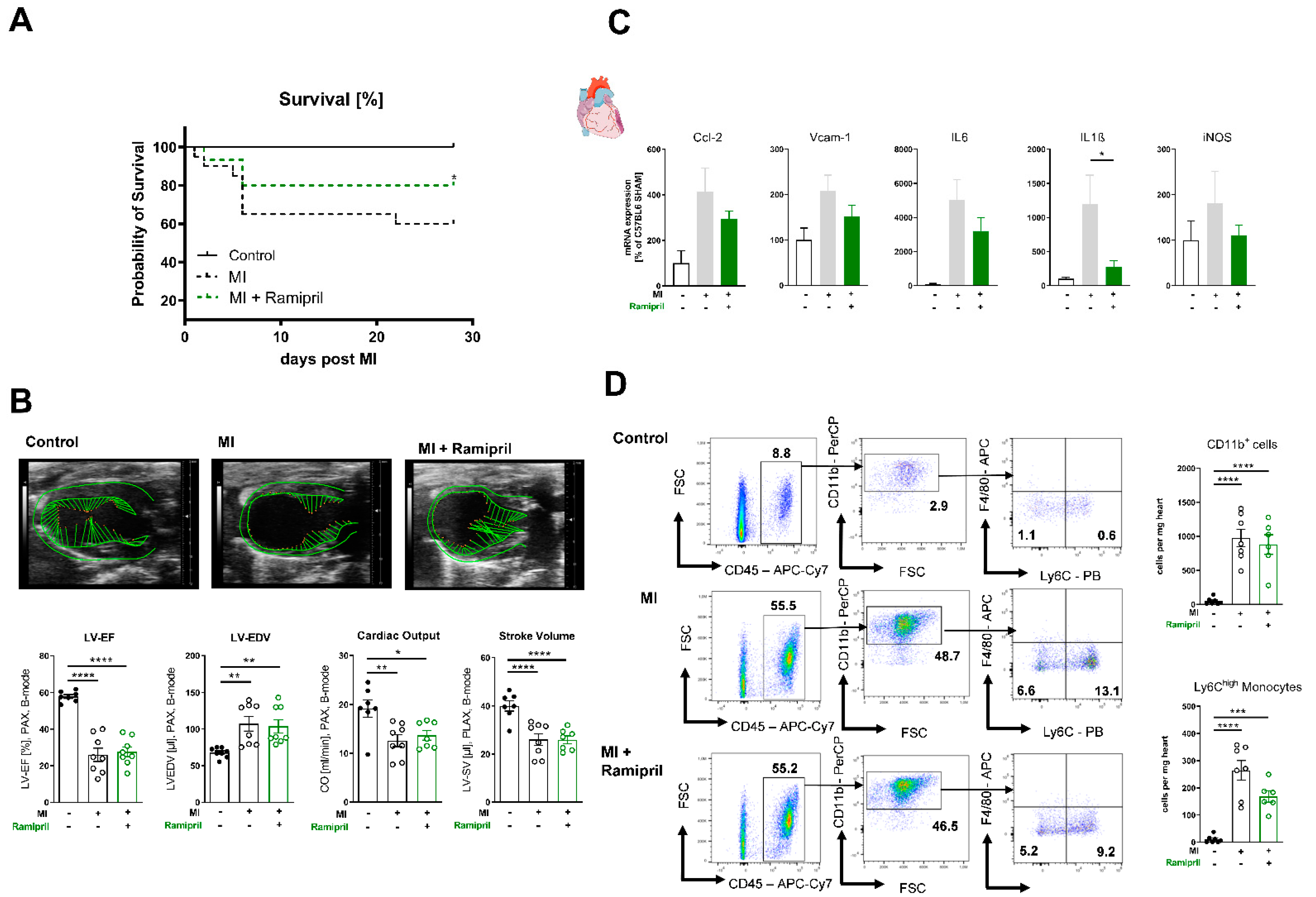{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Chemicals
2.2. Ethics Approval
2.3. Animals and In Vivo Treatment
2.4. Small Animal Echocardiography
2.5. Flow Cytometry Analysis of Immune Cells
2.6. Processing of Heart Tissue, Bone Marrow, Spleen and Aortic Vessels
2.7. Cell Staining
2.8. mRNA Expression Analysis
2.9. Enhanced Chemiluminescence
2.10. Flourescence Oxidative Microtopography
2.11. Vascular Tone Experiments
2.12. Statistical Analysis
3. Results
3.1. Immediate ACE Inhibition Post-MI Limits Infiltration of Inflammatory Monocytes in the Ischemic Myocardium due to Reduced Expression of Adhesion Molecules
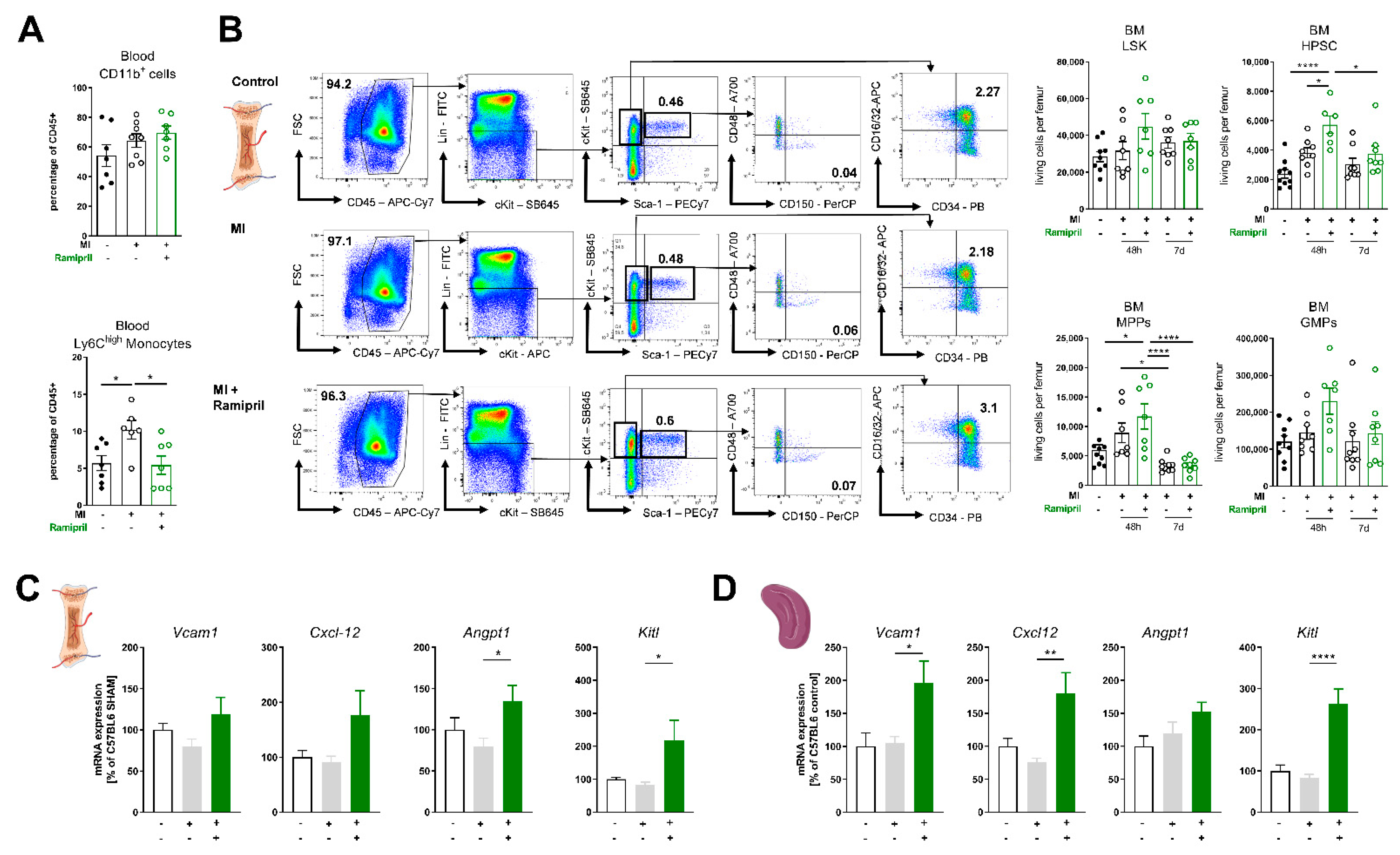
3.2. Ramipril Limits the Number of Circulating Monocytes and Retains HPSC Due to Upregulation of Retention Factors in the Bone Marrow and Spleen
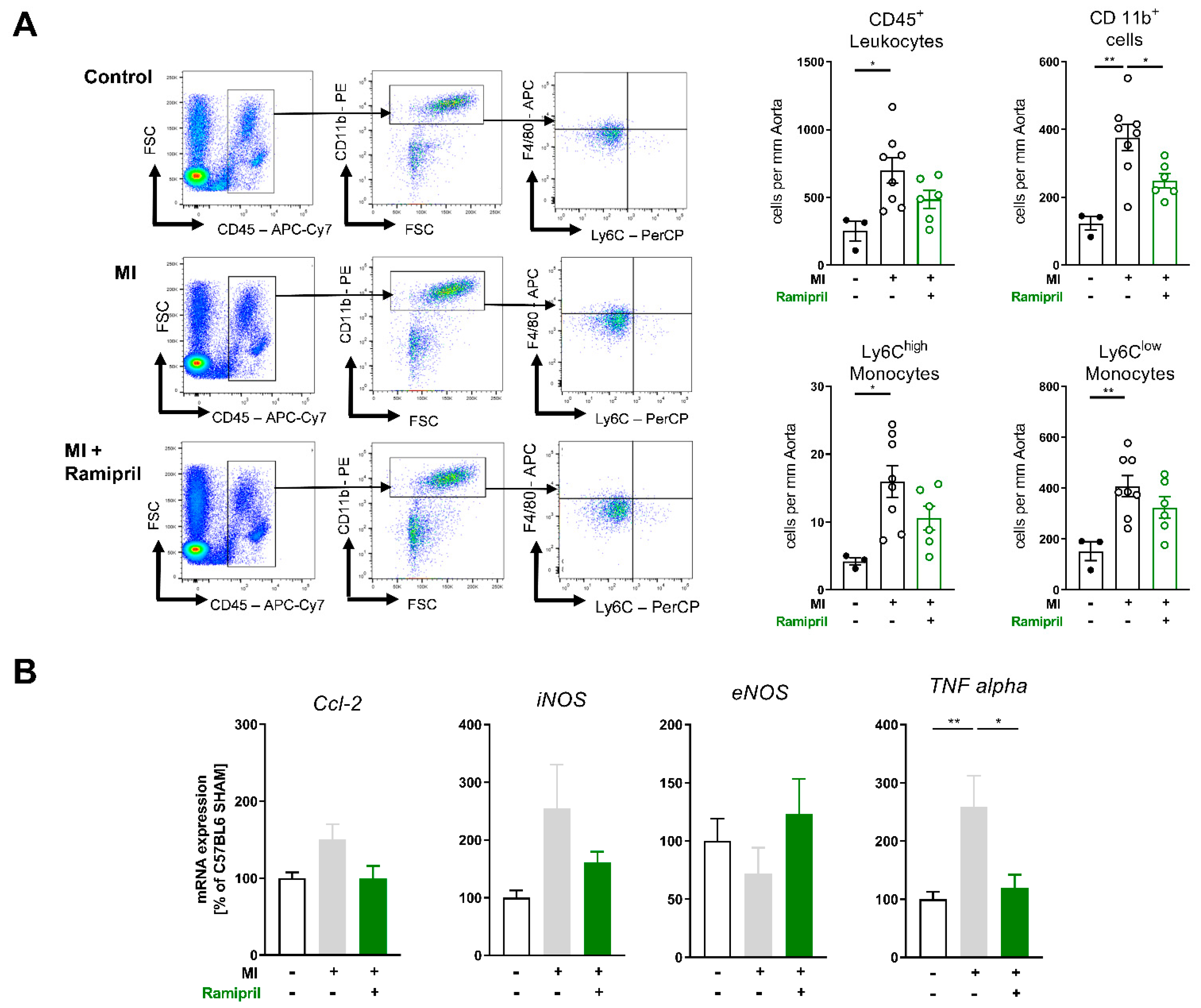
3.3. Early ACE Inhibitor Treatment Improves Vascular Endothelial Function and Leads to Attenuation of Vascular ROS and Inflammation in Ischemic Heart Failure
4. Discussion
5. Limitations of the Study
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Rev. Española Cardiol. 2016, 69, 1167. [Google Scholar]
- Poller, W.C.; Nahrendorf, M.; Swirski, F.K. Hematopoiesis and Cardiovascular Disease. Circ. Res. 2020, 126, 1061–1085. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Sager, H.B.; Stengel, K.R.; Naxerova, K.; Courties, G.; Saez, B.; Silberstein, L.; Heidt, T.; Sebas, M.; Sun, Y.; et al. Myocardial Infarction Activates CCR2(+) Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015, 16, 477–487. [Google Scholar] [CrossRef]
- Rasini, E.; Cosentino, M.; Marino, F.; Legnaro, M.; Ferrari, M.; Guasti, L.; Venco, A.; Lecchini, S. Angiotensin II type 1 receptor expression on human leukocyte subsets: A flow cytometric and RT-PCR study. Regul. Pept. 2006, 134, 69–74. [Google Scholar] [CrossRef]
