Abstract
Glutathione (GSH), a tripeptide particularly concentrated in the liver, is the most important thiol reducing agent involved in the modulation of redox processes. It has also been demonstrated that GSH cannot be considered only as a mere free radical scavenger but that it takes part in the network governing the choice between survival, necrosis and apoptosis as well as in altering the function of signal transduction and transcription factor molecules. The purpose of the present review is to provide an overview on the molecular biology of the GSH system; therefore, GSH synthesis, metabolism and regulation will be reviewed. The multiple GSH functions will be described, as well as the importance of GSH compartmentalization into distinct subcellular pools and inter-organ transfer. Furthermore, we will highlight the close relationship existing between GSH content and the pathogenesis of liver disease, such as non-alcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), chronic cholestatic injury, ischemia/reperfusion damage, hepatitis C virus (HCV), hepatitis B virus (HBV) and hepatocellular carcinoma. Finally, the potential therapeutic benefits of GSH and GSH-related medications, will be described for each liver disorder taken into account.
1. Introduction
Glutathione (GSH), or l-γ-glutamyl-l-cysteinyl-glycine, is a tripeptide present in mammalian cells at surprisingly high levels (1–10 mM) and particularly concentrated in the liver [1,2]. The thiol group of cysteine is responsible for GSH biological activity: GSH is considered the most important cellular redox buffer and major defender against oxidative stress. GSH is oxidized into glutathione disulfide (GSSG), which is actually two GSH molecules bound together at the sulfur atoms (Figure 1). The oxidized form is reconverted into GSH by the NADPH-dependent enzyme glutathione reductase (GR) [3]. The GSH:GSSG ratio is often used as a marker of cellular toxicity. Under normal conditions, the molar GSH:GSSG ratio is around 100:1; during stress, this ratio has been shown to decrease to 10:1 and even 1:1 [4,5,6,7,8].
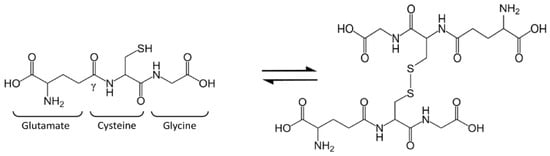
Figure 1.
Glutathione (GSH) structure and balance between GSH and glutathione disulfide (GSSG).
It has been demonstrated that GSH has multiple roles in cell physiology, including:
(i) direct scavenging of reactive oxygen species (ROS), nitric oxide (NO) and its derivatives (RNS), resulting in the protection of the electron transport chain, DNA, lipids and proteins; (ii) indirect neutralization of intoxicants; (iii) S-gluta-thionylation of protein thiol groups; (iv) regulation of cell cycle progression and apoptosis.
From these considerations, it emerges that GSH cannot be considered a mere free radical scavenger but that it has a weighty role in the network governing the choice between survival, necrosis and apoptosis [9,10] as well as in cell signaling [10,11] and metabolism [3,12].
The present review will focus on the molecular biology of the GSH system. GSH synthesis, metabolism and regulation will be reviewed; the multiple GSH functions will be described, as well as GSH compartmentalization and interorgan transfer. Finally, the role of GSH in pathogenesis and potential therapeutic approaches will be also highlighted.
2. Glutathione Synthesis and Metabolism
The main source of GSH is the liver, which has the unique ability to synthesize cysteine, a GSH precursor, from endogenous sources through the trans-sulphuration pathway [13], or to obtain it from protein breakdown and food. GSH availability in hepatocytes depends on the membrane transport of cysteine itself, cystine and methionine [2]. In fact, a good amount of GSH is indirectly obtained from methionine; methionine deficiency has been found to induce a reduction in GSH availability, especially in the liver [14,15]. As a consequence, the liver plays a central role in maintaining interorgan homeostasis of GSH by exporting nearly all of the synthesized GSH into plasma and bile; in fact, the liver has the highest capacity for GSH efflux through its basolateral and apical poles [13]. Consequently, an alteration of the hepatic ability to synthesize or export GSH has an impact on systemic GSH homeostasis [2].
The first step in GSH synthesis is catalyzed by l-glutamate l-cysteine γ-ligase (GCL), resulting in the formation of γ-glutamyl-cysteine. GCL is a heterodimer constituted by a catalytic monomer (GCLC) and a modifier monomer (GCLM). GCLC is sensitive to GSH-induced feedback inhibition. GCLM is enzymatically inert; however, in the dimer it reduces the inhibitory feedback response to GSH [2]. GCL expression responds to oxidative stress and is positively regulated by nuclear factor erythroid 2-related factor 2 (Nrf2) [16,17,18,19]. Nrf2 works in combination with small avian musculoaponeurotic fibrosarcoma oncogene homolog (MAF) proteins: together they recognize and bind antioxidant response elements (ARE) in promoters of antioxidant enzyme genes (phase 2 enzymes such as glutathione S-Transferase (GST), UDP-glucorosyl transferase and superoxide dismutase (SOD), GSH peroxidase (GPx) and catalase (CAT), sulfiredoxin 1 and Thioredoxin reductase 1 [16,17,18,19]. Nrf2 is inhibited by Kelch-like ECH-associated protein 1 (Keap1): specifically, it increases Nrf2 proteasome-mediated degradation [20,21]. Electrophilic agents and antioxidants induce conformational changes in Keap1, thus impeding the binding with Nrf2 leading to the consequent migration of the nuclear factor in the nucleus, where it can exert its function [22].
The second step in GSH synthesis is catalyzed by GSH synthetase (GS), which produces GSH by adding a glycine molecule to γ-glutamyl-cysteine. Both first and second steps require energy in the form of adenosine triphosphate (ATP); however, ATP is not usually considered a limiting factor in GSH production except in pathological conditions [10]. The rate-limiting enzyme is considered to be GCL [2]; in fact, by overexpressing GCL and not GS, a net increase in GSH was obtained in Saccharomyces cerevisiae [23].
Usually, amino acids are linked together by an α-carboxyl group; differently, in GSH, the peptide bond linking glutamate and cysteine is constituted by a γ-carbonyl group, which is resistant to the hydrolytic action of cellular peptidases. Consequently, GSH is highly stable intracellularly [24] and its degradation occurs only extracellularly. γ-glutamyl transpeptidase (GGT), the enzyme responsible for GSH degradation, is located on the external surface of epithelial cells in kidney tubules, biliary epithelium and brain capillaries, allowing GSH amino acidic constituents to be recycled in the so-called γ-glutamyl cycle for intracellular GSH synthesis [25].
The utilization of GSH is consequential to its chemical structure. In particular, the thiol group of GSH undergoes a one-electron reduction with radicals, producing an unstable GS•. This reaction is kinetically driven by two further reactions: the first one with thiolate anion GS¯, and the second one with oxygen, producing GSSG and the superoxide anion O2•¯. The process is then completed by SOD and CAT, converting O2•¯ into H2O and O2 [10].
It is widely believed that the reductive power needed to recycle GSSG into GSH is provided by cellular Nicotinamide Adenine Dinucleotide Phosphate (NADPH) generated in the pentose phosphate pathway (Figure 2); the first step is catalyzed by glucose-6-phosphate dehydrogenase (G6PDH) and results in the oxidation of a glucose-6-phosphate into 6-phosphoglucolactone with the release of one NADPH. The second reaction of the cycle is catalyzed by 6-phosphogluconate dehydrogenase (6-PGDH). NADPH-dependent malic enzyme (ME)-1 also has a role in hepatic GSH production. Cytosolic ME-1 is a homo-tetrameric enzyme catalyzing reversible decarboxylation of malate into pyruvate and carbon dioxide; in the process, ME-1 generates NADPH. However, hepatocytes do not strictly depend from ME-1 activity for NADPH synthesis: in ME-1 deficient mice, after an initial depletion, the GSH levels recover quickly and sensitivity to acetaminophen-induced liver injury was unaltered [26].
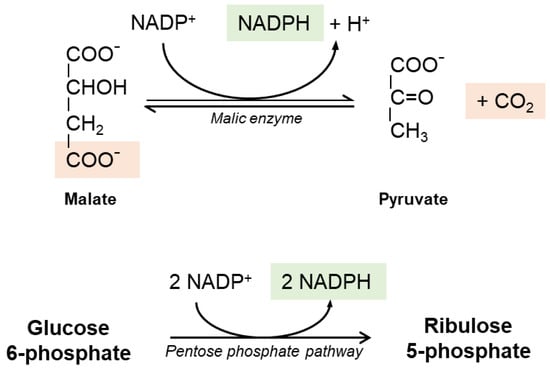
Figure 2.
The pentose phosphate pathway and the malic enzyme reaction. The pentose phosphate pathway is considered the main source of reducing equivalents used for GSH regeneration; it is also the sole source of NADPH in red blood cells. The malic enzyme is another relevant source of NADPH in hepatic cells.
3. Glutathione Functions
3.1. Direct Antioxidant Activity
Normally, the ROS-generating processes are counterbalanced by the antioxidant system, in which GSH is considered a key player. However, under pathological conditions, ROS level increases above the steady-state leading to oxidative stress, defined as a situation where the steady-state ROS concentration is transiently or chronically enhanced, disturbing cellular metabolism and its regulation and damaging cellular constituents [3]. When the cellular antioxidant defense system is overwhelmed by ROS production, the imbalance can culminate in cell death by apoptosis or necrosis [3]. In the process of counteracting the increase in oxidative stress, energy and amino acids are consumed resulting in the restoration of normal ROS level. Alternatively, in case of prolonged increase in ROS production, the instauration of a chronic condition may lead to various pathologies, such as cardiovascular diseases, chronic obstructive pulmonary disease, chronic kidney disease, neurodegenerative diseases, cancer and non-alcoholic steatohepatitis [27,28].
GSH directly scavenges ROS and RNS such as HO•, RO•, RO2•, HOCl, 1O2 and ONOO¯. NO was originally believed to be directly scavenged by GSH to produce S-nitroso-glutathione (GSNO). However, it has been demonstrated that NO firstly needs to be converted into a nitrosium ion (NO+) before being able to react with GSH to form GSNO [3]. It should be noted that GSNO is not a terminal GSH metabolite; it should rather be considered as a GSH and NO storage and transportation system. For example, NO is released from GSNO when reacting with nitrosylating enzymes, such as thioredoxin and GSNO reductase; the three of them are capable of protein nitrosylation/denitrosylation, depending on their oxidation state [29].
3.2. Detoxification of Endogenous Compounds and Xenobiotics
GSH and GSH-related enzymes have a relevant role in cellular detoxification. GSH is a nucleophile and therefore it can react with electrophilic species transforming them into more soluble and easily excretable molecules. Conjugation of GSH with electrophilic compounds is mainly mediated by the GSTs, a super family of Phase II detoxification enzymes classified, according to their localization and properties, into cytosolic, mitochondrial and microsomal GSTs [30].
The cytosolic GSTs have been divided into different classes based on their genesis, catalytic amino-acid residue, sequence similarity, and substrate specificity [31]. These distinct evolutionary classes include Alpha, Beta, Dehydro-ascorbate reductases (DHARs), Delta, Epsilon, Mu, Omega, Pi, Phi, Sigma, Tau, Theta, and Zeta. Among these classes, only six have been functionally characterized in plants (Figure 3). Lambda class has structural similarity with Omega GSTs of mammals; both the classes share the number of conserved residues in the N-terminal GSH binding domain and possess a distinct structural feature of N-terminal extension [32].
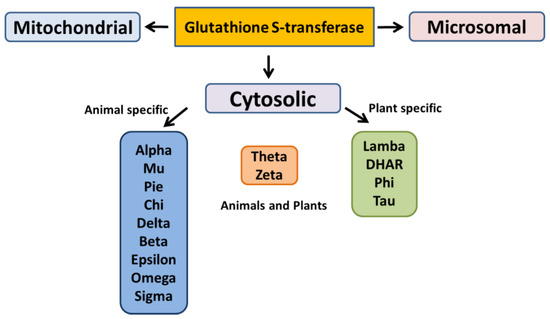
Figure 3.
Different classes of glutathione S-transferase.
The microsomal GSTs have been identified as the integral membrane proteins and are known as Membrane-Associated Proteins in Eicosanoid and Glutathione Metabolism (MAPEGs) while the mitochondrial GSTs are also referred as kappa-class GSTs [32].
GSTs are able to conjugate GSH to a wide range of endogenous electrophiles and xenobiotics. The endogenous 4-hydroxynonenal (4HNE) is an aldehyde resulting from lipid peroxidation that forms covalent adducts with DNA and proteins; high concentrations of 4HNE have been associated with apoptosis, altered gene expression, neurodegenerative and cardiovascular diseases, cancer and metabolic syndrome [33,34,35]. GSTs are involved in the detoxification of 4HNE by conjugation with GSH; the resulting GS-HNE is actively excreted by efflux transmembrane transporters [35]. Similarly, aflatoxin B1, a toxic metabolite produced by fungi of the genus Aspergillus, is activated by cytochromes P450 into highly reactive epoxides with mutagenic properties; GSTs are involved in the conjugation of aflatoxin B1-exo-8,9 epoxide with GSH resulting in its elimination through the mercapturic pathway [36]. Acrolein is a highly reactive aldehyde resulting from burning tobacco, wood and plastic and when animal or vegetal fats and oils are overheated; acrolein is also a metabolite of the anticancer drug cyclophosphamide [37]. Acrolein forms adducts with DNA, proteins and lipids inducing cellular damage. GSTs are responsible for the first step in acrolein metabolization, consisting in its conjugation with GSH; after this first step, other reactions follow, leading to the urinary excretion of mercapturic acid metabolites [37,38]. GSTs are also involved in the reduction of organic hydroperoxides, such as phospholipids, DNA hydroperoxides and fatty acids, to their corresponding alcohols [30].
GSH directly participates in the neutralization of a toxic acetaminophen metabolite. Acetaminophen is metabolized primarily by sulfation and glucuronidation; however, cytochrome P450 enzymes convert 5–9% of acetaminophen to N-acetyl-p-benzo-quinonimine (NAPQI), which causes hepatotoxicity by binding to cellular macromolecules. NAPQI detoxification occurs primarily by GSH conjugation [39]. N-acetylcysteine treatment can prevent or limit liver injury by restoring hepatic GSH concentrations and it is used as an antidote in acetaminophen poisoning [40].
3.3. GSH in Protein Glutathionylation
S-gluta-thionylation is a reversible post-translational modification of a protein cysteine thiol group (P-SH) into a protein-S glutathione group (PSSG). Spontaneous PSSG formation depends heavily on the local GSH:GSSG ratio. The physiological GSH:GSSG ratio is not able to promote the formation of PSSG; only a dramatic GSH:GSSG ratio decline (i.e., from 100:1 to 1:1) would result in a 50% conversion of PSH into PSSG. Such conditions are extremely rare in vivo, therefore, in most cases, spontaneous PSSG formation is uncommon [41]. Differently, P-SOH groups, produced when P-SH groups are oxidized by ROS, are rapidly neutralized by GSH. Without the gluta-thionylation, P-SOH groups may be further oxidized resulting in protein deactivation [42]. Different radicals or even weak oxidants, such as NO, are also able to promote S-gluta-thionylation through the formation of an unstable intermediate that is reduced by GSH. In addition, P-SH and P-SOH can be modified by GSNO to form PSNO and/or PSSG [43].
3.4. Hepatocyte Cell Death and GSH: Switch between Necrosis and Apoptosis
The crucial role of GSH content in the regulation of hepatocyte cell death has been documented when an increase in ROS or a depletion of GSH occur [44,45]. A common observation, in experimental models of liver injury, is that low levels of ROS induce apoptosis, whereas higher levels lead to necrosis [9]. While apoptosis involves caspase activation, chromatin fragmentation and the phagocytosis of cell bodies, which minimizes inflammation, necrosis is defined by cell swelling and lysis with release of intracellular components, which elicits an inflammatory response [9]. Marked redox changes in hepatocytes, by using oxidants or GSH depletion agents, cause necrosis, whereas modest redox changes sensitize hepatocytes to tumor necrosis factor (TNF)-induced apoptosis by inhibiting the expression of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB)-dependent survival genes [44]. Other evidence obtained during rewarming of cold-stored rat hepatocytes, demonstrated that changes in GSH levels play a role in the mechanisms regulating apoptosis and necrosis [46]. The temporal occurrence of GSH depletion, as well as thio-barbituric acid reactive substances (TBARS) and ROS formation, and intracellular ATP content, represent the factors driving cells toward necrosis or apoptosis. A controlled amount of oxidative stress balanced by a sufficient amount of ATP will cause the apoptotic cascade to be activated. Differently, a marked depletion of GSH and ATP levels, accompanied by a profound increase in TBARS and ROS production, are associated with hepatocyte necrosis (Figure 4) [9].
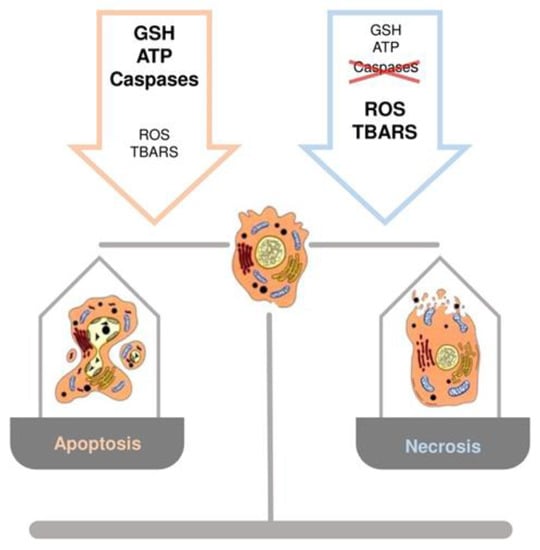
Figure 4.
Schematic representation of the role of GSH, ATP, thio-barbituric acid reactive substances (TBARS), reactive oxygen species (ROS) and caspases in the regulation of cell death, apoptosis or necrosis.
The intracellular ATP concentration is a critical factor directing cells toward apoptosis or necrosis; in fact, apoptosis requires energy in the form of ATP to assemble the apoptotic machinery. On the contrary, during necrosis, ATP is dissipated due to the depletion of energy stores and a reduction of mitochondrial energy-transducing capacity [47]. Furthermore, apoptosis is known to be an energy consuming process because the cell requires an adequate ATP content for the correct function of both caspases and the apoptosome [45]. Oxidation of caspases by ROS has been proposed as one of the mechanisms involved in cell necrosis [45]. Caspases are cysteine proteases that are inactivated by the oxidation of thiol groups to disulfides, as well as other proteins involved in cell survival are, such as NF-kB or c-Jun N-terminal kinases (JNK) [44]. Under moderate ROS formation, apoptosis can start with the activation of pro-apoptotic factors, i.e., JNK, and inhibition of pro-survival factor, i.e., NF-kB, without changing the redox status of the caspases. Evidence suggests that the mitochondrial GSH pool plays a critical role for cell survival [48]. In ethanol sensitized hepatocytes, TNF-α increased the susceptibility of hepatocytes after mitochondrial GSH depletion; the restoration of mitochondrial GSH levels had protective effects against TNF-α [49].
Moreover, GSH plays a crucial role in the regulation of events leading to apoptosis or necrosis in several hepatic pathologies. In most cases, GSH represents a causative factor for disease progression. The different pathological settings will be described below.
Despite its exclusive synthesis in the cytosol, GSH is distributed in intracellular organelles, including the nucleus, the mitochondria and the endoplasmic reticulum (ER) as depicted in Figure 5.