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Pastore, L.; Tessitore, A.; Martinotti, S.; Toniato, E.; Alesse, E.; Bravi, M.C.; Ferri, C.; Desideri, G.; Gulino, A.; Santucci, A. Angiotensin II stimulates intercellular adhesion molecule-1 (ICAM-1) expression by human vascular endothelial cells and increases soluble ICAM-1 release in vivo. Circulation 1999, 100, 1646–1652. [Google Scholar] [CrossRef]
- Tummala, P.E.; Chen, X.L.; Sundell, C.L.; Laursen, J.B.; Hammes, C.P.; Alexander, R.W.; Harrison, D.G.; Medford, R.M. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: A potential link between the renin-angiotensin system and atherosclerosis. Circulation 1999, 100, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Esteban, V.; Egido, J. Inflammation and angiotensin II. Int. J. Biochem. Cell Biol. 2003, 35, 881–900. [Google Scholar] [CrossRef]
- Molitor, M.; Rudi, W.S.; Garlapati, V.; Finger, S.; Schuler, R.; Kossmann, S.; Lagrange, J.; Nguyen, T.S.; Wild, J.; Knopp, T.; et al. Nox2+ Myeloid cells drive vascular inflammation and endothelial dysfunction in heart failure after myocardial infarction via angiotensin II receptor type 1. Cardiovasc. Res. 2021, 117, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Zingler, M.; Harrison, J.K.; Scott, E.W.; Cogle, C.R.; Luo, D.; Raizada, M.K. Angiotensin II Regulation of Proliferation, Differentiation, and Engraftment of Hematopoietic Stem Cells. Hypertension 2016, 67, 574–584. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Finger, S.; Knorr, M.; Molitor, M.; Schuler, R.; Garlapati, V.; Waisman, A.; Brandt, M.; Munzel, T.; Bopp, T.; Kossmann, S.; et al. A sequential interferon gamma directed chemotactic cellular immune response determines survival and cardiac function post myocardial infarction. Cardiovasc. Res. 2019, 115, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Takagawa, J.; Sievers, R.E.; Khan, M.F.; Viswanathan, M.N.; Springer, M.L.; Foster, E.; Yeghiazarians, Y. Validation of the wall motion score and myocardial performance indexes as novel techniques to assess cardiac function in mice after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1187–H1192. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.M.; Griendling, K.K. NADPH oxidases and angiotensin II receptor signaling. Mol. Cell Endocrinol. 2009, 302, 148–158. [Google Scholar] [CrossRef]