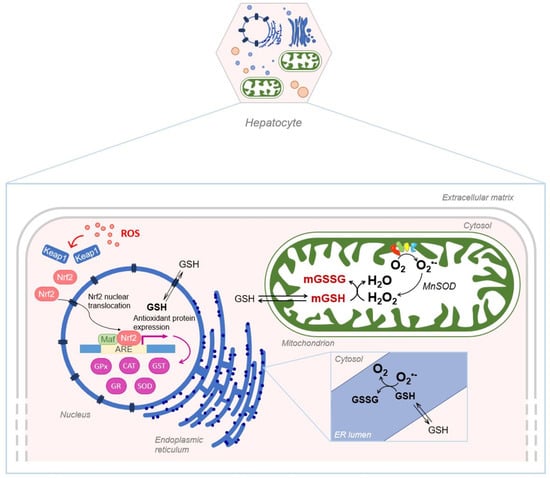
Figure 5.
Schematic representation of GSH compartmentalization, in the nucleus, mitochondria and endoplasmic reticulum. mGSH: mitochondrial GSH; mGSSG: mitochondrial GSSG.
4. Liver and Intracellular Glutathione Compartmentalization
4.1. Nuclear Glutathione
The first hypothesis of a possible hepatic nuclear localization of GSH has been supported by the immunohistochemical localization of human GST2 not only in the cytoplasm of hepatocytes but also in the nucleus [50]. Later, the hepatic nuclear localization of GSH was also documented: high concentrations of this tripeptide have been found in the nucleus of intact hepatocytes and a nuclear/cytoplasmic concentration gradient was maintained by active mechanisms [51]. The nucleus does not partake in the synthesis of GSH because it lacks GCL and GS [52]. GSH transport from cytosol to nucleus is ATP dependent: studies in isolated nuclei obtained from rat liver demonstrated both GSH-stimulated ATP hydrolysis and ATP-stimulated GSH accumulation [53]. More recently, it has been shown that the GSH pool in the nuclear matrix derives from the cytoplasmic pool, probably via a nuclear pore-mediated translocation [52].
Alpha and Mu classes of GST2 superfamily enzymes, able to protect the cell from toxic endogenous or xenobiotic compounds, are abundantly expressed in rat liver, where they represent 43% and 56%, respectively, of the entire pool of cytosolic GSTs [54]. In rat hepatocytes, a significant fraction of dinitrosyl-diglutathionyl-iron complex is found in subcellular components, mainly in the nucleus, apparently entirely bound to Alpha class GSTs [55]. It was also reported that a considerable amount of GST, corresponding to 10% of the cytosolic pool, is electrostatically associated with the outer nuclear membrane, and a similar quantity is compartmentalized inside the nucleus [56]. Mainly, Alpha class GSTs, and in particular GSTA1-1, GSTA2-2, and GSTA3-3, are involved in this interaction, suggesting the existence of a protective enzyme barrier at the nuclear envelope, in which GSH plays an important role [56].
The expression of GPx and GR has also been demonstrated in nuclear fractions from rat hepatocytes [57]; the expression of these proteins in the nucleus underscores the key role of glutathione in the regulation of the cell cycle and in chromosomal organization; in fact, nuclear proteins, mainly histones and other chromatin-related proteins, have to be maintained in a reduced state for optimal function [52,57].
Combining information obtained from in vitro and in vivo studies, the key role of GSH in many nuclear processes in the liver has been further consolidated. Studies carry out in isolated nuclei obtained from the liver demonstrated GSH role in the modulation of the thiol/disulfide redox status of nuclear proteins and in the control of chromatin compacting and decondensation [53]. In vivo studies demonstrated the effect of GSH on gene expression through chromatin remodeling [58]. It has been also demonstrated that GSH content fluctuations in the liver are involved in many of the processes in which c-myc gene is overexpressed [44]. Torres et al. documented that, at hepatic physiological GSH content, the presence of deacetylase complexes and Id2 bound to the c-myc promoter led to a closed chromatin structure inaccessible to transcription factors [58]. On the contrary, when an acute GSH depletion occurs, Id2 and deacetylase complexes are released from the c-myc promoter, allowing transcription factors such as STAT3 to gain access to the c-myc promoter, leading to mRNA transcription [58].
Under specific circumstances such as in the active phases of cell proliferation, the nucleus accumulates GSH to much greater extent than the cytoplasm [59]. Brown et al. (2007) showed for the first time in an in vivo model that high hepatic GSH (1.5-fold), cysteine (15-fold), and glutamate cysteine ligase activity (1.5-fold) levels correlate with increased telomerase activity; on the contrary, telomerase activity was inhibited by GSH depletion with N-ethylmaleimide [60].
4.2. Mitochondrial Glutathione
Mitochondrial matrix is characterized by a high GSH concentration that plays an important role in defending mitochondria against oxidants and electrophiles. Mitochondrial GSH is particularly relevant in the disposal of hydrogen peroxide generated from superoxide anion by MnSOD, which otherwise may oxidize protein SH groups or peroxidize mitochondrial lipids [61].
GSH is negatively charged at physiological pH so it is unable to cross both the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM). Studies on liver mitochondria have characterized GSH transport across mitochondrial membranes. Porins allow the diffusion of molecules such as GSH through the OMM [62]. As for the IMM, early findings in rat liver mitochondria suggested that active, energy-dependent processes inhibited by glutamate, carbonyl-cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP) and ophthalmic acid were involved [63]. GSH is transported into hepatic mitochondrial matrix by a high-affinity transporter, which is saturated at a GSH concentration of 1–2 mM and stimulated by ATP. Another component with lower affinity is stimulated by ATP and ADP [63,64]. Other studies showed that the incubation of rat liver mitochondria with butyl-malonate (a dicarboxylate carrier inhibitor) and phenyl-succinate (a 2-oxoglutarate carrier inhibitor) inhibits GSH transport across IMM; thus, it has been possible to identify two anion carriers, members of the mitochondrial carrier family (SLC25): the dicarboxylate carrier (DIC; SLC25A10), and the 2-oxoglutarate carrier (OGC; SLC25A1) [48,65]. These two transporters are responsible for 45%–50% GSH uptake into the mitochondrial matrix in rat liver mitochondria, suggesting the involvement of other transporters [65]. Armeni et al. demonstrated the existence of an alternative GSH source in mitochondria: S-d-lactyl-glutathione (SLG), an intermediate of the glyoxalase system, is hydrolyzed by the mitochondrial glyoxalase II enzyme into D-lactate and GSH [66].
The mitochondrial transport of GSH is also dependent on membrane fluidity. For example, the 2-oxoglutarate carrier capacity changes with mitochondrial membrane fluidity: in rat liver mitochondria enriched in cholesterol, a decrease in mitochondrial GSH uptake was found [67]. On the other hand, higher mitochondrial membrane fluidity resulted in the recovery of GSH uptake rate [68].
Mitochondrial GSH is the primary defense against oxidative damage to mitochondrial membranes by ensuring the reduction of hydroperoxide groups and other lipid peroxides on phospholipids through the action of mitochondrial GSTs. Furthermore, GSH can also work non-enzymatically by reacting with electrophiles that are generated because of metabolic processes, although this reaction is greatly improved in presence of GSTs. Several cytosolic GST isoforms have been identified and the class Kappa isoform is the only GST present in mitochondria [69].
Under oxidative stress conditions, the increased levels of mitochondrial GSSG and protein disulfides are controlled by gluta-redoxin (Grx), a GSH-dependent disulfide oxidoreductase that catalyzes dithiol reactions, reducing GSH-protein mixed disulfides in a coupled system with GSSG-reductase and NADPH [70].
Regarding hepatic pathological setting, mitochondrial glutathione plays a critical role in the survival of hepatocytes during hypoxia through the regulation of mitochondrial generation of oxidative stress [71]. Furthermore, GSH depletion compromises mitochondrial function, sensitizes cells to toxicants worsening drug-induced hepatotoxicity [61].
Mitochondrial glutathione also participates to hepatic disease progression such as alcoholic liver diseases and non-alcoholic liver disease (NAFLD), as described in the below sections.
4.3. Endoplasmatic Reticulum Glutathione
The ER plays a crucial role in many cellular functions, such as synthesis of macromolecules, regulation of cellular calcium homeostasis, and maturation, processing, and transport of secretory and membrane-associated proteins [72]. Proteins assembled in the ER are rich in disulfide bonds that help to promote efficient folding and oligomerization of nascent polypeptide chains [73]. The activities of ER folding enzymes are highly dependent on the reduction/oxidation (redox) environment, and even small perturbations in the redox status greatly affect enzymatic activity and, as a result, protein folding kinetics [74]. It is widely believed that high concentration of GSH in the ER keeps protein disulfide isomerases (PDIs) in the reduced form [75]; these enzymes are the most important constituent of the oxidative machinery, together with the endoplasmic reticulum thiol oxidase (ERO1) [52]. The hepatic GSH transport in ER has been investigated and a bi-directional, saturable GSH transport was found in rat liver microsomal vesicles. GSH is preferentially transported in the reduced form as GSSG is virtually impermeable and is entrapped and accumulated in the microsomal lumen creating the oxidizing environment necessary for the formation of disulfide bonds and for the proper folding of proteins [76]. In any case, the GSH and GSSG movement and concentration into ER remain debated [77].
5. Glutathione and Liver Diseases
5.1. Non-Alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease (NAFLD) is a chronic and multifactorial disorder characterized by an excessive fatty acid (FA) accumulation in hepatocytes [78], due to an imbalance between FA uptake and disposal [79]. Noteworthy, NAFLD is the hepatic manifestation of the so-called “metabolic syndrome”, and is associated with a series of metabolic abnormalities, such as insulin resistance (IR), obesity, dyslipidaemia, hyperglycaemia, and hypertension. The pathology proceeds through different stages: from simple hepatic steatosis, it progresses toward non-alcoholic steatohepatitis (NASH), fibrosis, cirrhosis and in the most severe cases, toward hepatocellular carcinoma (HCC) [79]. According to the “multi-hit hypothesis”, several factors contribute to NAFLD development and progression: sedentary lifestyle and high fat diet can be related to obesity, dyslipidaemia and IR; changes in intestinal microbiome, genetic and epigenetic factors can also contribute [80]. NAFLD is estimated to affect 25% of the population worldwide [81] but, to date, there are no specific therapies available for the treatment [82], although some pharmacological strategies used for related disorder (e.g., diabetes) are also recommended for the treatment of NAFLD patients [79].
The main feature of NAFLD is the FA accumulation in liver parenchyma, due to an enhancement in FA uptake from plasma, in addition to an overstimulation of the de novo lipogenesis via SREBP-1c transcription factor activation [83]. The increase in FAs in liver parenchyma leads to an increased rate of β-oxidation, with the consequent electron outflow from the electron transport chain ETC [84]. The loss of electrons from ETC is associated with ROS overproduction and oxidative stress generation. Indeed, it has been widely demonstrated that oxidative stress also has a role in NAFLD pathogenesis [85]. Koruk et al. in 2004 showed that serum levels of malondialdehyde (MDA), GSH, and SOD were higher in NASH patients than in the control group, suggesting that the antioxidant system has a key role in NAFLD pathogenesis and disease progression [86]. Nrf2 seems to play a significant role in NAFLD development and oxidative stress response and is considered a novel potential target for the treatment of NAFLD. The role of Nrf2 in liver fibrosis has been clarified by Lu, who showed that GCL subunits and GS are regulated byNrf2, which binds AREs present in their promoters [2] (Figure 6). Furthermore, Zhang et al. have evidenced that an increase in the Nrf2 expression results in hepatic steatosis attenuation [21]. However, Nrf2 is not only implicated in the antioxidant response, but also plays a role in lipid metabolism [87,88]. Genes that are involved in lipid accumulation are negatively regulated by Nrf2 [87], and this finding supports the relevance of Nrf2 in NAFLD patients.
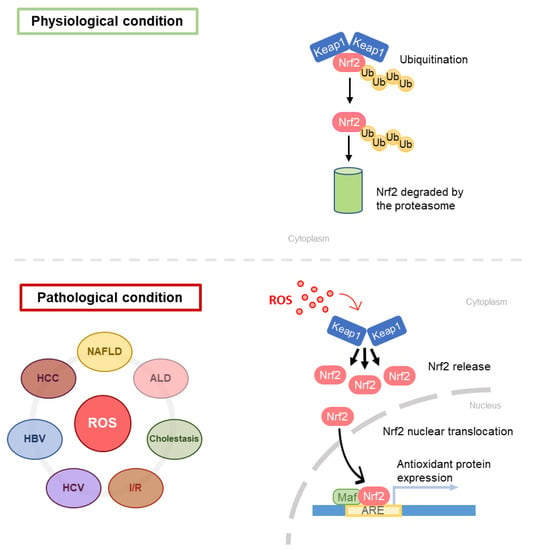
Figure 6.
Schematic representation of Nrf2 pathway in different liver diseases. ARE, antioxidant response elements.
The role of ferroptosis has been evaluated as a determining factor for NASH onset [89]. Qi et al. showed that ferroptosis leads to a diminished hepatic expression of GPx4: experiments performed on MCD-fed mice revealed a reduced GPx4 expression after RSL-3 (a ferroptosis inducer) administration. Moreover, a higher amount of apoptosis inducing factors were found in mice liver, indicating that ferroptosis plays a key role in NASH-associated cell death [90]. On the other hand, the administration of a ferroptosis inhibitor, Liproxstatin-1, reduces hepatic lipid peroxidation and cell death [90].
GPx4 is highly expressed in liver mitochondria; Se, which has a role in GPx function, shows an antioxidant effect on liver; in fact, Se depletion is implied in the pathogenesis of hepatic inflammation and fibrosis [91], which are NAFLD-related injuries. In a case-control study with NASH patients, a positive correlation was found between the increase in Se levels and the GSH/GSSG ratio. This means that Se may have an important impact on GSH pathway, increasing the antioxidant capacity of the whole system [92]. It is worth noting that a diminished GSH level was observed in rats fed with a Se-deficient diet; specifically, an increase in n-6/n-3 fatty acid ratio has been shown that is one of the hits involved in NAFLD pathogenesis [93]. On the other hand, some authors have recently reported that high Se intake is associated with NAFLD progression [94]. Clearly, further exploration is needed to better elucidate the mechanisms at the basis of selenium role in the NAFLD establishment, considering its importance for GPx functioning.
S-adenosyl-l-methionine (SAMe) is a methyl donor involved in methionine metabolism. It is produced by the activity of methionine adenosyltransferase-1A (MAT1A), which is specifically present in the liver. It has been observed that MAT1A null mice present low GSH levels, combined with a reduced expression of lipid peroxidation genes. These mice develop steatohepatitis after eight months [95]. Furthermore, Montfort and colleagues showed in MAT1A−/− mice a reduced SOD activity, possibly due to an increase in oxidative stress [96].
Cholesterol concentration in membranes is an essential factor for their fluidity and permeability. The mitochondrial GSH transporter 2-oxoglutarate carrier is involved in NASH development [97]. High amounts of free cholesterol in the liver reduce the activity of 2-oxoglutarate carrier. As a result, less GSH is translocated from the cytosol into mitochondria, favouring ROS accumulation and the consequent lipid peroxidation [98]. It has been demonstrated that the re-establishment of mitochondrial membrane fluidity at normal levels leads to the restoration of mitochondrial GSH levels [61]. Interestingly, ob/ob mice treated with atorvastatin did not show free cholesterol accumulation in mitochondria and the consequent mGSH reduction [98].
GSH involvement in NAFLD and NASH progression has been studied in vitro. In a nutritional model of steatosis in rats, Grattigliano et al. have evidenced an initial GSH increase, followed by a progressive depletion [99]. Also Yang et al. [100] noticed that fatty liver mitochondria from obese mice displayed enhanced GSH concentration. Probably, this result may be explained considering the increased GSH production in relation to a consistent increase in FAs in the liver. It has been also shown that in HepG2/C3A cells, the gene expression associated to GSH production is enhanced [101]. This response can be due to the rapid activation of the antioxidant system to protect the cell.
In vitro experiments evidenced that GPx was enhanced in steatotic immortalized human hepatocytes [102]; on the contrary, Videla et al. did not observe any difference in GPx expression between NAFLD patients and controls [103]. On the other hand, Videla and colleagues have demonstrated a robust correlation between NAFLD onset and GSH depletion in patients when compared with healthy individuals. Specifically, in NAFLD patients, they found an increase in protein oxidation/GSH ratio in comparison with controls in parallel with a significant decrease in SOD activity. In addition, low levels of CAT activity were observed in NASH patients, suggesting a reduced scavenging capacity of the antioxidant system. Changes in SOD and CAT activities in NAFLD and NASH patients are also accompanied by an increase in CYP2E1 activity [103]. CYP2E1 belongs to the cytochrome P450 family; it is involved in fatty acid oxidation [104] and is associated with NAFLD progression since it possesses an extremely high NADPH oxidase activity that leads to the production of ROS (H2O2, hydroxyl radical, and superoxide anion) [105]. Remarkably, the overexpression of CYP2E1 gene in an immortalized rat cell line results in GSH depletion and lipid peroxidation [106]. On the other hand, other authors reported an enhancement in GSH levels in HepG2 cells overexpressing CYP2E1 [107].
GSH in the Therapy of NAFLD
Through an open-label, single arm, multi-center, pilot study, Honda et al. proved a beneficial effect of oral GSH in NAFLD patients. They considered the reduction of ALT levels as the primary endpoint of the study since it is one of the principal NAFLD markers. The patients were treated for four months by oral GSH, and, after this period, blood ALT levels were considerably decreased, as well as the triglyceride content. Finally, the pilot study has evidenced that oral administration of GSH has positive effects on hepatic metabolism, improving NAFLD [108]. According to several studies, exogenous GSH seems to be degraded into its amino acidic constituents during digestive processes, which are used for the synthesis of endogenous GSH. Additional studies have clarified that the administration of the GSH precursors serine and glycine leads to an attenuation of NAFLD both in humans and in animal models [109,110]. The effect of glutamine administration on GSH levels has also been evaluated in a 5-fluorouracil-induced model of liver injury. In this model, endogenous glutamine production is not sufficient to counteract GSH depletion due to oxidative stress, and exogenous administration attenuates GSH depletion and MDA levels [111]. A more recent work investigated the supplementation of glutamine in combination with Western diet (WD), which resulted in a protective action against NASH development [112]. Rats fed with WD and glutamine showed less macro-vesicular fat accumulation, a decrease in lipid peroxidation and lower liver inflammation [112]. In another study on C57BL/6J mice fed with WD, liver morphology, blood enzymes and proinflammatory markers were improved in mice administered with Gln [113]. In a similar model, glutamine supplementation reduced oxidative stress in rats fed with a high-fat diet and induced an increase in GSH [114]. It is known that supplementation of serine and N-acetylcysteine (NAC) induces an increase in GSH levels. Indeed, NAC is a GSH precursor; it stimulates GSH synthesis, increases GST activity and promotes detoxification, scavenging ROS [103,115]. Its administration has been shown to restore GSH levels in NAFLD rats [116]. An experiment by Thong-Ngam et al. has shown that 20 mg/Kg NAC reduces oxidative stress in rats fed with high fat diet for 6 weeks to induce NASH development. In particular, serum GSH content was restored, and liver histology showed a normal morphological appearance in NAC-treated rats, with respect to controls [117]. An alternative pharmacological approach to counteract the oxidative stress related with NAFLD progression to NASH, consists in silymarin administration, which has been shown to restore GSH and GPx levels in serum [118]. It has also been proven that the supply of silymarin, a well-known compound with antioxidant properties extracted from the edible plant Silybum marianum [119,120], can help in terms not only of antioxidant effect, but also anti-inflammatory and antifibrotic effect [121]. It is worth noting that research into plant extracts to counteract oxidative stress are more and more common. Açai plant extract, for example, common name for Enturpe oleracea, contains significant amounts of polyphenols and anthocyanins that have shown an interesting antioxidant activity. De Freitas Carvalho et al. [122] investigated the contribution of açai extract in a murin model of NAFLD induced by a high-fat diet. They demonstrated an increase in GSH content, due to the augmented transcription of GCL. Moreover, açai extract administration led to a lower activity of GPx, SOD and CAT enzymes in the model used [122]. Another therapeutic plant extract that seems to have a beneficial effect on NAFLD-related oxidative stress is the blue honeysuckle (BH), common name for Lonicera caerulea. Kim et al. demonstrated that BHe (BH extract) administration led to an increase of the antioxidant defense system in high-fat diet NAFLD induced mice. In particular, in the groups treated with the BHe, the GSH content resulted increased, in a dose-dependent manner [123]. Codonopsis lanceolata plant extract were found to increase GSH/GSSG ratio [124], and, remarkably, Cunningham et al. proved that curcumin prevention led to a significant increase of GSH levels in female Wistar rats [125].
Not only plant extracts are useful to counteract oxidative damage in NAFLD condition, but also probiotics. For instance, treatments with 1, 2, 4 × 1010 cfu Jlus66 (strain of Lactobacillus paracasei) daily for 20 weeks in high-fat diet-fed rats ameliorated the SOD activity, as well as GPx activity [126].
5.2. Alcoholic Liver Disease (ALD)
Alcoholic liver disease (ALD) is one of the commonest forms of liver disease globally. This is largely due to the ever-increasing consumption of alcoholic beverages, combined with a progressively sedentary lifestyle and wrong eating habits [127]. Under the spectrum of ALD a wide range of pathological conditions are addressed: simple steatosis, alcoholic steatohepatitis (ASH), fibrosis, cirrhosis, and HCC [128]. Among individuals who consume more than 60 g of alcohol per day and develop steatosis (about 90%), 25% progress to ASH and 10–20% of patients eventually develop cirrhosis [129]. ALD often fits into a picture already compromised by other factors such as hepatitis C virus (HCV) infection, or coexisting NAFLD, which accelerate the fibrotic process [130]. Several other factors can play an important role in the progression of ALD: environmental, genetic or behavioural causes can greatly influence the development of advanced stages of the disease [131]. Noteworthy, all these factors act by promoting an increase in oxidative stress via mitochondrial defects and the production of ROS and RNS [132]. Indeed, among the factors contributing to the progression of ALD to ASH and cirrhosis, oxidative stress certainly plays a key role [133]. Several works have shown that the accumulation of fat is toxic per se and causes lipo-toxicity [134]; the resulting fatty acid oxidation produces ROS, which can bind mitochondrial proteins, resulting in mitochondrial ROS production and mitochondrial dysfunction [135]. Moreover, it is well known that ethanol metabolism induces in the liver the production of ROS and RNS, altering mitochondrial structure and function and, concomitantly, decreasing many antioxidant mechanisms. This process, in turn, produces a dramatic decrease in ATP production, which alters the energy state of the cell, triggering the cell death process, in patients who chronically consume alcohol [136]. There are three main pathways employed by hepatocytes to metabolize ethanol and all produce ROS. The first pathway is catalyzed by alcohol dehydrogenase (ADH) and produces acetaldehyde from ethanol, leading to ROS and NADH generation [137]. The NADH produced can interfere with the mitochondrial electron transfer system producing ROS [138,139]. An additional amount of ROS is obtained by the microsomal ethanol-oxidizing system (MEOS) activity. This set of enzymes is located in the ER and the main player is the ethanol-inducible cytochrome P450 2E1 (CYP2E1), which converts ethanol to acetaldehyde, generating ROS [140,141]. Lastly, acetate is produced from ethanol in a reaction catalyzed by CAT in the peroxisomes [142]. Once acetaldehyde is obtained from ethanol metabolism, aldehyde dehydrogenase (ALDH) converts it into acetate, but if the system is overloaded, the excess of acetaldehyde reacts with mitochondrial GSH (mGSH), depleting the mitochondrial pool [143]. Since the mGSH acts in various molecular pathways involved in ROS detoxification, it is clear that the depletion of its stores by about 45–60% is strictly associated with the progression and development of ASH [144]. In in vivo rat models of ALD by intragastric ethanol feeding, it has been shown that chronic alcohol abuse causes a depletion of mGSH stores [145]. In chronic alcohol consumption, mGSH depletion is also caused by the disruption of GSH transport across the inner mitochondrial membrane, due to increased micro-viscosity caused by the accumulation of unesterified cholesterol on this site. A partial recovery in membrane fluidity of isolated mitochondria from ethanol-fed rat livers was obtained by the administration of SAMe, which restores the GSH mitochondrial pool [64,146]. A similar result was achieved employing taurourso-deoxycholic acid [147] or the fatty acid analogue 2-(2-methoxyethoxy)ethyl 8-(cis-2-n-octylcyclopropyl) (A2C), a fluidizing agent, in the same model of ethanol-fed rats [68]. Prolonged ethanol exposure, in fact, produces a reduction in the transport rate of GSH from cytosol to mitochondria, but not from mitochondria to cytosol, emptying the mitochondrial stores. The taurourso-deoxycholic acid administration restores GSH transport rate in hepatic mitochondria, replenishing the mGSH pool necessary to counteract the TNF-α mediated apoptosis [147]. The altered dynamic properties of the mitochondrial membrane, which affects GSH transport, could also be restored by A2C administration, as demonstrated in isolated mitochondria from long-term ethanol-fed rats [68]. Lastly, the depletion in the stores of mGSH may also originate from the activity of CYP2E1. CYP2E1 is located predominantly in the microsomal fraction of the cytosol, but is also expressed in mitochondria [148] and a three-fold increase in this enzyme was found in hepatic mitochondria isolated from ethanol-fed rats [149]. Not surprisingly, liver injury and oxidative stress are reduced in Cyp2e1−/− ethanol-fed mice or when CYP2E1 is pharmacologically inhibited [150]. On the other hand, the oxidative stress was higher in transgenic mice overexpressing CYP2E1 [151], or in mice infected with an adenovirus to overexpress Cyp2e1 gene and in HepG2 cells transduced with an adenovirus encoding the human CYP2E1 gene [152]. An interesting finding by Cederbaum et al. showed that ROS produced by CYP2E1 induces an increase in Nrf2 expression, which promotes the upregulation of GSH-related genes as a compensatory mechanism [153]. Moreover, Nrf2 also upregulates the expression of the xCT anti-porter system that favors the further restoration of GSH stocks through the entry into hepatocytes of extracellular cystine and the efflux of intracellular glutamate [154]. In this context, the increased extracellular glutamate can bind to its receptor, the metabotropic glutamate receptor 5 (mGluR5) on the stellate cell surface, stimulating it to synthesize 2-arachidonoylglycerol (2-AG) [155], which in turn, once released into the extracellular space, binds to the cannabinoid receptor-1 (CB1R) on hepatocytes, triggering a mechanism of de novo lipogenesis and therefore of fat accumulation [156] (Figure 7). It is therefore clear that some components of this cross-talk between stellate cells and hepatocytes could be good pharmacological targets to be exploited in the treatment of steatosis. In fact, as demonstrated by Ferrigno et al., the selective blockade of the mGlu5 receptor attenuates fat accumulation in an in vitro model of mild steatosis [157].
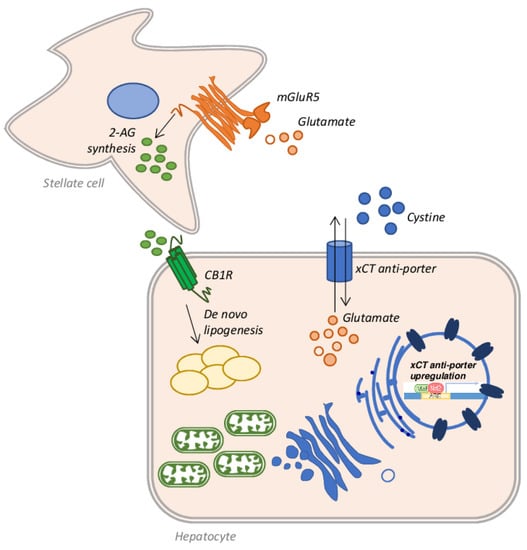
Figure 7.
Schematic representation of the cross-talk between hepatocytes and stellate cells in alcoholic liver disease (ALD). Nrf2 activation leads to the upregulation of the xCT anti-porter system, which introduces cysteine within the hepatocytes and favors the efflux of glutamate in the extracellular space. Glutamate binds its mGluR5 receptor on the stellate cell surface, which synthetizes and releases 2-AG. 2-AG, once bound to its receptor CB1R, promotes the de novo lipogenesis mechanism activation and consequent lipid accumulation in the hepatic cells.
Antioxidant Approach to the Treatment of ALD
At present, no pharmacological treatments for ALD and its progressive stages have been approved. The ideal treatment should be able to target all the issues of this pathology: increased inflammatory parameters, lipid accumulation, and oxidative stress generation. Obviously, given the enormous importance of oxidative stress in the progression of the disease, various treatments with antioxidant agents have been employed. As already mentioned, silymarin is a compound exhibiting antioxidant features [119,120]. In particular, silymarin shows antioxidant, anti-inflammatory, and anti-fibrotic properties useful in the treatment of ALD because it can reduce lipid peroxidation and increase GSH levels in cirrhotic patients after six months of treatment [158]. N-acetylcysteine (NAC), when used in animal models of early ALD, reduced lipid peroxidation, preventing hepatic GSH depletion [159,160]. However, these effects were not observed in severe stages of ALD, when used both alone or in combination with corticosteroids [161,162]. Among the emerging therapeutic strategies to reduce oxidative stress in ALD, several studies have shown the beneficial effect of folate, betaine, and metadoxine administration. Folate increases the conversion of homocysteine to methionine triggering the synthesis of SAM, useful to restore glutathione levels. Betaine acts as a methyl donor, providing a methyl group for the conversion of homocysteine to methionine. The synthetic drug metadoxine can restore NAD, ATP, glutathione, and adenosine levels both in the liver and brain, decreasing alcohol and acetaldehyde accumulation [163]. In a double-blind randomized trial, patients with ALD treated with 150 mg metadoxine for three months showed a considerable decrease in transaminase and γ-GT levels, as well as a reduction of steatosis, after the first month of treatment [164].
Several plant extracts have drawn attention for their antioxidant role in ALD. Quercetin, a compound belonging to the family of flavonoids, ameliorates lipid metabolism and ethanol-induced liver injury in animal models of ALD [165]. It has been demonstrated that it can induce antioxidant enzymes, increase GSH levels, reducing at the same time CYP2E1 activity and avoiding mitochondrial dysfunction and mGSH store depletion in rodents [166,167]. Similarly, honokiol, extracted from Magnolia officinalis, has been reported to improve ALD symptoms increasing hepatic levels of GSH and SOD activity in rats [168]; whereas barberine, a benzyl iso-quinoline alkaloid, increase levels of GSH and normalizes CYP2E1 hepatic expression in mice [169]. A similar replenishment of GSH stores was observed in primary rat hepatocytes exposed to ethanol and pretreated with curcumin [170].
It is well known that alcohol consumption affects the intestinal flora composition, so in recent years many groups have begun to pay attention on the role of probiotics in alcoholic liver disease. In particular, studies employing Lactobacillus rhamnosus GG in rats showed a reduction in alcohol-induced gut leakiness, oxidative stress and inflammation, both in the liver and intestine [171]. Moreover, Lactobacillus rhamnosus GG administration in ethanol-fed rats decreased both TNF-a and CYP2E1 protein and mRNA expression, enhancing Nrf2 protein expression [172].
5.3. Chronic Cholestatic Liver Injury
Cholestasis is a common impairment in bile formation, secretion, or flow. occurring in a plethora of human liver diseases [173] and is classified into two kinds: extrahepatic and intrahepatic. The first is the result of a mechanical blockade occurring in the biliary duct system caused by the presence of a gallstone, or a consequence of a pressure on bile ducts caused by tumor mass, bile duct tumors themselves, pancreatic tumors, pancreatitis, cysts, or primary sclerosing cholangitis. The intrahepatic cholestasis, instead, occurs inside the liver and could derive from ALD, amyloidosis, bacterial abscess, lymphoma, liver cancer, primary sclerosing cholangitis, viral hepatitis, or sepsis [174]. Moreover, cholestasis could be a metabolic impairment induced by genetic defects or scarring as seen in biliary atresia [175], or acquired as a side effect of many medications. In any case, the most commonly shared hallmark of different forms of cholestasis is an interruption of the enterohepatic circulation of bile acids and consequent retention of toxic bile salts in the bloodstream and at the intracellular level, leading to fibrosis and cirrhosis [173]. Toxic bile salts retention in the liver is accompanied by intracellular metabolic disorders, inflammation and reduced detoxification capacity [176], which may enhance the generation of oxidative stress and the accumulation of ROS and RNS in hepatocytes [177], leading to generalized systemic damage to major organs, starting from the liver and reaching the brain, heart, intestine, and kidney [178,179,180].
In the altered scenario of cholestasis, the retention of more hydrophobic bile salts, such as the glyco- and tauro-conjugates of deoxycholate, plays a major part in the onset of liver injury, causing hepatotoxicity due to mitochondrial dysfunction and production of ROS, with consequent oxidative damage [181,182]. Regarding GSH concentration during chronic cholestasis, conflicting data have been reported. However, many teams have demonstrated that a progressive decrease in GSH hepatocellular concentration occurs as the cholestasis progresses. Experiments conducted by Krahenbuhl et al. in a bile duct ligation (BDL) rat model of chronic cholestasis confirmed that the concentration of GSH and ubiquinone decreases during cholestasis, whereas an increase in lipid peroxidation is observed [183]. Moreover, GSH stores are consumed in animals subjected to experimental cholestasis or in hepatocytes exposed to toxic bile acids [184,185]. As shown by Neuschwander-tetri et al. and Serviddio et al., this fall in GSH stores is in part due to a decrease in GCL activity [184,186], a condition that, in rats subjected to the BDL procedure, can be prevented by the administration of urso-deoxycholic acid (UDCA) [186]. In fact, it has been demonstrated that UDCA can increase GCL expression in cultured rat hepatocytes [187]. Although the complete molecular mechanism underlying changes in GCL expression during chronic cholestasis or in response to UDCA has not yet been clarified, Yang and colleagues showed a transient and early increase in GSH-related enzymes, followed by their dramatic decrease in a second phase of cholestasis. In fact, they showed an early increase in both mRNA and protein expression of the two subunits of GCL (GCLC) and (GCLM), and also of GS from day 1 to day 7 after BDL, but these indices decrease below the baseline by day 14 to day 28 after BDL [188].
It is now well established that Nrf2, AP-1, NF-kB, and c-Myc are transcription factors able to induce the synthesis of GSH producing enzymes, both in humans and in rodents [189,190,191,192]. The mRNA and protein expression of these transcription factors remains unchanged after the BDL procedure, but GSH synthesis falls after 14 days of BDL. This occurrence is due to the impairment in Nrf2 ability to bind to AREs [188]. The binding of Nrf2 to AREs is controlled by the small Maf proteins (MafG, MafK, and MafF), which form heterodimers with Nrf2, effective in the activation of AREs, but at the same time, Maf proteins can homodimerize with each other, preventing the activation of AREs by Nrf2 [193,194]. Another protein able to block the Nrf2 activity is the Batch-1 protein. When an overexpression of Bach-1 occurs, as in high oxidative stress, it competes with Nrf2 to heterodimerize with small Maf proteins, repressing ARE-mediated gene expression [195]. Then, to understand why GSH synthesis decreases after BDL, it is necessary to know what happens to Maf and Bach-1 proteins. From day 14 to day 28, the hepatic nuclear content of small Maf proteins increases significantly. They form homodimers and prevent the bond between Nrf2 and AREs, which occurs early in the first period after the BDL procedure [188]. This fine regulatory mechanism at the level of gene transcription can therefore partly explain why the key enzymes in the synthesis of GSH decrease during cholestasis leading to an emptying of the GSH stores.
Another pathway involved in GSH depletion in cholestasis is strictly related to inflammation. In fact, in the compromised scenario of cholestasis, an inflammatory process is established, with a concomitant increase in levels of acute-phase proteins (APP) like serum TNF-α and biliary phospholipase A2, resulting in a further increase in oxidative stress highlighted by raised levels of MDA in the bile [176]. In rat models of extrahepatic cholestasis, it has been demonstrated that the regulation of some proteins of the acute phase depends on GSH, whose depletion causes beta-fibrinogen and haptoglobin up-regulation [196]. Lastly, another condition that affects GSH content is the bile regurgitation into the blood, caused by the intra-biliary pressure increase that causes the canalicular and Disse’s spaces able to establish a deep communication during prolonged obstructive cholestasis [197]. Thus, the toxic molecules can re-circulate between the bile and the hepatocytes, causing further depletion of GSH stores.
Antioxidant Approach for the Treatment of Cholestasis
Cholestasis affects not only the liver, but is a pathological condition that spreads in different organs. Unfortunately, currently no specific pharmacological strategy is optimal to treat cholestasis-induced organ injury. Nevertheless, several attempts have been made over the years to try to decrease the oxidative stress that plays a great part in its progression. Among them, UDCA has been demonstrated to be a valid candidate. In fact, it has been shown that UDCA pre-treatment protects cultured rat hepatocytes against oxidative stress induced by ROS, significantly increasing the total amount of GSH and thiol-containing proteins, via the up-regulation of GCL mRNA expression and metallothionein [187]. Serviddio et al. showed similar data on increased levels of GCL and γ-cystathionase mRNA in a BDL rat model [186]. Another compound that has been demonstrated to be useful in a rat model of extrahepatic cholestasis is silymarin. Indeed, silymarin-loaded gold nanoparticle, orally administered in BDL-rats for 7 days, improved liver function, reducing transaminase, γGT, and direct and total bilirubin serum levels, diminished oxidative stress markers both in serum and liver and, lastly, increased antioxidant support [198]. Agmatine has also shown antioxidant properties in a rat model of obstructive cholestasis. Agmatine is naturally produced in humans by arginine decarboxylation; it plays modulatory effects in various molecular pathways, including neurotransmitter systems, nitric oxide (NO) synthesis and polyamine metabolism [199]. Ommati et al. showed that the administration of agmatine in BDL rats for seven days after bile duct ligation was able to reduce ROS and nitric oxide formation and lipid peroxidation, and to increase mitochondrial GSH both in the liver and in the kidney in a dose-dependent way [200]. Moreover, the same research group recently obtained interesting data also on edaravone antioxidant capacities in the BDL rat model of obstructive cholestasis [201]. Edaravone showed potent antioxidant and radical scavenging activities in various pathologies, such as cerebral ischemia, stroke and amyotrophic lateral sclerosis [202,203,204], also improving oxidative stress-induced mitochondrial impairment in various diseases [205,206]. In their work, Ommati et al. demonstrated that BDL-rats treated with edaravone, at a dose of 1–10 mg/kg/day intraperitoneally for 14 days, show significant improvements in cholestasis-associated hepatic and renal injury, via reduction of ROS levels, lipid peroxidation, protein carbonylation, and GSSG levels. Antioxidant defenses, such as the total amount of GSH, and mitochondrial function were restored both in the liver and kidney [201]. Lastly, it is well known that prolonged cholestasis can induce cirrhosis and consequent disruption of gut barrier function and integrity [207]. This occurrence can favor bacterial lipopolysaccharide (LPS) translocation to the systemic circulation, leading to higher morbidity and mortality in cirrhotic patients [208]. In this case also, oxidative stress plays a notable role; in fact, NAC and dithiothreitol (DTT) induced a significant decrease in oxidative stress and restored intestinal antioxidant capacity in a BDL-induced model of cirrhosis in rats [209]. Finally, bombesin (BBS) and neurotensin (NT), two regulatory gut peptides, have been demonstrated to play a role in reducing hepatic oxidative stress in rats treated for 10 days with BBS and NT after BDL [210].