- Mollnau, H.; Wendt, M.; Szocs, K.; Lassegue, B.; Schulz, E.; Oelze, M.; Li, H.; Bodenschatz, M.; August, M.; Kleschyov, A.L.; et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 2002, 90, E58–E65. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Interleukin-1 in cardiac injury, repair, and remodeling: Pathophysiologic and translational concepts. Discoveries 2015, 3. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Inflammation in cardiac injury, repair and regeneration. Curr. Opin. Cardiol. 2015, 30, 240–245. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Pittet, M.J.; Nahrendorf, M.; Swirski, F.K. The journey from stem cell to macrophage. Ann. N. Y. Acad. Sci. 2014, 1319, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial infarction accelerates atherosclerosis. Nature 2012, 487, 325–329. [Google Scholar] [CrossRef]
- Cremer, S.; Schloss, M.J.; Vinegoni, C.; Foy, B.H.; Zhang, S.; Rohde, D.; Hulsmans, M.; Fumene Feruglio, P.; Schmidt, S.; Wojtkiewicz, G.; et al. Diminished Reactive Hematopoiesis and Cardiac Inflammation in a Mouse Model of Recurrent Myocardial Infarction. J. Am. Coll. Cardiol. 2020, 75, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Leuschner, F.; Panizzi, P.; Chico-Calero, I.; Lee, W.W.; Ueno, T.; Cortez-Retamozo, V.; Waterman, P.; Gorbatov, R.; Marinelli, B.; Iwamoto, Y.; et al. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ. Res. 2010, 107, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Hoyer, F.F.; Grigoryeva, L.S.; Sager, H.B.; Leuschner, F.; Courties, G.; Borodovsky, A.; Novobrantseva, T.; Ruda, V.M.; Fitzgerald, K.; et al. Macrophages retain hematopoietic stem cells in the spleen via VCAM-1. J. Exp. Med. 2015, 212, 497–512. [Google Scholar] [CrossRef]