As regards plant extracts, quercetin also showed beneficial effects in the treatment of cholestasis. In fact, it has been shown in rats, that quercetin alleviates BDL-induced oxidative stress, leukocyte recruitment, NF-kB activation and pro-inflammatory cytokine secretion [211]. Moreover, another compound that showed antioxidant properties is hydroalcoholic extract of watercress, which is able to reduce oxidative stress in rats subjected to a BDL procedure, preventing hepatic protein oxidation and promoting GPx activity via antioxidant effect and free-radical scavenging [212].
5.4. Hepatic Ischemia/Reperfusion Injury
Hepatic Ischemia/Reperfusion (I/R) injury occurs during liver transplantation, resection surgery, and hypovolemic shock. The reperfusion occurring after ischemia elicits pathogenic processes, exacerbating injury caused by ischemia per se [213]. During ischemia, hepatocytes show morphological changes at the plasmatic membrane level, consisting in bleb formation. When blebs burst out, a release of enzymes and intracellular catabolites occurs, followed by the collapse of the ionic and electrochemical gradients, mitochondrial impairment, cellular acidosis, and Kupffer cells activation [214]. Instead, during the reperfusion period, reactive oxygen species, oxidative stress, mitochondrial dysfunction, and systemic inflammatory response take place [213]. After the I/R process, ROS are produced from different sources. Several protective mechanisms are triggered at the cellular level, many of them controlled by the Nrf2 transcription factor, which is normally inhibited by Keap1 [215,216]. When oxidative stress increases, Nrf2 moves to the nucleus enhancing the expression of antioxidant genes, such as NAD(P)H: quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HO-1), GCL, microsomal epoxide hydrolase, GST, and sulfiredoxin 1 [217]. GSH levels are controlled by Nrf2 via transcriptional activation of the GCL gene [189]. In a model of Nrf2−/− null mice subject to partial ischemia and reperfusion, the expression of several Nrf2-dependent genes was compromised, including GSTm1, NQO1, and GCL, resulting in the worsening of the redox state in comparison with livers from wild-type mice [218]. Moreover, a continuous release of GSH from hepatocytes to extracellular/vascular space detoxifies ROS generated by Kupffer cells, protecting the hepatic vasculature, as demonstrated in a in vivo model of partial no-flow ischemia/reperfusion injury [219].
Antioxidant Approach for the Treatment of Ischemia/Reperfusion Injury
Considering the relevant part played by oxidative stress in hepatic I/R injury, many attempts have been made to counteract this impaired condition. Different antioxidant agents showed clinical or experimental advantages if employed in I/R injury. For example, exogenous GSH administration in liver I/R at the dose of 100 μmol/h/kg showed significant beneficial effects both in cold and warm liver ischemia in rats [220,221]. Furthermore, in a model of isolated rat liver perfusion, GSH administration during cold preservation prevented hepatocyte damage, deterioration of hepatic circulation, and the fall of intracellular GSH levels during the reperfusion period [222]. Similar results were achieved by Schauer and colleagues, in a model of warm ischemia, followed by isolated rat liver perfusion [221]. In addition, many studies have also demonstrated the beneficial effects of NAC administration in hepatic ischemia, both in experimental and clinical settings. In in vivo and ex vivo models of hepatic ischemia and reperfusion injury [223,224], NAC administration had protective effects, increasing GSH levels and reducing ROS [225]. Moreover, when administered in a high dose, NAC is also able to support mitochondrial energy metabolism [226].
In a randomized Phase II study in patients undergoing liver transplantation, 30 mg/kg NAC administered 1 h before liver procurement and 150 mg/kg via portal vein in the recipient before liver implantation improved transplant survival by about 20% after one year and limited the risk of primary non-function. The authors argue that these findings should be kept in mind when suboptimal grafts are used, such as livers obtained from older people; moreover, early NAC systemic infusion is needed to reach a positive effect, filling the GSH stores before transplantation. All these beneficial effects of NAC administration go hand in hand with low cost and absence of adverse side effects [227]. Lastly, another attractive and innovative strategy in glutathione management is gene transfer. Considering that GSH has a very short half-life [228], gene transfer of glutathione synthesis components such as GCLC, GCLM, and GS could be a useful therapeutic approach in I/R injury through an increase in GSH intracellular concentration [215,216]. Gene therapy applied in hepatic I/R injury consists mainly in viral vectors, like recombinant virus transfection. In a rat model of I/R injury, the administration of high doses of mitochondrial SOD, via viral vector, inactivated NF-kB and activator protein-1, preventing I/R [229]. In addition, in a rat liver transplantation model using healthy and steatotic livers, the cytosolic and mitochondrial SOD delivered with adenovirus ameliorated the graft survival [230]. Lastly, in an in vitro study using murine NIH-3T3 cells, the transduction of human genes encoding for the peroxidase peroxiredoxin protected cells from oxidative stress [231]. Gene therapy, thus, could improve the effect of I/R and in particular could be applied in liver resection for tumors, or living donor liver transplantation. However, further studies are needed to clarify the effective efficacy of gene therapy in treating I/R injury [232].
Concerning plant derived compounds, Hypericum perforatum L. extract (HPE) showed antioxidant properties in studies focusing on I/R injury. In particular, rats subjected to I/R and pretreated with HPE displayed a significant decrease in hepatic damage markers such as ALT, AST, LDH and MDA. Moreover, a marked increase in CAT and GPx activity was found in livers from HPE-treated rats [233]. A similar effect was obtained also using rosmarinic acid, which showed anti-inflammatory and antioxidant role in rats subjected to ischemia and reperfusion procedure [234]. Intestinal microflora plays a role in many kinds of liver disease and also in I/R injury. It has been found that rats, treated with Bifidobacterium Catenulatum ZYB0401 and Lactobacillus Fermentum ZYL040, and then subjected to I/R, showed a decrease in TNF-a and hepatic MDA levels, accompanied by increased liver SOD activity [235].
5.5. Hepatitis C Virus (HCV) and Hepatitis B Virus (HBV)
According to the World Health Organization report, Hepatitis C virus infections worldwide total 71 million, with 400,000 deaths per year. After a six month period of acute infection, undiagnosed hepatitis C becomes chronic in about 80% of infected patients, leading to steatosis, fibrosis and cirrhosis; one patient out of five develop end-stage hepatocellular carcinoma [236,237]. Briefly, HCV is a positive single-stranded RNA virus belonging to the Flaviviridae family. Its genome is about 9600 bases in length and the translated polypeptide encodes for both structural (core, E1 and E2) and non-structural (NS1, NS2, NS3, NS4 A/B, NS5 A/B) proteins [238] (Figure 8A). Even though oxidative stress is considered a particular hallmark of hepatitis in both acute and chronic infection [239], contradictory data have been reported about the effect of HCV on intracellular GSH metabolism. In Huh7.5 cells infected by HCV genotype 2 an increase in GSSG and a decrease of GSH has been observed by Roe et al. [240], whereas de Mochel, using HCV-infected Huh7 cells, found an increase in GSH [241]. In the acute phase, viral replication takes place in the so called “membranous web” in the ER [242]. Moreover, ER stress induced by NS5A and core HCV proteins causes calcium release from the ER into the cytoplasm. Through mitochondrial transition pore (MTP), calcium enters into mitochondria, enhancing ROS production and misbalancing electron transport chain [243]. Oxidative stress can also be caused by the mitochondrial localization of viral proteins. Viral NS3/NS4 complex, associated with the outer mitochondrial membrane, cleaves the mitochondrial antiviral signalling proteins, abrogating the innate immune response pathway [244]. A ROS increase has also been associated to the presence of viral core proteins within the outer and inner mitochondrial membrane and in the mitochondria-associated membrane, which links the ER to mitochondria [243]. Korenaga et al. demonstrated that transgenic mice expressing HCV structural genes were characterized by an altered mitochondrial oxidative status. In transgenic mice, mitochondria GSH levels decreased compared to controls. Similarly, normal mitochondria treated with different concentrations of HCV core protein showed a significant oxidation of GSH. Moreover, the core protein increased calcium influx, and reduced electron transport chain complex I activity and consequent ROS production [245]. In line with these findings, hepatocytes overexpressing HCV core protein were also found to have decreased GSH levels and increased thioredoxin oxidation; while hepatocytes overexpressing viral NS5A protein have increased levels of GSH, CAT and SOD [246].
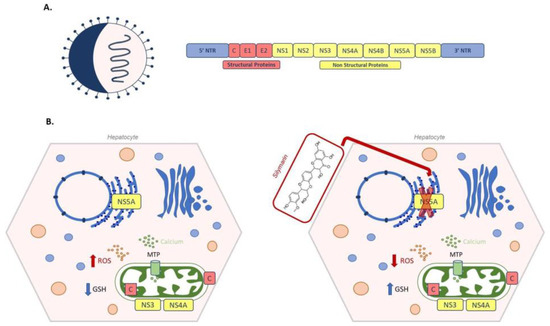
Figure 8.
Schematic representation of hepatitis C virus (HCV) and silymarin treatment. (A) On the left a viral particle of HCV, on the right a HCV genome, with structural (Core, Envelope 1 and Envelope (2) and non-structural proteins (NS1, NS2, NS3, NS4A, NS4B, NS5A and NS5B). (B) On the left, an example of oxidative stress caused by viral proteins: core protein C binds to the inner and to the outer mitochondrial membrane; viral NS3/NS4 complex is associated with the outer mitochondrial membrane, while NS5A localizes on the endoplasmic reticulum. Viral NS5A increases calcium release from the ER into the cytoplasm. Through mitochondrial transition pore (MTP), calcium enters into the mitochondria, enhancing ROS production, together with C and NS3/NS4 proteins. On the right, silymarin administration blocks NS5A polymerase activity, ROS decreases and GSH levels increase.
HCV also impairs redox state signalling pathways, such as Nrf2/ARE. It has been shown that, in the early phase of HCV infection, ROS-induced MAPK/JNK or PKC signalling mediates the activation of Nrf2 and Nrf2-activated antioxidant genes [243,247]. Other authors have reported an inhibitory effect on Nrf2 signalling due to the binding of sMaf to NS3 viral protein [248]. One of the side effects of Nrf2-downregulation is the inhibition of GPx4: in this case, lipid peroxides accumulate, worsening the damage and causing ferroptosis [249]. Conversely, NS5A viral protein is able to induce GPx4 by PI3K/AKT/mTOR pathway activation, protecting HCV infected cells against lipid peroxidation [250]. Another mechanism by which HCV causes oxidative stress is the induction of NADPH oxidases (NOX), which generate superoxide anions and hydrogen peroxide starting from molecular oxygen and NADPH [251]. In particular, during the acute phase, in which HCV replicates at a high rate, a marked oxidative stress due to the NOX4 isoform, ubiquitously expressed in parenchymal and non-parenchymal liver cells, has been observed, as well as a concomitant decrease in GSH. On the contrary, during the chronic phase, ROS production returns to basal levels and GSH content is restored [241]. In fact, in chronically HCV-infected patients, GSH content was found both in serum and hepatic tissue [252]. Furthermore, the HCV genotypes also affect glutathione levels in a different way. Patients affected by genotype 1b exhibited lower levels of hepatic and plasmatic glutathione, which correlates with increased lipoperoxidation markers as well as serum ferritin compared to patients with genotype 2a/2c and 3a and to controls [252]. Besides higher ferritin levels, probably due to activation of Nrf2-induced ferritin chains [253], patients can present higher iron content, which could lead to ferroptosis. Thus, the subtype 1a/1b, having not only low GSH but higher GSSG, cause more serious disease in contrast to the subtypes 2a/c, 3a and 4 [254]. GSH levels are re-established after antioxidant treatment (with a mixture of vitamin E, C, and zinc) alone or in combination with peg-interferon and ribavirin. Moreover, GPx and other antioxidant enzymatic idexesalso ameliorate after antioxidant supplementation. Concomitantly, peroxidation and iron content decrease, while zinc levels increase [255,256].
It is estimated that Hepatitis B virus (HBV) chronically infects 260 million people globally, causing almost one million annual deaths [257]. Besides chronic infection, HBV can cause acute, fulminant and occult infection [258]. HBV is a member of the Hepadnaviridae family. Its genome is a circular, partially double-stranded DNA, which encodes four overlapping open reading frames: C (core, HbcAg), X (HBx), P (DNA polymerase) and S (surface antigen, HbsAg). While C, P, S proteins have specific roles, HBx function is not completely understood: in fact, it seems to be implicated in signal transduction, DNA repair, transcriptional activation, as well as protein degradation inhibition. Moreover, it plays a central role in the induction of oxidative and inflammatory responses and in the progression to HCC [259].
The mechanisms of reactive oxygen species generation in HBV infections are several. ROS are useful for HBV assembly, by heat shock protein-90 (HSP-90) interaction with the core protein [260]. The presence of GSH, instead, changes HSP-90 conformation, abrogating HBV assembly [261]. HBV-induced ROS also enhance TNF-α, which activates JNK. In turn, JNK down-regulates both the expression and the activity of GPx in HBV-transgenic mice [262]. HBV-infected cell cultures and liver tissues have been demonstrated to induce antioxidant defence by the Nrf2/ARE pathway [263,264]. In particular, it has been shown that HBx protein forms a complex with p62 and Keap1; then, in HBV-infected HepG2.2.15 cells, the activated Nrf2 translocates to the nucleus [265] and stimulates ARE-regulated genes [264]. Moreover, HBV-infected HepG2.2.15 showed increased expression of GS, GR and GCL subunits when compared to non-infected HepG2 cells. Surprisingly, HepG2 cells exhibited higher levels of GSH with respect to infected HepG2.2.15, probably because GSH is engaged in facing HBV-induced ROS [266]. Short-term studies on HBV-inducible cell line HepAD38 also confirmed that HBV replication stimulates oxidative stress, as revealed by higher MDA levels after 24-h induction. Even though total GSH did not change during HBV infection, GSSG/GSH ratio has been observed to triple after 72 h HBV-induction, indicating that GSH depletion occurred in favour of its oxidized form [267]. However, in contrast with these findings, Nrf2/ARE downstream genes could be epigenetically suppressed. Hence, in HBV-hepatoma cells, DNA methyltransferase 3A (DNMT3A) hyper-methylates NQO1 promoter, silencing it in a HBx-dependent manner [268]. Another example regards two isoforms of glutathione-S-transferase: M3 (GSTM3) and pi (GSTP1) [269]. Patients with chronic hepatitis B displayed a positive correlation between oxidative stress (MDA), methyltransferase expression (DNMT1 and DNMT3A) and methylation of GSTM3 and GSTP1. In HepG2.2.15, GSTP1 protein expression decreases, while, curiously, glutathione-S-transferase omega 1 (GSTO1) expression increases [270]. This could be traced back to the fact that GSTO1 is not under Nrf2/ARE control [271]: it acts more like glutaredoxins than GSTs [272] and is involved in the activation of inflammasome [273]. Similarly, in HBV mice transgenic for the antigen HbsAG, GSTP1 protein expression decreases, while Nrf2/ARE-independent glutathione-S-transferase kappa 1 (GSTK1) increases [274]. Furthermore, HBV infection is able to change the expression levels of antioxidant enzymes in an Nrf2-/ARE independent fashion. For example, HBx inhibited GSTA2, GSTM1 and GSTM2 gene expression by C/EBP-mediated mechanism [275]. Another way for HBx to induce oxidative stress is the suppression of proteins indirectly involved in the antioxidant defence, as observed in HBx-transfected HepG2 cells for the inhibition of selenoprotein P (SeP) [276] and in HBV-transgenic mice for selenium-binding protein 2 (Selenbp2) [274]. These data, in addition to low selenium content in HBV patients [277], exacerbate liver damage, hindering the activity of GPx, GST and thioredoxin reductase. However, as seen previously for HCV, the genotype affects the induced pathway. For example, genotype A presents a higher activation of Nrf2/ARE pathway than genotype G [263]. Similarly, in HepG2 cells infected by genotype D, GSTP1 is inhibited, but is not in genotypes A, B and C [278]. Even though much effort has been made to clarify the mechanisms at the basis of redox status changes in HBV in in vitro and in vivo models, the role of antioxidants has not been fully elucidated. However, several studies in plasma patients seem to agree on the increase in oxidative stress and on the decline of GSH and GSH-related enzymes [279]. In fact, increased MDA and transaminase levels in plasma of chronic HBV patients and diminished levels of GSH and GPx were reported, while GST content was upregulated [280,281]. In a study on Egyptian patients, along with MDA, GR also increases, whereas GSH, GPx and GST diminished [282]. GSH intravenous injections improved liver function, as indicated by reduced levels of transaminases and cytokines [283].
Antioxidant Approach for the Treatment of Viral Hepatitis C and B
Regarding viral hepatitis, several trials suggest that antioxidant therapy might be beneficial for patients with chronic infection. For instance, the supplementation of vitamins has been associated with the protection against free radical damage, HCV- or HBV-induced. The administration of GSH, a precursor or a derivative, inhibits the replication both in vitro and in vivo. The administration of a mixture of antioxidants, including GSH, decreased transaminase levels in comparison with placebo treatment and was well tolerated by chronic HVC patients [284]. Nigella sativa, both seeds and oil, has been demonstrated to have beneficial effects on HCV patients, thanks to its antioxidant properties [285]. In HCV patients the administration of sofosbuvir and ribavirin, along with black cumin seeds, derived from N. sativa, and ascorbate, reduced transaminase levels, increasing GSH and SOD, and decreasing GSSG, GGT, and MDA, when compared to sofosbuvir and ribavirin-treated patients [286]. Similar results were obtained for HBV treatment. In fact, an intravenous drip of 1200 mg of GSH was successful in decreasing the serum levels of transaminases, TNF-α, interleukin-6, and interleukin-8, compared with the control group, suggesting that GSH treatment can ameliorate liver function and suppress inflammation and hepatic fibrosis in chronic hepatitis B patients [283]. Even though a 48-h NAC treatment has been associated with about a 50-fold decrease in HBV assembly in HepG2-A5 cells [247], NAC has been revealed to be ineffective in acute viral B hepatitis treatment [287], However, other studies reported that NAC alone or in combination with other antiviral had no effects on plasma concentration of GSH, SOD and CAT for HBV [288,289] or HCV [290].
Silymarin treatment has been evaluated in viral hepatitis. Silymarin exerts its antioxidant effects in vitro not only by blocking NS5B polymerase activity, but mainly by inhibition of viral entry, stabilizing the membranes and yielding them less prone to fusion. Moreover, silymarin inhibits MTP activity and apoB secretion, limiting the viral spread [291] (Figure 8B). Similarly, silibinin, the major constituent of silymarin, has been demonstrated to inhibit HBV entry into hepatocytes in in vitro studies [292]. RT-PCR analysis revealed that the administration of silymarin upregulates genes involved in GSH metabolism, as well as in glutamate and citrate metabolisms, which indirectly could increase GSH [293]. Unfortunately, there are no univocal data about in vivo hepatoprotection mediated by silymarin [294]. In fact, the administration of silymarin formula MediHerb 600–1200 mg daily for 12 weeks did not change transaminase levels nor HCV RNA titer versus placebo [295]. Similar results were observed by Hawke [296] and Fried [297]. The supplementation of several antioxidants to 250 mg of silymarin demonstrated histological improvements as well as ameliorated ALT content [284]. As for HCV treatment with silymarin for HBV, there are also confusing reports. Wei et al., in their meta-analysis, concluded that silymarin seems to be safe and well tolerated by patients and, in combination with antiviral drug or protection liver drugs, serum transaminases were reduced [298].