- Hess, C.N.; James, S.; Lopes, R.D.; Wojdyla, D.M.; Neely, M.L.; Liaw, D.; Hagstrom, E.; Bhatt, D.L.; Husted, S.; Goodman, S.G.; et al. Apixaban Plus Mono Versus Dual Antiplatelet Therapy in Acute Coronary Syndromes: Insights From the APPRAISE-2 Trial. J. Am. Coll. Cardiol. 2015, 66, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Roe, M.T.; Ohman, E.M. A new era in secondary prevention after acute coronary syndrome. N. Engl. J. Med. 2012, 366, 85–87. [Google Scholar] [CrossRef]
- Mega, J.L.; Braunwald, E.; Murphy, S.A.; Plotnikov, A.N.; Burton, P.; Kiss, R.G.; Parkhomenko, A.; Tendera, M.; Widimsky, P.; Gibson, C.M. Rivaroxaban in patients stabilized after a ST-segment elevation myocardial infarction: Results from the ATLAS ACS-2-TIMI-51 trial (Anti-Xa Therapy to Lower Cardiovascular Events in Addition to Standard Therapy in Subjects with Acute Coronary Syndrome-Thrombolysis In Myocardial Infarction-51). J. Am. Coll. Cardiol. 2013, 61, 1853–1859. [Google Scholar] [PubMed]
- Latini, R.; Staszewsky, L.; Maggioni, A.P.; Marino, P.; Hernandez-Bernal, F.; Tognoni, G.; Labarta, V.; Gramenzi, S.; Bianchi, F.; Sarcina, G.; et al. Beneficial effects of angiotensin-converting enzyme inhibitor and nitrate association on left ventricular remodeling in patients with large acute myocardial infarction: The Delapril Remodeling after Acute Myocardial Infarction (DRAMI) trial. Am. Heart J. 2003, 146, 133. [Google Scholar] [CrossRef]
- Latini, R.; Tognoni, G.; Maggioni, A.P.; Baigent, C.; Braunwald, E.; Chen, Z.M.; Collins, R.; Flather, M.; Franzosi, M.G.; Kjekshus, J.; et al. Clinical effects of early angiotensin-converting enzyme inhibitor treatment for acute myocardial infarction are similar in the presence and absence of aspirin: Systematic overview of individual data from 96,712 randomized patients. Angiotensin-converting Enzyme Inhibitor Myocardial Infarction Collaborative Group. J. Am. Coll. Cardiol. 2000, 35, 1801–1807. [Google Scholar] [PubMed]