Another strategy to counteract oxidative stress in viral hepatitis could be the use of probiotics. In fact, according to several studies, dysbiosis occurred, leading to a decrease in Bifidobacteria and Lactobacilli and the proliferation of Bacteroidetes and Enterobacteriaceae [299]. The lipopoli-saccharides of these Gram-negative bacteria causes endotoxemia in HCV and HBV patients [300], with the consequent decline in SOD and MDA levels [301] and increase in pro-inflammatory cytokines and ROS [302]. However, the administration of Bifidobacteria, as well as lactitol [301], or feces transplant [303], is associated with gut microbioma re-establishment [301].
5.6. Hepatocellular Carcinoma (HCC)
HCC represents about 75–85% of primary liver cancers cases, followed by intrahepatic cholangiocarcinoma (10–20%) and rare tumors, including hepatoblastoma [304]. HCC is a complex and heterogeneous tumor characterized by multiple genetic mutations in genes controlling cell proliferation, cell death, homeostasis and oxidative stress [305]. However, it is well known that the main risk factors for HCC development and progression are chronic infections due to viral hepatitis and alcoholic and non-alcoholic fatty liver disease, along with metabolic syndrome [306]. As reported in this review, since the liver diseases previously described have oxidative stress in common for their pathogenesis, it follows that the worsening of oxidative damage contributes to HCC development. Transient induction of Nrf2 has been observed to provide protection against oxidative stress. However, persistent activation of Nrf2, due to Keap1 or Nrf2 gene mutations, can drive HCC pathogenesis [307]. In line with these considerations, an Nrf2-dependent increase in GCLC expression levels and activity was observed in tumors, in comparison with peritumoral tissues [308]. Moreover, Nrf2 overexpression contributes to sorafenib-induced ferroptosis resistance both in in vitro and in in vivo models. In HCC cell lines, upon sorafenib administration, p62 inhibited Nrf2 degradation and enhanced its nuclear accumulation. On the contrary, C57BL/6 mice injected subcutaneously with Nrf2 knockdown Hepa1–6 cells showed a decrease in tumor size and diminished NQO1 and HO-1 mRNA expression after sorafenib administration when compared to Nrf2-shRNA control mice [309]. In contrast, the administration of curcumin in HepG2 cells treated with the conditioned medium derived from hepatic stellate cells prevents HIF-1α stabilization, suppresses angiogenesis and upregulates GSH through the Nrf2 pathway [310]. Other contrasting data were reported on selenium dependent GPx. Se deficiency increases cancer incidence [311]. Hence, low Se levels have been observed in HCC patients’ blood, while its supplementation decreased the risk of cancer [311]. However, the seleno-protein GPx2 plays an important role in the progression of malignant tumors [312]. In HCC tissue, immunohistochemical analyses detected GPx2 overexpression when compared to adjacent non-tumor tissues [313]. Similarly, GPx4 and GPx7 were found to be overexpressed in HCC tissues and their levels correlate with the malignancy grade [314]. Several studies investigated GSH and GSH-related enzymes in HCC tissues. GSH content was lower in HCC tissues when compared to healthy livers, as well as selenium-dependent GPx, GST and GR [315,316]. Several studies report a decrease in GSTP1 in HCC tissues when compared to healthy or HBV patients [317], probably due to the hypermethylation of its promoter [318]. Likewise, the opposite has been shown. GSH content doubled in HCC biopsies as compared to normal liver, due to increase in GCL heavy subunit and GS. The presence of regulatory elements like ARE, activator protein-1 and NF-kB on GCL heavy subunits promoter enhances its transcription. Moreover, GSH increase has been positively correlated with HepG2 cell growth: inhibiting or increasing cellular GSH, DNA synthesis and cell proliferation were, respectively, reduced or improved [319]. Thus, GSH increase seems to promote tumour growth. In fact, GSH nuclear pool has also been reported to be fundamental for the advancement of cell cycle from G1 to S-phase in healthy tissues. A shift toward GSSG redox state, instead, keeps the cell cycle blocked into G1 phase [320]. Recently, Cheng et al. confirmed the presence of increased GSH content in HCC tissue, when compared to neighbouring healthy tissues. Interestingly, they found a difference in oxidative markers in plasma before and after the tumor resection: after surgical intervention, MDA concentration lowered and GSH levels were restored [321]. Besides the nuclear GSH pool, in HCC cells and human HCC tumors, mitochondrial GSH increase has also been observed [322]. Indeed, the 2-oxoglutarate carrier is over-expressed in HCC, ensuring unlimited mGSH levels, useful to counteract ROS. Silencing this carrier, instead, is associated with decreased liver tumorigenesis in vivo.
Antioxidant Approach for the Treatment of Hepatocellular Carcinoma
Contrasting data 0n the efficacy of GSH in HCC have been reported. GSH and antioxidant agents are often recommended in hepatic chronic diseases to prevent hepatocellular carcinoma [121,323]. In preclinical studies, silymarin treatment has been associated with ROS decrease in HCC cell culture [324], and in HepG2 cells it decreased cellular proliferation [325]. Similarly, in N-nitroso-diethylamine-induced rat hepatocarcinogenesis, silymarin induced a decrease in lipid peroxidation concomitantly to an increase in GSH [325,326]. The administration of quercetin, a plant polyphenol, was associated with enhanced GSH levels and a reduction in lipid peroxidation, resulting in the inhibition of cancer development, both in vitro and in vivo models [327,328]. Tea polyphenols seem to have positive effects on gut microbioma, repressing pathogenic bacteria in favour of friendly ones. The same role is exerted also by pro-anthocyanidins, found for example in almonds, grapes and chocolate [329]. Phenols contained in curcumin were reported to be able to arrest cell cycle and inhibit proliferation and metastasis [330].
However, it is also well known that the administration of antioxidants such as GSH or its precursors, as well as vitamins, increases chemoresistance [331]. In fact, elevated GSH and GCLC levels were found in different malignant tumors, masking the tissue more resistant to chemotherapeutic drugs such as cisplatin [332,333]. GST isoforms have also been found to increase HCC cells’ resistance to chemotherapy [334]. For this reason, GSH depletion has been considered a potential strategy to sensitize tumor cells to therapy [331]. Tompkins et al. demonstrated that the disruption of mitochondrial pyruvate carrier downregulates GSH metabolism, with lethal effects on carcinoma-induced hepatocytes [335]. The generation of oxidative stress could also be used as a therapy. Indeed, three hepatocellular cancer cell lines (Huh-7, HepG2 and PLC-PRF-5), treated with the anticancer sorafenib in combination with the anti-inflammatory diclofenac, showed a greater oxidative stress and cell death, associated with GSH decrease [336]. However, it has been shown that the use of amifostine, an antioxidant scavenger, could exert a protective effect on normal tissues [337]. In fact, because its thiol is susceptible to pH and alkaline phosphatase, it acts only in normal tissues, without affecting the anticancer effects of chemotherapy and ionizing radiation [338,339,340].
6. Conclusions
GSH represents one of the most commonly investigated redox-active molecules, as changes in its content contribute to the pathogenesis of many diseases. It is now evident that GSH is not merely a ROS and RNS scavenger. In fact, GSH has additional roles in cell physiology, such the metabolization of toxic substances, cell signaling through protein S-glutathionylation and cell survival. Oxidative stress contributes to the pathogenesis of many liver diseases: it is involved in SEC activation and fibrotic progression in NAFL and ALD; it induces mitochondrial dysfunction in cholestasis and in ischemia/reperfusion injury; it is also a hallmark of viral hepatitis and hepatocellular carcinoma. Consequently, the supplementation of GSH or GSH precursors has been demonstrated to be a suitable contribution to the therapy for many hepatic conditions.
Author Contributions
Conceptualization, A.F. and M.V.; resources, P.R.; writing—original draft preparation, M.V., A.F., L.G.D.P., C.B. and M.C.; writing—review and editing, A.F., C.B. and P.R.; supervision, A.F. and M.V. All authors have read and agreed to the published version of the manuscript.
Funding
No external funding was used for this work.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 2-AG | 2-arachidonoylglycerol |
| 4-HNE | 4-hydroxynonenal |
| ADH | Alcohol dehydrogenase |
| Akt | Protein kinase B |
| ALD | Alcoholic liver disease |
| ALDH | Aldehyde dehydrogenase |
| ALT | Alanine aminotransferase |
| AP-1 | Activator protein 1 |
| APP | Acute phase protein |
| ARE | Antioxidant response element |
| ASH | Alcoholic steatohepatitis |
| ATP | Adenosine triphosphate |
| BBS | Bombesin |
| BDL | Bile duct ligation |
| Bhe | Blue honeysuckle extract |
| CAT | Catalase |
| CB1R | Cannabinoid receptor-1 |
| CHB | Chronic hepatitis B |
| CYP2E1 | Cytochrome P450 2E1 |
| DIC | Dicarboxylate carrier |
| DILI | Drug-induced liver injury |
| DNMT | DNA methyltransferase |
| DTT | Dithiothreitol |
| ER | Endoplasmic reticulum |
| ETC | Electron transport chain |
| FA | Fatty acid |
| FCCP | carbonylcyanide p-(trifluoromethoxy)phenylhydrazone |
| GCL | l-glutamate l-cysteine γ-ligase |
| GCLC | GCL catalytic monomer |
| GCLM | GCL modifier monomer |
| GPx | Glutathione peroxide |
| GR | Glutathione reductase |
| Grx | Glutaredoxin |
| GS | GSH synthetase |
| GSH | Reduced glutathione |
| GSSH | Oxidized glutathione |
| GST | Glutathione S transferase |
| HBV | Hepatitis B virus |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| HO-1 | Heme oxygenase-1 |
| HPE | Hypericum perforatum L. |
| HSP90 | Heat shock protein 90 |
| I/R | Ischemia/reperfusion |
| IL-6 | Interleukin-6 |
| IMM | Inner mitochondrial membrane |
| IR | Insulin resistance |
| JNK | c-Jun N-terminal kinases |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LPS | Lipopolysaccharide |
| MAF | Musculoaponeurotic fibrosarcoma oncogene homolog (avian) |
| MAPK | Mitogen-activated protein kinase |
| MAT1A | Methionine adenosyltransferase-1 |
| Mcp1 | Monocyte chemoattractant protein 1 |
| MDA | Malondialdehyde |
| MEOS | Microsomal ethanol-oxidizing system |
| mGSH | Mitochondrial glutathione |
| MnSOD | Manganese-dependent superoxide dismutase |
| mTOR | Mammalian target of rapamycin |
| MTP | Microsomal triglyceride transfer protein |
| NAC | N-acetylcysteine |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NAFLD | Non-alcoholic fatty liver disease |
| NAPQI | N-acetyl-p-bemzoquinone imine |
| NASH | Non-alcoholic steatohepatitis |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| NQO1 | NAD(P)H:quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NT | Neurotensin |
| OGC | 2-oxoglutarate carrier |
| OMM | Outer mitochondrial membrane |
| PAI1 | Plasminogen activator inhibitor 1 |
| PDI | Disulfide isomerase protein family |
| PI3K | Phosphoinositide 3-kinase |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SAM | S-adenosyl-l-methionine |
| SLG | S-d-lactoylglutathione |
| SOD | Superoxide dismutase |
| SREBP-1c | Sterol regulatory element-binding protein 1C |
| TBARS | Thiobarbituric acid reactive substances |
| TNF-α | Tumor necrosis factor alpha |
| UDCA | Ursodeoxycholic acid |
| WD | Western diet |
| xCT | Cystine/glutamate antiporter |
| γ-GT | Gamma-glutamyltransferase |
References
- Pizzorno, J. Glutathione! Integr. Med. 2014, 13, 8–12. [Google Scholar]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 1–26. [Google Scholar] [CrossRef]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.C.; Ashraf, S.S.; Rokutan, K.; Johnston, R.B.; Thomas, J.A. S-thiolation of individual human neutrophil proteins including actin by stimulation of the respiratory burst: Evidence against a role for glutathione disulfide. Arch. Biochem. Biophys. 1994, 310, 273–281. [Google Scholar] [CrossRef]
- Samuele, A.; Mangiagalli, A.; Armentero, M.-T.; Fancellu, R.; Bazzini, E.; Vairetti, M.; Ferrigno, A.; Richelmi, P.; Nappi, G.; Blandini, F. Oxidative stress and pro-apoptotic conditions in a rodent model of Wilson’s disease. Biochim. Biophys. Acta 2005, 1741, 325–330. [Google Scholar] [CrossRef]
- Ferrigno, A.; Rizzo, V.; Bianchi, A.; Di Pasqua, L.G.; Berardo, C.; Richelmi, P.; Vairetti, M. Changes in ADMA/DDAH Pathway after Hepatic Ischemia/Reperfusion Injury in Rats: The Role of Bile. Biomed. Res. Int. 2014, 2014, 627434. [Google Scholar] [CrossRef]
- Vairetti, M.; Ferrigno, A.; Rizzo, V.; Richelmi, P.; Cillo, U.; Imberti, R. Liver damage during ischemia/reperfusion and glutathione: Implications for potential organ donors. Transplant. Proc. 2007, 39, 1768–1770. [Google Scholar] [CrossRef]
- Vairetti, M.; Ferrigno, A.; Bertone, R.; Richelmi, P.; Bertè, F.; Freitas, I. Apoptosis vs. necrosis: Glutathione-mediated cell death during rewarming of rat hepatocytes. Biochim. Biophys. Acta 2005, 1740, 367–374. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signalling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The role of glutathione in protecting against the severe inflammatory response triggered by covid-19. Antioxidants 2020, 9, 624. [Google Scholar] [CrossRef] [PubMed]
- Ookhtens, M.; Kaplowitz, N. Role of the Liver in Interorgan Homeostasis of Glutathione and Cyst(e)ine. Semin. Liver Dis. 1998, 18, 313–329. [Google Scholar] [CrossRef]
- Richie, J.P.; Komninou, D.; Leutzinger, Y.; Kleinman, W.; Orentreich, N.; Malloy, V.; Zimmerman, J.A. Tissue glutathione and cysteine levels in methionine-restricted rats. Nutrition 2004, 20, 800–805. [Google Scholar] [CrossRef]
- Maddineni, S.; Nichenametla, S.; Sinha, R.; Wilson, R.P.; Richie, J.P. Methionine restriction affects oxidative stress and glutathione-related redox pathways in the rat. Exp. Biol. Med. 2013, 238, 392–399. [Google Scholar] [CrossRef]
- Solis, W.A.; Dalton, T.P.; Dieter, M.Z.; Freshwater, S.; Harrer, J.M.; He, L.; Shertzer, H.G.; Nebert, D.W. Glutamate-cysteine ligase modifier subunit: Mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem. Pharmacol. 2002, 63, 1739–1754. [Google Scholar] [CrossRef]
- Neumann, C.A.; Cao, J.; Manevich, Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 2009, 8, 4072–4078. [Google Scholar] [CrossRef]
- Soriano, F.X.; Baxter, P.; Murray, L.M.; Sporn, M.B.; Gillingwater, T.H.; Hardingham, G.E. Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin. Mol. Cells 2009, 27, 279–282. [Google Scholar] [CrossRef]
- Hayes, J.D.; Chanas, S.A.; Henderson, C.J.; McMahon, M.; Sun, C.; Moffat, G.J.; Wolf, C.R.; Yamamoto, M. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. In Proceedings of the Biochemical Society Transactions; Portland Press Ltd.: London, UK, 2000; Volume 28, pp. 33–41. [Google Scholar]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef]
- Li, L.; Fu, J.; Sun, J.; Liu, D.; Chen, C.; Wang, H.; Hou, Y.; Xu, Y.; Pi, J. Is Nrf2-ARE a potential target in NAFLD mitigation? Curr. Opin. Toxicol. 2019, 13, 35–44. [Google Scholar] [CrossRef]
- Grant, C.M.; MacIver, F.H.; Dawes, I.W. Glutathione synthetase is dispensable for growth under both normal and oxidative stress conditions in the yeast Saccharomyces cerevisiae due to an accumulation of the dipeptide γ-glutamylcysteine. Mol. Biol. Cell 1997, 8, 1699–1707. [Google Scholar] [CrossRef]
- Zhang, H.; Forman, H.J.; Choi, J. γ-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005, 401, 468–483. [Google Scholar] [PubMed]
- Pompella, A.; Corti, A.; Paolicchi, A.; Giommarelli, C.; Zunino, F. γ-Glutamyltransferase, redox regulation and cancer drug resistance. Curr. Opin. Pharmacol. 2007, 7, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Mumick, S.; Nizner, P.; Tota, M.R.; Menetski, J.; Reitman, M.L.; MacNeil, D.J. Deficiency in cytosolic malic enzyme does not increase acetaminophen- induced hepato-toxicity. Basic Clin. Pharmacol. Toxicol. 2008, 103, 36–42. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef]
- Sengupta, R.; Holmgren, A. Thioredoxin and thioredoxin reductase in relation to reversible S-nitrosylation. Antioxid. Redox Signal. 2013, 18, 259–269. [Google Scholar] [CrossRef]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef]
- Labrou, N.E.; Papageorgiou, A.C.; Pavli, O.; Flemetakis, E. Plant GSTome: Structure and functional role in xenome network and plant stress response. Curr. Opin. Biotechnol. 2015, 32, 186–194. [Google Scholar] [CrossRef]
- Kumar, S.; Trivedi, P.K. Glutathione S-transferases: Role in combating abiotic stresses including arsenic detoxification in plants. Front. Plant Sci. 2018, 9, 751. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress:4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Dohnal, V.; Wu, Q.; Kuča, K. Metabolism of aflatoxins: Key enzymes and interindividual as well as interspecies differences. Arch. Toxicol. 2014, 88, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.F.; Maier, C.S. Acrolein: Sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 2008, 52, 7–25. [Google Scholar] [CrossRef]
- Carmella, S.G.; Chen, M.; Zhang, Y.; Zhang, S.; Hatsukami, D.K.; Hecht, S.S. Quantitation of acrolein-derived (3-hydroxypropyl)mercapturic acid in human urine by liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry: Effects of cigarette smoking. Chem. Res. Toxicol. 2007, 20, 986–990. [Google Scholar] [CrossRef]
- Moyer, A.M.; Fridley, B.L.; Jenkins, G.D.; Batzler, A.J.; Pelleymounter, L.L.; Kalari, K.R.; Ji, Y.; Chai, Y.; Nordgren, K.K.S.; Weinshilboum, R.M. Acetaminophen-NAPQI hepatotoxicity: A cell line model system genome-wide association study. Toxicol. Sci. 2011, 120, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Marzullo, L. An update of N-acetylcysteine treatment for acute acetaminophen toxicity in children. Curr. Opin. Pediatr. 2005, 17, 239–245. [Google Scholar] [CrossRef]
- Gilbert, H.F. Molecular and cellular aspects of thiol-disulfide exchange. Adv. Enzymol. Relat. Areas Mol. Biol. 1990, 63, 69–172. [Google Scholar]
- Mailloux, R.J.; Willmore, W.G. S-glutathionylation reactions in mitochondrial function and disease. Front. Cell Dev. Biol. 2014, 2, 68. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef]
- Han, D.; Hanawa, N.; Saberi, B.; Kaplowitz, N. Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G1–G7. [Google Scholar] [CrossRef]
- Yuan, L.; Kaplowitz, N. Glutathione in liver diseases and hepatotoxicity. Mol. Asp. Med. 2009, 30, 29–41. [Google Scholar] [CrossRef]
- Vairetti, M.; Griffini, P.; Pietrocola, G.; Richelmi, P.; Freitas, I. Cold-induced apoptosis in isolated rat hepatocytes: Protective role of glutathione. Free Radic. Biol. Med. 2001, 31, 954–961. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Cadenas, E. Mitochondrial thiols in the regulation of cell death pathways. Antioxid. Redox Signal. 2012, 17, 1714–1727. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 909. [Google Scholar] [CrossRef]
- Colell, A.; Garcia-Ruiz, C.; Miranda, M.; Ardite, E.; Mari, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernandez-Checa, J.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998, 115, 1541–1551. [Google Scholar] [CrossRef]
- Abei, M.; Harada, S.; Tanaka, N.; McNeil, M.; Osuga, T. Immunohistochemical localization of human liver glutathione S-transferase (GST) isozymes with special reference to polymorphic GST1. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. 1989, 995, 279–284. [Google Scholar] [CrossRef]
- Bellomo, G.; Vairetti, M.; Stivala, L.; Mirabelli, F.; Richelmi, P.; Orrenius, S. Demonstration of nuclear compartmentalization of glutathione in hepatocytes. Proc. Natl. Acad. Sci. USA 1992, 89, 4412–4416. [Google Scholar] [CrossRef]
- Scirè, A.; Cianfruglia, L.; Minnelli, C.; Bartolini, D.; Torquato, P.; Principato, G.; Galli, F.; Armeni, T. Glutathione compartmentalization and its role in glutathionylation and other regulatory processes of cellular pathways. BioFactors 2019, 45, 152–168. [Google Scholar] [CrossRef]
- Bellomo, G.; Palladini, G.; Vairetti, M. Intranuclear distribution, function and fate of glutathione and glutathione-S-conjugate in living rat hepatocytes studied by fluorescence microscopy. Microsc. Res. Tech. 1997, 36, 243–252. [Google Scholar] [CrossRef]
- Yeh, H.I.; Hsieh, C.H.; Wang, L.Y.; Tsai, S.P.; Hsu, H.Y.; Tam, M.F. Mass spectrometric analysis of rat liver cytosolic glutathione S-transferases: Modifications are limited to N-terminal processing. Biochem. J. 1995, 308, 69–75. [Google Scholar] [CrossRef][Green Version]
- Pedersen, J.Z.; De Maria, F.; Turella, P.; Federici, G.; Mattei, M.; Fabrini, R.; Dawood, K.F.; Massimi, M.; Caccuri, A.M.; Ricci, G. Glutathione transferases sequester toxic dinitrosyl-iron complexes in cells: A protection mechanism against excess nitric oxide. J. Biol. Chem. 2007, 282, 6364–6371. [Google Scholar] [CrossRef]
- Stella, L.; Pallottini, V.; Moreno, S.; Leoni, S.; De Maria, F.; Turella, P.; Federici, G.; Fabrini, R.; Dawood, K.F.; Lo Bello, M.; et al. Electrostatic association of glutathione transferase to the nuclear membrane: Evidence of an enzyme defense barrier at the nuclear envelope. J. Biol. Chem. 2007, 282, 6372–6379. [Google Scholar] [CrossRef] [PubMed]
- Soboll, S.; Grundel, S.; Harris, J.; Kolb-Bachofen, V.; Ketterer, B.; Sies, H. The content of glutathione and glutathione S-transferases and the glutathione peroxidase activity in rat liver nuclei determined by a non-aqueous technique of cell fractionation. Biochem. J. 1995, 311, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.; Sandoval, J.; Penella, E.; Zaragozá, R.; García, C.; Rodríguez, J.L.; Viña, J.R.; García-Trevijano, E.R. In vivo GSH depletion induces c-myc expression by modulation of chromatin protein complexes. Free Radic. Biol. Med. 2009, 46, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- García-Giménez, J.L.; Markovic, J.; Dasí, F.; Queval, G.; Schnaubelt, D.; Foyer, C.H.; Pallardó, F.V. Nuclear glutathione. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3304–3316. [Google Scholar] [CrossRef]
- Brown, K.E.; Meleah Mathahs, M.; Broadhurst, K.A.; Coleman, M.C.; Ridnour, L.A.; Schmidt, W.N.; Spitz, D.R. Increased hepatic telomerase activity in a rat model of iron overload: A role for altered thiol redox state? Free Radic. Biol. Med. 2007, 42, 228–235. [Google Scholar] [CrossRef][Green Version]
- Fernandez-Checa, J.C.; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Kojer, K.; Bien, M.; Gangel, H.; Morgan, B.; Dick, T.P.; Riemer, J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 2012, 31, 3169–3182. [Google Scholar] [CrossRef] [PubMed]
- Martensson, J.; Lai, J.C.K.; Meister, A. High-affinity Transport of Glutathione is Part of a Multicomponent System Essential for Mitochondrial Function. Proc. Natl. Acad. Sci. USA 1990, 87, 7185–7189. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; García-Ruiz, C.; Morales, A.; Ballesta, A.; Ookhtens, M.; Rodés, J.; Kaplowitz, N.; Fernández-Checa, J.C. Transport of reduced glutathione in hepatic mitochondria and mitoplasts from ethanol-treated rats: Effect of membrane physical properties and S-adenosyl-L-methionine. Hepatology 1997, 26, 699–708. [Google Scholar] [CrossRef]
- Zhong, Q.; Putt, D.A.; Xu, F.; Lash, L.H. Hepatic mitochondrial transport of glutathione: Studies in isolated rat liver mitochondria and H4IIE rat hepatoma cells. Arch. Biochem. Biophys. 2008, 474, 119–127. [Google Scholar] [CrossRef]
- Armeni, T.; Cianfruglia, L.; Piva, F.; Urbanelli, L.; Luisa Caniglia, M.; Pugnaloni, A.; Principato, G. S-D-Lactoylglutathione can be an alternative supply of mitochondrial glutathione. Free Radic. Biol. Med. 2014, 67, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Lluis, J.M.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology 2003, 124, 708–724. [Google Scholar] [CrossRef]
- Coll, O.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology 2003, 38, 692–702. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Liu, A.; Novoselov, S.V.; Krysan, K.; Sun, Q.A.; Kryukov, V.M.; Kryukov, G.V.; Lou, M.F. Identification and Characterization of a New Mammalian Glutaredoxin (Thioltransferase), Grx2. J. Biol. Chem. 2001, 276, 30374–30380. [Google Scholar] [CrossRef]
- Lluis, J.M.; Morales, A.; Blasco, C.; Colell, A.; Mari, M.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Critical role of mitochondrial glutathione in the survival of hepatocytes during hypoxia. J. Biol. Chem. 2005, 280, 3224–3232. [Google Scholar] [CrossRef]
- Görlach, A.; Klappa, P.; Kietzmann, T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Ellgaard, L. Catalysis of disulphide bond formation in the endoplasmic reticulum. Biochem. Soc. Trans. 2004, 32, 663–667. [Google Scholar] [CrossRef]
- Molteni, S.N.; Fassio, A.; Ciriolo, M.R.; Filomeni, G.; Pasqualetto, E.; Fagioli, C.; Sitia, R. Glutathione limits Ero1-dependent oxidation in the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 32667–32673. [Google Scholar] [CrossRef]
- Kojer, K.; Riemer, J. Balancing oxidative protein folding: The influences of reducing pathways on disulfide bond formation. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1383–1390. [Google Scholar] [CrossRef]
- Bánhegyi, G.; Lusini, L.; Puskás, F.; Rossi, R.; Fulceri, R.; Braun, L.; Mile, V.; Di Simplicio, P.; Mandl, J.; Benedetti, A. Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. J. Biol. Chem. 1999, 274, 12213–12216. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C. Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. J. Cell Sci. 2011, 124, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and prevention of hepatic steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Jeznach-Steinhagen, A.; Ostrowska, J.; Czerwonogrodzka-Senczyna, A.; Boniecka, I.; Shahnazaryan, U.; Kuryłowicz, A. Dietary and pharmacological treatment of nonalcoholic fatty liver disease. Medicina 2019, 55, 166. [Google Scholar] [CrossRef]
- Moon, Y.-A. The SCAP/SREBP Pathway: A Mediator of Hepatic Steatosis. Endocrinol. Metab. 2017, 32, 6. [Google Scholar] [CrossRef] [PubMed]
- Seifert, E.L.; Estey, C.; Xuan, J.Y.; Harper, M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 2010, 285, 5748–5758. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef] [PubMed]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar]
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The dual role of Nrf2 in nonalcoholic fatty liver disease: Regulation of antioxidant defenses and hepatic lipid metabolism. Biomed. Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.-W.; Zhou, Z.; Lim, C.-W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation–Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Goulas, A.; Duntas, L. Selenium and selenoprotein P in nonalcoholic fatty liver disease. Hormones 2020, 19, 61–72. [Google Scholar] [CrossRef]
- Machado, M.V.; Ravasco, P.; Jesus, L.; Marques-Vidal, P.; Oliveira, C.R.; Proença, T.; Baldeiras, I.; Camilo, M.E.; Cortez-Pinto, H. Blood oxidative stress markers in non-alcoholic steatohepatitis and how it correlates with diet. Scand. J. Gastroenterol. 2008, 43, 95–102. [Google Scholar] [CrossRef]
- Schäfer, K.; Kyriakopoulos, A.; Gessner, H.; Grune, T.; Behne, D. Effects of selenium deficiency on fatty acid metabolism in rats fed fish oil-enriched diets. J. Trace Elem. Med. Biol. 2004, 18, 89–97. [Google Scholar] [CrossRef]
- Wu, J.; Zeng, C.; Yang, Z.; Li, X.; Lei, G.; Xie, D.; Wang, Y.; Wei, J.; Yang, T. Association Between Dietary Selenium Intake and the Prevalence of Nonalcoholic Fatty Liver Disease: A Cross-Sectional Study. J. Am. Coll. Nutr. 2020, 39, 103–111. [Google Scholar] [CrossRef]
- London, R.M.; George, J. Pathogenesis of NASH: Animal models. Clin. Liver Dis. 2007, 11, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Montfort, C.V.; Matias, N.; Fernandez, A.; Fucho, R.; De La Rosa, L.C.; Martinez-Chantar, M.L.; Mato, J.M.; MacHida, K.; Tsukamoto, H.; Murphy, M.P.; et al. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. J. Hepatol. 2012, 57, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Grattagliano, I.; Caraceni, P.; Calamita, G.; Ferri, D.; Gargano, I.; Palasciano, G.; Portincasa, P. Severe liver steatosis correlates with nitrosative and oxidative stress in rats. Eur. J. Clin. Investig. 2008, 38, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhu, H.; Li, Y.; Lin, H.; Gabrielson, K.; Trush, M.A.; Diehl, A.M. Mitochondrial adaptations to obesity-related oxidant stress. Arch. Biochem. Biophys. 2000, 378, 259–268. [Google Scholar] [CrossRef]
- Lee, J.I.; Dominy, J.E.; Sikalidis, A.K.; Hirschberger, L.L.; Wang, W.; Stipanuk, M.H. HepG2/C3A cells respond to cysteine deprivation by induction of the amino acid deprivation/integrated stress response pathway. Physiol. Genom. 2008, 33, 218–229. [Google Scholar] [CrossRef]
- De Gottardi, A.; Vinciguerra, M.; Sgroi, A.; Moukil, M.; Ravier-Dall’Antonia, F.; Pazienza, V.; Pugnale, P.; Foti, M.; Hadengue, A. Microarray analyses and molecular profiling of steatosis induction in immortalized human hepatocytes. Lab. Investig. 2007, 87, 792–806. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef]
- Cobbina, E.; Akhlaghi, F. Non-Alcoholic Fatty Liver Disease (NAFLD)-Pathogenesis, Classification, and Effect on Drug Metabolizing Enzymes and Transporters HHS Public Access. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef]
- Caro, A.A.; Cederbaum, A.I. Oxidative Stress, Toxicology, and Pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 27–42. [Google Scholar] [CrossRef]
- Singh, R.; Wang, Y.; Schattenberg, J.M.; Xiang, Y.; Czaja, M.J. Chronic oxidative stress sensitizes hepatocytes to death from 4-hydroxynonenal by JNK/c-Jun overactivation. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297. [Google Scholar] [CrossRef]
- Wu, D.; Cederbaum, A.I. Oxidative stress mediated toxicity exerted by ethanol-inducible CYP2E1. Toxicol. Appl. Pharmacol. 2005, 207 (Suppl. S2), 70–76. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Kessoku, T.; Sumida, Y.; Kobayashi, T.; Kato, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; et al. Efficacy of glutathione for the treatment of nonalcoholic fatty liver disease: An open-label, single-arm, multicenter, pilot study. BMC Gastroenterol. 2017, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Söderlund, S.; Ståhlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD+ and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, D.; Xu, R.; Wu, H.; Qu, C.; Wang, F.; Wang, X.; Zhao, Y. Glycine protects against high sucrose and high fat-induced non-alcoholic steatohepatitis in rats. Oncotarget 2016, 7, 80223–80237. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.C.; Jiang, Z.M.; Li, D.M. Glutamine: A precursor of glutathione and its effect on liver. World J. Gastroenterol. 1999, 5, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Jin, C.J.; Degen, C.; De Bandt, J.P.; Bergheim, I. Oral Glutamine Supplementation Protects Female Mice from Nonalcoholic Steatohepatitis1-3. J. Nutr. 2015, 145, 2280–2286. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Baumann, A.; Brandt, A.; Jin, C.J.; Nier, A.; Bergheim, I. Oral supplementation of glutamine attenuates the progression of nonalcoholic steatohepatitis in C57BL/6J mice. J. Nutr. 2017, 147, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Cai, F.; Lin, N.; Ye, J.; Zheng, Q.; Ding, G. Effects of glutamine on oxidative stress and nuclear factor-κB expression in the livers of rats with nonalcoholic fatty liver disease. Exp. Ther. Med. 2014, 7, 365–370. [Google Scholar] [CrossRef] [PubMed]
- De Vries, N.; De Flora, S. N-acetyl-l-cysteine. J. Cell. Biochem. 1993, 53, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Ural, D.; Zeybel, M.; Yuksel, H.H.; Uhlén, M.; Borén, J. The potential use of metabolic cofactors in treatment of NAFLD. Nutrients 2019, 11, 1578. [Google Scholar] [CrossRef]
- Thong-Ngam, D.; Samuhasaneeto, S.; Kulaputana, O.; Klaikeaw, N. N-acetylcysteine attenuates oxidative stress and liver pathology in rats with non-alcoholic steatohepatitis. World J. Gastroenterol. 2007, 13, 5127–5132. [Google Scholar] [CrossRef]
- Wellington, K.; Adis, B.J. Silymarin: A review of its clinical properties in the management of hepatic disorders. BioDrugs 2001, 15, 465–489. [Google Scholar] [CrossRef]
- Dixit, N.; Baboota, S.; Kohli, K.; Ahmad, S.; Ali, J. Silymarin: A review of pharmacological aspects and bioavailability enhancement approaches. Indian J. Pharmacol. 2007, 39, 172–179. [Google Scholar] [CrossRef]
- Frazier, T.H.; Mcclain, C.J.; Kershner, N.A.; Marsano, L.S. Treatment of alcoholic liver disease. Therap. Adv. Gastroenterol. 2011, 4, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Feher, J.; Lengyel, G. Silymarin in the Prevention and Treatment of Liver Diseases and Primary Liver Cancer. Curr. Pharm. Biotechnol. 2011, 13, 210–217. [Google Scholar] [CrossRef]
- De Freitas Carvalho, M.M.; Lage, N.N.; de Souza Paulino, A.H.; Pereira, R.R.; de Almeida, L.T.; da Silva, T.F.; de Brito Magalhães, C.L.; de Lima, W.G.; Silva, M.E.; Pedrosa, M.L.; et al. Effects of açai on oxidative stress, ER stress, and inflammation-related parameters in mice with high fat diet-fed induced NAFLD. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, Y.S.; Seol, D.J.; Cho, I.J.; Kwang Ku, S.; Choi, J.S.; Lee, H.J. Anti-obesity and fatty liver-preventing activities of Lonicera caerulea in high-fat diet-fed mice. Int. J. Mol. Med. 2018, 42, 3047–3064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Zhang, L.; Yuan, Y.; Yu, D. Codonopsis lanceolata polysaccharide CLPS alleviates high fat/high sucrose diet-induced insulin resistance via anti-oxidative stress. Int. J. Biol. Macromol. 2020, 145, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.P.; Moore, M.P.; Moore, A.N.; Healy, J.C.; Roberts, M.D.; Rector, R.S.; Martin, J.S. Curcumin supplementation mitigates NASH development and progression in female Wistar rats. Physiol. Rep. 2018, 6. [Google Scholar] [CrossRef]
- Wang, W.; Li, Q.; Chai, W.; Sun, C.; Zhang, T.; Zhao, C.; Yuan, Y.; Wang, X.; Liu, H.; Ye, H. Lactobacillus paracasei Jlus66 extenuate oxidative stress and inflammation via regulation of intestinal flora in rats with non alcoholic fatty liver disease. Food Sci. Nutr. 2019, 7, 2636–2646. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Arora, A. Clinical presentation of alcoholic liver disease and non-alcoholic fatty liver disease: Spectrum and diagnosis. Transl. Gastroenterol. Hepatol. 2020, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef]
- Teli, M.R.; Day, C.P.; James, O.F.W.; Burt, A.D.; Bennett, M.K. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995, 346, 987–990. [Google Scholar] [CrossRef]