- Pfeffer, M.A.; Braunwald, E.; Moye, L.A.; Basta, L.; Brown, E.J., Jr.; Cuddy, T.E.; Davis, B.R.; Geltman, E.M.; Goldman, S.; Flaker, G.C.; et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N. Engl. J. Med. 1992, 327, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.A.; Yang, D.; Kida, K.; Molotkova, N.; Yeo, S.J.; Varki, N.; Iwata, M.; Dalton, N.D.; Peterson, K.L.; Siems, W.E.; et al. Effects of ACE2 inhibition in the post-myocardial infarction heart. J. Card. Fail. 2010, 16, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Group, C.T. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudi, W.-S.; Molitor, M.; Garlapati, V.; Finger, S.; Wild, J.; Münzel, T.; Karbach, S.H.; Wenzel, P. ACE Inhibition Modulates Myeloid Hematopoiesis after Acute Myocardial Infarction and Reduces Cardiac and Vascular Inflammation in Ischemic Heart Failure. Antioxidants 2021, 10, 396. https://doi.org/10.3390/antiox10030396
Rudi W-S, Molitor M, Garlapati V, Finger S, Wild J, Münzel T, Karbach SH, Wenzel P. ACE Inhibition Modulates Myeloid Hematopoiesis after Acute Myocardial Infarction and Reduces Cardiac and Vascular Inflammation in Ischemic Heart Failure. Antioxidants. 2021; 10(3):396. https://doi.org/10.3390/antiox10030396
Chicago/Turabian StyleRudi, Wolf-Stephan, Michael Molitor, Venkata Garlapati, Stefanie Finger, Johannes Wild, Thomas Münzel, Susanne H. Karbach, and Philip Wenzel. 2021. "ACE Inhibition Modulates Myeloid Hematopoiesis after Acute Myocardial Infarction and Reduces Cardiac and Vascular Inflammation in Ischemic Heart Failure" Antioxidants 10, no. 3: 396. https://doi.org/10.3390/antiox10030396
APA StyleRudi, W.-S., Molitor, M., Garlapati, V., Finger, S., Wild, J., Münzel, T., Karbach, S. H., & Wenzel, P. (2021). ACE Inhibition Modulates Myeloid Hematopoiesis after Acute Myocardial Infarction and Reduces Cardiac and Vascular Inflammation in Ischemic Heart Failure. Antioxidants, 10(3), 396. https://doi.org/10.3390/antiox10030396