- Poynard, T.; Bedossa, P.; Opolon, P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet 1997, 349, 825–832. [Google Scholar] [CrossRef]
- Singal, A.K.; Bataller, R.; Ahn, J.; Kamath, P.S.; Shah, V.H. ACG clinical guideline: Alcoholic liver disease. Am. J. Gastroenterol. 2018, 113, 175–194. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Leung, T.M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Landar, A.; Zmijewski, J.W.; Dickinson, D.A.; Le Goffe, C.; Johnson, M.S.; Milne, G.L.; Zanoni, G.; Vidari, G.; Morrow, J.D.; Darley-Usmar, V.M. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am. J. Physiol. Hear. Circ. Physiol. 2006, 290, H1777–H1787. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M. A review of the role of reactive oxygen and nitrogen species in alcohol-induced mitochondrial dysfunction. Free Radic. Res. 2003, 37, 585–596. [Google Scholar] [CrossRef] [PubMed]
- McKillop, I.H.; Schrum, L.W. Alcohol and liver cancer. Alcohol 2005, 35, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.C.; Bailey, S.M. Ethanol Consumption and Liver Mitochondria Function. Neurosignals 2001, 10, 271–282. [Google Scholar] [CrossRef]
- 1Lieber, C.S. Role of Oxidative Stress and Antioxidant Therapy in Alcoholic and Nonalcoholic Liver Diseases. Adv. Pharmacol. 1996, 38, 601–628. [Google Scholar] [CrossRef]
- Adachi, M.; Ishii, H. Role of mitochondria in alcoholic liver injury. Free Radic. Biol. Med. 2002, 32, 487–491. [Google Scholar] [CrossRef]
- Dupont, I.; Lucas, D.; Clot, P.; Ménez, C.; Albano, E. Cytochrome P4502E1 inducibility and hydroxyethyl radical formation among alcoholics. J. Hepatol. 1998, 28, 564–571. [Google Scholar] [CrossRef]
- Crabb, D.W.; Bosron, W.F.; Li, T.K. Ethanol metabolism. Pharmacol. Ther. 1987, 34, 59–73. [Google Scholar] [CrossRef]
- Seronello, S.; Sheikh, M.Y.; Choi, J. Redox regulation of hepatitis C in nonalcoholic and alcoholic liver. Free Radic. Biol. Med. 2007, 43, 869–882. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione: Hepatocellular survival–death switch. J. Gastroenterol. Hepatol. 2006, 21, S3–S6. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Morales, A.; Ballesta, A.; Rodés, J.; Kaplowitz, N.; Fernández-Checa, J.C. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J. Clin. Investig. 1994, 94, 193–201. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Morales, A.; Colell, A.; Ballesta, A.; Rodés, J.; Kaplowitz, N.; Fernandez-Checa, J.C. Feeding S-adenosyl-l-methionine attenuates both ethanol-induced depletion of mitochondrial glutathione and mitochondrial dysfunction in periportal and perivenous rat hepatocytes. Hepatology 1995, 21, 207–214. [Google Scholar] [CrossRef]
- Colell, A.; Coll, O.; García-Ruiz, C.; París, R.; Tiribelli, C.; Kaplowitz, N.; Fernández-Checa, J.C. Tauroursodeoxycholic acid protects hepatocytes from ethanol-fed rats against tumor necrosis factor-induced cell death by replenishing mitochondrial glutathione. Hepatology 2001, 34, 964–971. [Google Scholar] [CrossRef]
- Neve, E.P.A.; Ingelman-Sundberg, M. Molecular basis for the transport of cytochrome P450 2E1 to the plasma membrane. J. Biol. Chem. 2000, 275, 17130–17135. [Google Scholar] [CrossRef]
- Bai, J.; Cederbaum, A.I. Overexpression of CYP2E1 in mitochondria sensitizes HepG2 cells to the toxicity caused by depletion of glutathione. J. Biol. Chem. 2006, 281, 5128–5136. [Google Scholar] [CrossRef]
- Bardag-Gorce, F.; Yuan, Q.X.; Li, J.; French, B.A.; Fang, C.; Ingelman-Sundberg, M.; French, S.W. The effect of ethanol-induced cytochrome p4502E1 on the inhibition of proteasome activity by alcohol. Biochem. Biophys. Res. Commun. 2000, 279, 23–29. [Google Scholar] [CrossRef]
- Butura, A.; Nilsson, K.; Morgan, K.; Morgan, T.R.; French, S.W.; Johansson, I.; Schuppe-Koistinen, I.; Ingelman-Sundberg, M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2009, 50, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Cederbaum, A.I. Adenovirus-Mediated Expression of CYP2E1 Produces Liver Toxicity in Mice. Toxicol. Sci. 2006, 91, 365–371. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Nrf2 and antioxidant defense against CYP2E1 toxicity. Subcell. Biochem. 2013, 67, 105–130. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Osei-Hyiaman, D.; Park, O.; Liu, J.; Bátkai, S.; Mukhopadhyay, P.; Horiguchi, N.; Harvey-White, J.; Marsicano, G.; Lutz, B.; et al. Paracrine Activation of Hepatic CB1 Receptors by Stellate Cell-Derived Endocannabinoids Mediates Alcoholic Fatty Liver. Cell Metab. 2008, 7, 227–235. [Google Scholar] [CrossRef]
- Choi, W.M.; Kim, H.H.; Kim, M.H.; Cinar, R.; Yi, H.S.; Eun, H.S.; Kim, S.H.; Choi, Y.J.; Lee, Y.S.; Kim, S.Y.; et al. Glutamate Signaling in Hepatic Stellate Cells Drives Alcoholic Steatosis. Cell Metab. 2019, 30, 877–889.e7. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, A.; Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Siciliano, V.; Richelmi, P.; Vairetti, M. The selective blockade of metabotropic glutamate receptor-5 attenuates fat accumulation in an in vitro model of benign steatosis. Eur. J. Histochem. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Andrade, R.J.; de la Cruz, J.P.; Rodriguez-Mendizabal, M.; Blanco, E.; Sánchez de la Cuesta, F. Effects of silymarin MZ-80 on oxidative stress in patients with alcoholic cirrhosis. Int. J. Clin. Pharmacol. Ther. 2002, 40, 2–8. [Google Scholar] [CrossRef]
- Ronis, M.J.J.; Butura, A.; Sampey, B.P.; Shankar, K.; Prior, R.L.; Korourian, S.; Albano, E.; Ingelman-Sundberg, M.; Petersen, D.R.; Badger, T.M. Effects of N-acetylcysteine on ethanol-induced hepatotoxicity in rats fed via total enteral nutrition. Free Radic. Biol. Med. 2005, 39, 619–630. [Google Scholar] [CrossRef]
- Ozaras, R.; Tahan, V.; Aydin, S.; Uzun, H.; Kaya, S.; Senturk, H. N-acetylcysteine attenuates alcohol-induced oxidative stress in the rat. World J. Gastroenterol. 2003, 9, 125–128. [Google Scholar] [CrossRef]
- Nguyen-Khac, E.; Thevenot, T.; Piquet, M.-A.; Benferhat, S.; Goria, O.; Chatelain, D.; Tramier, B.; Dewaele, F.; Ghrib, S.; Rudler, M.; et al. Glucocorticoids plus N -Acetylcysteine in Severe Alcoholic Hepatitis. N. Engl. J. Med. 2011, 365, 1781–1789. [Google Scholar] [CrossRef]
- Stewart, S.; Prince, M.; Bassendine, M.; Hudson, M.; James, O.; Jones, D.; Record, C.; Day, C.P. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J. Hepatol. 2007, 47, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Abdelmalek, M.F.; Barve, S.; Benevenga, N.J.; Halsted, C.H.; Kaplowitz, N.; Kharbanda, K.K.; Liu, Q.Y.; Lu, S.C.; McClain, C.J.; et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am. J. Clin. Nutr. 2007, 86, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Caballería, J.; Parés, A.; Brú, C.; Mercader, J.; Plaza, A.G.; Caballería, L.; Clemente, G.; Rodrigo, L.; Rodés, J. Metadoxine accelerates fatty liver recovery in alcoholic patients: Results of a randomized double-blind, placebo-control trial. J. Hepatol. 1998, 28, 54–60. [Google Scholar] [CrossRef]
- Han, K.H.; Hashimoto, N.; Fukushima, M. Relationships among alcoholic liver disease, antioxidants, and antioxidant enzymes. World J. Gastroenterol. 2016, 22, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gao, C.; Xing, M.; Li, Y.; Zhu, L.; Wang, D.; Yang, X.; Liu, L.; Yao, P. Quercetin prevents ethanol-induced dyslipidemia and mitochondrial oxidative damage. Food Chem. Toxicol. 2012, 50, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, Y.; Yu, H.; Gao, C.; Liu, L.; Xing, M.; Yao, P. Quercetin attenuates chronic ethanol hepatotoxicity: Implication of “free” iron uptake and release. Food Chem. Toxicol. 2014, 67, 131–138. [Google Scholar] [CrossRef]
- Yin, H.Q.; Kim, Y.C.; Chung, Y.S.; Kim, Y.C.; Shin, Y.K.; Lee, B.H. Honokiol reverses alcoholic fatty liver by inhibiting the maturation of sterol regulatory element binding protein-1c and the expression of its downstream lipogenesis genes. Toxicol. Appl. Pharmacol. 2009, 236, 124–130. [Google Scholar] [CrossRef]
- Zhang, P.; Ma, D.; Wang, Y.; Zhang, M.; Qiang, X.; Liao, M.; Liu, X.; Wu, H.; Zhang, Y. Berberine protects liver from ethanol-induced oxidative stress and steatosis in mice. Food Chem. Toxicol. 2014, 74, 225–232. [Google Scholar] [CrossRef]
- Bao, W.; Li, K.; Rong, S.; Yao, P.; Hao, L.; Ying, C.; Zhang, X.; Nussler, A.; Liu, L. Curcumin alleviates ethanol-induced hepatocytes oxidative damage involving heme oxygenase-1 induction. J. Ethnopharmacol. 2010, 128, 549–553. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Farhadi, A.; Jakate, S.M.; Tang, Y.; Shaikh, M.; Keshavarzian, A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol 2009, 43, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Kirpich, I.; Ma, Z.; Wang, C.; Zhang, M.; Suttles, J.; McClain, C.; Feng, W. Lactobacillus rhamnosus GG reduces hepatic TNFα production and inflammation in chronic alcohol-induced liver injury. J. Nutr. Biochem. 2013, 24, 1609–1615. [Google Scholar] [CrossRef]
- Samant, H.; Manatsathit, W.; Dies, D.; Shokouh-Amiri, H.; Zibari, G.; Boktor, M.; Alexander, J.S. Cholestatic liver diseases: An era of emerging therapies. World J. Clin. Cases 2019, 7, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Delemos, A.S.; Friedman, L.S. Systemic Causes of Cholestasis. Clin. Liver Dis. 2013, 17, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Sticova, E.; Jirsa, M.; Pawłowska, J. New Insights in Genetic Cholestasis: From Molecular Mechanisms to Clinical Implications. Can. J. Gastroenterol. Hepatol. 2018, 2018, 2313675. [Google Scholar] [CrossRef] [PubMed]
- Shoda, J.; Kano, M.; Oda, K.; Kamiya, J.; Nimura, Y.; Suzuki, H.; Sugiyama, Y.; Miyazaki, H.; Todoroki, T.; Stengelin, S.; et al. The expression levels of plasma membrane transporters in the cholestatic liver of patients undergoing biliary drainage and their association with the impairment of biliary secretory function. Am. J. Gastroenterol. 2001, 96, 3368–3378. [Google Scholar] [CrossRef]
- Portincasa, P.; Grattagliano, I.; Testini, M.; Caruso, M.L.; Wang, D.Q.-H.; Moschetta, A.; Calamita, G.; Vacca, M.; Valentini, A.M.; Renna, G.; et al. Parallel intestinal and liver injury during early cholestasis in the rat: Modulation by bile salts and antioxidants. Free Radic. Biol. Med. 2007, 42, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.-M.; Huang, L.-T.; Hsieh, C.-S.; Chen, C.-C.; Wang, J.-Y.; Tain, Y.-L. Bile duct ligation in developing rats: Temporal progression of liver, kidney, and brain damage. J. Pediatr. Surg. 2010, 45, 1650–1658. [Google Scholar] [CrossRef]
- Ljubuncic, P.; Tanne, Z.; Bomzon, A. Evidence of a systemic phenomenon for oxidative stress in cholestatic liver disease. Gut 2000, 47, 710–716. [Google Scholar] [CrossRef]
- Grintzalis, K.; Papapostolou, I.; Assimakopoulos, S.F.; Mavrakis, A.; Faropoulos, K.; Karageorgos, N.; Georgiou, C.; Chroni, E.; Konstantinou, D. Time-related alterations of superoxide radical levels in diverse organs of bile duct-ligated rats. Free Radic. Res. 2009, 43, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Krähenbühl, S.; Talos, C.; Reichen, J. Mechanisms of impaired hepatic fatty acid metabolism in rats with long-term bile duct ligation. Hepatology 1994, 19, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; McKim, J.; Goff, M.C.; Ruyle, S.Z.; Devereaux, M.W.; Han, D.; Packer, L.; Everson, G. Vitamin E reduces oxidant injury to mitochondria and the hepatotoxicity of taurochenodeoxycholic acid in the rat. Gastroenterology 1998, 114, 164–174. [Google Scholar] [CrossRef]
- Krähenbühl, S.; Talos, C.; Lauterburg, B.H.; Reichen, J. Reduced antioxidative capacity in liver mitochondria from bile duct ligated rats. Hepatology 1995, 22, 607–612. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Nicholson, C.; Wells, L.D.; Tracy, T.F. Cholestatic liver injury down-regulates hepatic glutathione synthesis. J. Surg. Res. 1996, 63, 447–451. [Google Scholar] [CrossRef]
- Gumpricht, E.; Devereaux, M.W.; Dahl, R.H.; Sokol, R.J. Glutathione status of isolated rat hepatocytes affects bile acid-induced cellular necrosis but not apoptosis. Toxicol. Appl. Pharmacol. 2000, 164, 102–111. [Google Scholar] [CrossRef]
- Serviddio, G.; Pereda, J.; Pallardó, F.V.; Carretero, J.; Borras, C.; Cutrin, J.; Vendemiale, G.; Poli, G.; Viña, J.; Sastre, J. Ursodeoxycholic Acid Protects against Secondary Biliary Cirrhosis in Rats by Preventing Mitochondrial Oxidative Stress. Hepatology 2004, 39, 711–720. [Google Scholar] [CrossRef]
- Mitsuyoshi, H.; Nakashima, T.; Sumida, Y.; Yoh, T.; Nakajima, Y.; Ishikawa, H.; Inaba, K.; Sakamoto, Y.; Okanoue, T.; Kashima, K. Ursodeoxycholic acid protects hepatocytes against oxidative injury via induction of antioxidants. Biochem. Biophys. Res. Commun. 1999, 263, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ramani, K.; Xia, M.; Ko, K.S.; Li, T.W.H.; Oh, P.; Li, J.; Lu, S.C. Dysregulation of glutathione synthesis during cholestasis in mice: Molecular mechanisms and therapeutic implications. Hepatology 2009, 49, 1982–1991. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zeng, Y.; Lee, T.D.; Yang, Y.; Ou, X.; Chen, L.; Haque, M.; Rippe, R.; Lu, S.C. Role of AP-1 in the coordinate induction of rat glutamate-cysteine ligase and glutathione synthetase by tert-butylhydroquinone. J. Biol. Chem. 2002, 277, 35232–35239. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Magilnick, N.; Ou, X.; Lu, S.C. Tumour necrosis factor α induces co-ordinated activation of rat GSH synthetic enzymes via nuclear factor κB and activator protein-1. Biochem. J. 2005, 391, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell 2006, 21, 509–519. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Jaiswal, A.K. Small Maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:Quinone oxidoreductase1 gene. J. Biol. Chem. 2000, 275, 40134–40141. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef]
- Roeb, E.; Purucker, E.; Gartung, C.; Geier, A.; Jansen, B.; Winograd, R.; Matern, S. Effect of glutathione depletion and hydrophilic bile acids on hepatic acute phase reaction in rats with extrahepatic cholestasis. Scand. J. Gastroenterol. 2003, 38, 878–885. [Google Scholar] [CrossRef]
- Ker, C.-G.; Huang, T.-J.; Sheen, P.-C. Electron Microscopic Assessment of Bile Regurgitation of Human in Extrahepatic Obstructive Jaundice. Eur. Surg. Res. 1985, 17, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Clichici, S.; David, L.; Moldovan, B.; Baldea, I.; Olteanu, D.; Filip, M.; Nagy, A.; Luca, V.; Crivii, C.; Mircea, P.; et al. Hepatoprotective effects of silymarin coated gold nanoparticles in experimental cholestasis. Mater. Sci. Eng. C 2020, 115, 111117. [Google Scholar] [CrossRef] [PubMed]
- Piletz, J.E.; Aricioglu, F.; Cheng, J.T.; Fairbanks, C.A.; Gilad, V.H.; Haenisch, B.; Halaris, A.; Hong, S.; Lee, J.E.; Li, J.; et al. Agmatine: Clinical applications after 100 years in translation. Drug Discov. Today 2013, 18, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Ommati, M.M.; Farshad, O.; Mousavi, K.; Taghavi, R.; Farajvajari, S.; Azarpira, N.; Moezi, L.; Heidari, R. Agmatine alleviates hepatic and renal injury in a rat model of obstructive jaundice. PharmaNutrition 2020, 13, 100212. [Google Scholar] [CrossRef]
- Ommati, M.M.; Attari, H.; Siavashpour, A.; Shafaghat, M.; Azarpira, N.; Ghaffari, H.; Moezi, L.; Heidari, R. Mitigation of cholestasis-associated hepatic and renal injury by edaravone treatment: Evaluation of its effects on oxidative stress and mitochondrial function. Liver Res. 2020. [Google Scholar] [CrossRef]
- Yoshida, H.; Yanai, H.; Namiki, Y.; Fukatsu-Sasaki, K.; Furutani, N.; Tada, N. Neuroprotective effects of edaravone: A novel free radical scavenger in cerebrovascular injury. CNS Drug Rev. 2006, 12, 9–20. [Google Scholar] [CrossRef]
- Shinohara, Y.; Saito, I.; Kobayashi, S.; Uchiyama, S. Edaravone (Radical Scavenger) versus sodium ozagrel (Antiplatelet Agent) in acute noncardioembolic ischemic stroke (EDO Trial). Cerebrovasc. Dis. 2009, 27, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.; Heiman-Patterson, T.; Kittrell, P.; Baranovsky, T.; McAnanama, G.; Bower, L.; Agnese, W.; Martin, M. Radicava (edaravone) for amyotrophic lateral sclerosis: US experience at 1 year after launch. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Okatani, Y.; Wakatsuki, A.; Enzan, H.; Miyahara, Y. Edaravone protects against ischemia/reperfusion-induced oxidative damage to mitochondria in rat liver. Eur. J. Pharmacol. 2003, 465, 163–170. [Google Scholar] [CrossRef]
- Hassan, M.Q.; Akhtar, M.S.; Afzal, O.; Hussain, I.; Akhtar, M.; Haque, S.E.; Najmi, A.K. Edaravone and benidipine protect myocardial damage by regulating mitochondrial stress, apoptosis signalling and cardiac biomarkers against doxorubicin-induced cardiotoxicity. Clin. Exp. Hypertens. 2020, 42, 381–392. [Google Scholar] [CrossRef]
- Fukui, H.; Wiest, R. Changes of Intestinal Functions in Liver Cirrhosis. Inflamm. Intest. Dis. 2016, 1, 24–40. [Google Scholar] [CrossRef]
- Nolan, J.P. The role of intestinal endotoxin in liver injury: A long and evolving history. Hepatology 2010, 52, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Ommati, M.M.; Farshad, O.; Niknahad, H.; Mousavi, K.; Moein, M.; Azarpira, N.; Mohammadi, H.; Jamshidzadeh, A.; Heidari, R. Oral administration of thiol-reducing agents mitigates gut barrier disintegrity and bacterial lipopolysaccharide translocation in a rat model of biliary obstruction. Curr. Res. Pharmacol. Drug Discov. 2020, 1, 10–18. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Vagianos, C.E.; Zervoudakis, G.; Filos, K.S.; Georgiou, C.; Nikolopoulou, V.; Scopa, C.D. Gut regulatory peptides bombesin and neurotensin reduce hepatic oxidative stress and histological alterations in bile duct ligated rats. Regul. Pept. 2004, 120, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Wang, Y.Y.; Chen, W.Y.; Chuang, Y.H.; Pan, P.H.; Chen, C.J. Beneficial effect of quercetin on cholestatic liver injury. J. Nutr. Biochem. 2014, 25, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, H.; Azarmehr, N.; Razmkhah, F.; Sadeghi, H.; Danaei, N.; Omidifar, N.; Vakilpour, H.; Pourghadamyari, H.; Doustimotlagh, A.H. The hydroalcoholic extract of watercress attenuates protein oxidation, oxidative stress, and liver damage after bile duct ligation in rats. J. Cell. Biochem. 2019, 120, 14875–14884. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, A.; Di Pasqua, L.G.; Palladini, G.; Berardo, C.; Verta, R.; Richelmi, P.; Perlini, S.; Collotta, D.; Collino, M.; Vairetti, M. Transient expression of reck under hepatic ischemia/reperfusion conditions is associated with mapk signaling pathways. Biomolecules 2020, 10, 747. [Google Scholar] [CrossRef]
- Lemasters, J.J.V. Necrapoptosis and the mitochondrial permeability transition: Shared pathways to necrosis and apoptosis. Am. J. Physiol. 1999, 276, G1–G6. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Reisman, S.A. Nrf2 the rescue: Effects of the antioxidative/electrophilic response on the liver. Toxicol. Appl. Pharmacol. 2010, 244, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef]
- Kudoh, K.; Uchinami, H.; Yoshioka, M.; Seki, E.; Yamamoto, Y. Nrf2 activation protects the liver from ischemia/reperfusion injury in Mice. Ann. Surg. 2014, 260, 118–127. [Google Scholar] [CrossRef]
- Jaeschke, H.; Farhood, A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 1991, 260, G355–G362. [Google Scholar] [CrossRef]
- Schauer, R.J.; Kalmuk, S.; Gerbes, A.L.; Leiderer, R.; Meissner, H.; Schildberg, F.W.; Messmer, K.; Bilzer, M. Intravenous administration of glutathione protects parechymal and non-paranchymal liver cells against reperfusion injury following rat liver transplantation. World J. Gastroenterol. 2004, 10, 864–870. [Google Scholar] [CrossRef]
- Schauer, R.J.; Gerbes, A.L.; Vonier, D.; Meissner, H.; Michl, P.; Leiderer, R.; Schildberg, F.W.; Messmer, K.; Bilzer, M. Glutathione Protects the Rat Liver Against Reperfusion Injury after Prolonged Warm Ischemia. Ann. Surg. 2004, 239, 220–231. [Google Scholar] [CrossRef]
- Bilzer, M.; Paumgartner, G.; Gerbes, A.L. Glutathione protects the rat liver against reperfusion injury after hypothermic preservation. Gastroenterology 1999, 117, 200–210. [Google Scholar] [CrossRef]
- Smyrniotis, V.; Arkadopoulos, N.; Kostopanagiotou, G.; Theodoropoulos, T.; Theodoraki, K.; Farantos, C.; Kairi, E.; Paphiti, A. Attenuation of ischemic injury by N-acetylcysteine preconditioning of the liver. J. Surg. Res. 2005, 129, 31–37. [Google Scholar] [CrossRef]
- Nagasaki, H.; Nakano, H.; Boudjema, K.; Jaeck, D.; Alexandre, E.; Baek, Y.; Kitamura, N.; Yamaguchi, M.; Kumada, K. Efficacy of preconditioning with n-acetylcysteine against reperfusion injury after prolonged cold ischaemia in rats liver in which glutathione had been reduced by buthionine sulphoximine. Eur. J. Surg. 1998, 164, 139–146. [Google Scholar] [CrossRef]
- Sener, G.; Tosun, O.; Şehirli, A.Ö.; Kaçmaz, A.; Arbak, S.; Ersoy, Y.; Ayanoǧlu-Dülger, G. Melatonin and N-acetylcysteine have beneficial effects during hepatic ischemia and reperfusion. Life Sci. 2003, 72, 2707–2718. [Google Scholar] [CrossRef]
- Saito, C.; Zwingmann, C.; Jaeschke, H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 2010, 51, 246–254. [Google Scholar] [CrossRef]
- D’Amico, F.; Vitale, A.; Piovan, D.; Bertacco, A.; Ramirez Morales, R.; Chiara Frigo, A.; Bassi, D.; Bonsignore, P.; Gringeri, E.; Valmasoni, M.; et al. Use of N-acetylcysteine during liver procurement: A prospective randomized controlled study. Liver Transplant. 2013, 19, 135–144. [Google Scholar] [CrossRef]
- Wendel, A.; Cikryt, P. The level and half-life of glutathione in human plasma. FEBS Lett. 1980, 120, 209–211. [Google Scholar] [CrossRef]
- Zwacka, R.M.; Zhou, W.; Zhang, Y.; Darby, C.J.; Dudus, L.; Halldorson, J.; Oberley, L.; Engelhardt, J.F. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-κB activation. Nat. Med. 1998, 4, 698–704. [Google Scholar] [CrossRef]
- Lehmann, T.G.; Wheeler, M.D.; Froh, M.; Schwabe, R.F.; Bunzendahl, H.; Samulski, J.R.; Lemasters, J.J.; Brenner, D.A.; Thurman, R.G. Effects of three superoxide dismutase genes delivered with an adenovirus on graft function after transplantation of fatty livers in the rat1. Transplantation 2003, 76, 28–37. [Google Scholar] [CrossRef]
- Shau, H.; Merino, A.; Chen, L.; Shih, C.C.-Y.; Colquhoun, S.D. Induction of Peroxiredoxins in Transplanted Livers and Demonstration of Their In Vitro Cytoprotection Activity. Antioxid. Redox Signal. 2000, 2, 347–354. [Google Scholar] [CrossRef]
- Glantzounis, G.K.; Salacinski, H.J.; Yang, W.; Davidson, B.R.; Seifalian, A.M. The contemporary role of antioxidant therapy in attenuating liver ischemia-reperfusion injury: A review. Liver Transplant. 2005, 11, 1031–1047. [Google Scholar] [CrossRef]
- Bayramoglu, G.; Bayramoglu, A.; Engur, S.; Senturk, H.; Ozturk, N.; Colak, S. The hepatoprotective effects of Hypericum perforatum L. on hepatic ischemia/reperfusion injury in rats. Cytotechnology 2014, 66, 443–448. [Google Scholar] [CrossRef]
- Ramalho, L.N.; Pasta, Â.A.; Terra, V.A.; Augusto, M.; Sanches, S.C.; Souza-Neto, F.P.; Cecchini, R.; Gulin, F.; Ramalho, F.S. Rosmarinic acid attenuates hepatic ischemia and reperfusion injury in rats. Food Chem. Toxicol. 2014, 74, 270–278. [Google Scholar] [CrossRef]
- Xing, H.-C.; Li, L.-J.; Xu, K.-J.; Shen, T.; Chen, Y.-B.; Sheng, J.-F.; Chen, Y.; Fu, S.-Z.; Chen, C.-L.; Wang, J.-G.; et al. Protective role of supplement with foreign Bifidobacterium and Lactobacillus in experimental hepatic ischemia-reperfusion injury. J. Gastroenterol. Hepatol. 2006, 21, 647–656. [Google Scholar] [CrossRef]
- Westbrook, R.H.; Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 2014, 61, S58–S68. [Google Scholar] [CrossRef]
- Hoofnagle, J.H. Course and outcome of hepatitis C. Hepatology 2002, 36, s21–s29. [Google Scholar]
- Ruggieri, A.; Anticoli, S.; Nencioni, L.; Sgarbanti, R.; Garaci, E. Interplay between Hepatitis C Virus and Redox Cell Signaling. Int. J. Mol. Sci. 2013, 14, 4705–4721. [Google Scholar] [CrossRef]
- Paracha, U.Z.; Fatima, K.; Alqahtani, M.; Chaudhary, A.; Abuzenadah, A. Oxidative stress and hepatitis C virus. Virol. J. 2013, 10, 1. [Google Scholar] [CrossRef]
- Roe, B.; Kensicki, E.; Mohney, R.; Hall, W.W. Metabolomic Profile of Hepatitis C Virus-Infected Hepatocytes. PLoS ONE 2011, 6, 1–8. [Google Scholar] [CrossRef]
- De Mochel, N.S.R.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2011, 52, 209–228. [Google Scholar] [CrossRef]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C Virus RNA Replication and Assembly: Living on the Fat of the Land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef]
- Medvedev, R.; Ploen, D.; Hildt, E. HCV and Oxidative Stress: Implications for HCV Life Cycle and HCV-Associated Pathogenesis. Oxid. Med. Cell. Longev. 2016, 2016, 13. [Google Scholar] [CrossRef]
- Li, X.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef]
- Korenaga, M.; Showalter, L.; Chan, T.; Sun, J. Hepatitis C Virus Core Protein Inhibits Mitochondrial Electron Transport and Hepatitis C Virus Core Protein Inhibits Mitochondrial Electron (ROS) Production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef]
- Abdalla, M.Y.; Ahmad, I.M.; Spitz, D.R.; Schmidt, W.N.; Britigan, B.E. Hepatitis C Virus-Core and Non Structural Proteins Lead to Different Effects on Cellular Antioxidant Defenses. J. Med. Virol. 2005, 497, 489–497. [Google Scholar] [CrossRef]
- Bender, D.; Hildt, E. Effect of Hepatitis Viruses on the Nrf2 / Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis. Int. J. Mol. Sci. 2019, 18, 4659. [Google Scholar] [CrossRef]
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis C Virus Impairs the Induction of Cytoprotective Nrf2 Target Genes by Delocalization of Small Maf Proteins. J. Biol. Chem. 2011, 286, 8941–8951. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Brault, C.; Lévy, P.; Duponchel, S.; Michelet, M.; Sallé, A.; Pécheur, E.; Plissonnier, M.; Parent, R.; Véricel, E.; Ivanov, A.V.; et al. Glutathione peroxidase 4 is reversibly induced by HCV to control lipid peroxidation and to increase virion infectivity. Gut 2016, 65, 144–154. [Google Scholar] [CrossRef]
- Choi, J.; Corder, N.L.B.; Koduru, B.; Wang, Y. Oxidative stress and hepatic Nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free Radic. Biol. Med. 2014, 72, 267–284. [Google Scholar] [CrossRef]
- Barbaro, G.; Di Lorenzo, G.; Ribersani, M.; Soldini, M.; Giancaspro, G.; Bellomo, G.; Belloni, G.; Grisorio, B.; Barbarini, G. Serum ferritin and hepatic glutathione concentrations in chronic hepatitis C patients related to the hepatitis C virus genotype. J. Hepatol. 1999, 30, 174–182. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef]
- Ansari, M.K.H.; Omrani, M.; Kheradmand, F. Oxidative Stress Response in Patients Infected by Diverse Hepatitis C Virus Genotypes. Hepat. Mon. 2015, 15, e22069. [Google Scholar] [CrossRef]
- Razzaq, Z.; Malik, A. Viral load is associated with abnormal serum levels of micronutrients and glutathione and glutathione-dependent enzymes in genotype 3 HCV patients. BBA Clin. 2014, 2, 72–78. [Google Scholar] [CrossRef]
- Farias, M.S.; Budni, P.; Ribeiro, C.M.; Parisotto, E.B.; Santos, C.E.I.; Ferraz Dias, J.; Dalmarco, E.M.; Frode, S.T.; Pedrosa, R.C.; Filho, W.D. Antioxidant supplementation attenuates oxidative stress in chronic hepatitis C patients. Gastroenterol. Hepatol. 2012, 35, 386–394. [Google Scholar] [CrossRef]
- Revill, P.A.; Penicaud, C.; Brechot, C.; Zoulim, F. Meeting the Challenge of Eliminating Chronic Hepatitis b Infection. Genes 2019, 10, 260. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef]
- Liang, T.J. Hepatitis B: The virus and disease. Hepatology 2009, 49 (Suppl. S5), S13–S21. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.Y.; Quan, X.; Yi, Y.S.; Jung, G. Heat shock protein 90 facilitates formation of the HBV capsid via interacting with the HBV core protein dimers. Virology 2011, 410, 161–169. [Google Scholar] [CrossRef]
- Kim, Y.S.; Seo, H.W.; Jung, G. Reactive oxygen species promote heat shock protein 90-mediated HBV capsid assembly. Biochem. Biophys. Res. Commun. 2015, 457, 328–333. [Google Scholar] [CrossRef]
- Wang, Q.; Na, B.; Ou, J. hsiung J.; Pulliam, L.; Yen, T.S.B. Hepatitis B virus alters the antioxidant system in transgenic mice and sensitizes hepatocytes to fas signaling. PLoS ONE 2012, 7, e36818. [Google Scholar] [CrossRef]
- Peiffer, K.H.; Akhras, S.; Himmelsbach, K.; Hassemer, M.; Finkernagel, M.; Carra, G.; Nuebling, M.; Chudy, M.; Niekamp, H.; Glebe, D.; et al. Intracellular accumulation of subviral HBsAg particles and diminished Nrf2 activation in HBV genotype G expressing cells lead to an increased ROI level. J. Hepatol. 2015, 62, 791–798. [Google Scholar] [CrossRef]
- Schaedler, S.; Krause, J.; Himmelsbach, K.; Carvajal-Yepes, M.; Lieder, F.; Klingel, K.; Nassal, M.; Weiss, T.S.; Werner, S.; Hildt, E. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J. Biol. Chem. 2010, 285, 41074–41086. [Google Scholar] [CrossRef]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef]
- Li, H.; Zhu, W.; Zhang, L.; Lei, H.; Wu, X.; Guo, L.; Chen, X.; Wang, Y.; Tang, H. The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment. Sci. Rep. 2015, 5, 8421. [Google Scholar] [CrossRef]
- Severi, T.; Ying, C.; Vermeesch, J.R.; Cassiman, D.; Cnops, L.; Verslype, C.; Fevery, J.; Arckens, L.; Neyts, J.; van Pelt, J.F. Hepatitis B virus replication causes oxidative stress in HepAD38 liver cells. Mol. Cell. Biochem. 2006, 290, 79–85. [Google Scholar] [CrossRef]
- Wu, Y.L.; Wang, D.; Peng, X.E.; Chen, Y.L.; Zheng, D.L.; Chen, W.N.; Lin, X. Epigenetic silencing of NAD(P)H:quinone oxidoreductase 1 by hepatitis B virus X protein increases mitochondrial injury and cellular susceptibility to oxidative stress in hepatoma cells. Free Radic. Biol. Med. 2013, 65, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.-P.; Ji, X.-F.; Li, X.-Y.; Gao, S.; Fan, Y.-C.; Wang, K. Methylation of the Glutathione-S-Transferase P1 Gene Promoter Is Associated with Oxidative Stress in Patients with Chronic Hepatitis B. Tohoku J. Exp. Med. 2016, 238, 57–64. [Google Scholar] [CrossRef]
- Tong, A.; Wu, L.; Lin, Q.; Quek, C.L.; Zhao, X.; Li, J.; Chen, P.; Chen, L.; Tang, H.; Huang, C.; et al. Proteomic analysis of cellular protein alterations using a hepatitis B virus-producing cellular model. Proteomics 2008, 8, 2012–2023. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Effect of graded nrf2 activation on phase-i and -ii drug metabolizing enzymes and transporters in mouse liver. PLoS ONE 2012, 7, e39006. [Google Scholar] [CrossRef]
- Board, P.G.; Coggan, M.; Chelvanayagam, G.; Easteal, S.; Jermiin, L.S.; Schulte, G.K.; Danley, D.E.; Hoth, L.R.; Griffor, M.C.; Kamath, A.V.; et al. Identification, characterization, and crystal structure of the omega class glutathione transferases. J. Biol. Chem. 2000, 275, 24798–24806. [Google Scholar] [CrossRef]
- Hughes, M.M.; Hooftman, A.; Angiari, S.; Tummala, P.; Zaslona, Z.; Runtsch, M.C.; McGettrick, A.F.; Sutton, C.E.; Diskin, C.; Rooke, M.; et al. Glutathione Transferase Omega-1 Regulates NLRP3 Inflammasome Activation through NEK7 Deglutathionylation. Cell Rep. 2019, 29, 151–161.e5. [Google Scholar] [CrossRef]
- Ding, C.; Wei, H.; Sun, R.; Zhang, J.; Tian, Z. Hepatocytes proteomic alteration and seroproteome analysis of HBV-transgenic mice. Proteomics 2009, 9, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.J.; Ki, S.H.; Brooks, C.; Kim, S.G. Role of hepatitis B virus X repression of C/EBPβ activity in the down-regulation of glutathione S-transferase A2 gene: Implications in other phase II detoxifying enzyme expression. Xenobiotica 2009, 39, 182–192. [Google Scholar] [CrossRef]
- Yi, Y.S.; Park, S.G.; Byeon, S.M.; Kwon, Y.G.; Jung, G. Hepatitis B virus X protein induces TNF-α expression via down-regulation of selenoprotein P in human hepatoma cell line, HepG2. Biochim. Biophys. Acta Mol. Basis Dis. 2003, 1638, 249–256. [Google Scholar] [CrossRef]
- Himoto, T.; Masaki, T. Current trends of essential trace elements in patients with chronic liver diseases. Nutrients 2020, 12, 2084. [Google Scholar] [CrossRef]
- Niu, D.; Zhang, J.; Ren, Y.; Feng, H.; Chen, W.N. HBx genotype D represses GSTP1 expression and increases the oxidative level and apoptosis in HepG2 cells. Mol. Oncol. 2009, 3, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Alavian, S.M.; Showraki, A. Hepatitis B and its relationship with oxidative stress. Hepat. Mon. 2016, 16, e37973. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fan, Y.C.; Sun, F.K.; Zhao, Z.H.; Wang, L.Y.; Hu, L.H.; Yin, Y.P.; Li, T.; Gao, S.; Wang, K. Peripheral type i interferon receptor correlated with oxidative stress in chronic hepatitis b virus infection. J. Interf. Cytokine Res. 2013, 33, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Fisgin, N.T.; Aydin, B.K.; Sarikaya, H.; Tanyel, E.; Esen, S.; Sunbul, M.; Leblebicioglu, H. Oxidative stress and antioxidant defense in patients with chronic hepatitis B. Clin. Lab. 2012, 58, 273–280. [Google Scholar]
- Shaban, N.Z.; Salem, H.H.; Elsadany, M.A.; Ali, B.A.; Hassona, E.M.; Mogahed, F.A.K. Alterations in Lipid Peroxidation and Antioxidants in Patients’ with Different Stages of Hepatitis B Virus Infection in Egypt. Life Sci. J. 2014, 11, 960–967. [Google Scholar]
- Qian, L.; Wang, W.; Zhou, Y.; Ma, J. Effects of reduced glutathione therapy on chronic hepatitis B. Cent. Eur. J. Immunol. 2017, 42, 2015–2018. [Google Scholar] [CrossRef]
- Melhem, A.; Stern, M.; Shibolet, O.; Israeli, E.; Ackerman, Z.; Pappo, O.; Hemed, N.; Rowe, M.; Ohana, H.; Zabrecky, G.; et al. Treatment of chronic hepatitis C virus infection via antioxidants: Results of a phase I clinical trial. J. Clin. Gastroenterol. 2005, 39, 737–742. [Google Scholar] [CrossRef]
- Barakat, E.M.F.; El Wakeel, L.M.; Hagag, R.S. Effects of Nigella sativa on outcome of hepatitis C in Egypt. World J. Gastroenterol. 2013, 19, 2529–2536. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Zahoor, A.; Ibrahim, M.; Younus, M.; Nawaz, S.; Naseer, R.; Akram, Q.; Deng, C.L.; Ojha, S.C. Enhanced Efficacy of Direct-Acting Antivirals in Hepatitis C Patients by Coadministration of Black Cumin and Ascorbate as Antioxidant Adjuvants. Oxid. Med. Cell. Longev. 2020, 2020, 7087921. [Google Scholar] [CrossRef] [PubMed]
- Gunduz, H.; Karabay, O.; Tamer, A.; Özaras, R.; Mert, A.; Tabak, Ö.F. N-acetyl cysteine therapy in acute viral hepatitis. World J. Gastroenterol. 2003, 9, 2698–2700. [Google Scholar] [CrossRef]
- Grant, P.R.; Black, A.; Garcia, N.; Prieto, J.; Garson, J.A. Combination therapy with interferon-alpha plus N-acetyl cysteine for chronic hepatitis C: A placebo controlled double-blind multicentre study. J. Med. Virol. 2000, 442, 439–442. [Google Scholar] [CrossRef]
- Shohrati, M.; Dermanaki, F.; Babaei, F.; Alavian, S.M. Evaluation of the effects of Oral N-Acetylcysteine and a Placebo in Paraclinical and Oxidative Stress Parameters of Patients with Chronic Hepatitis B. Hepat. Mon. 2010, 10, 95–100. [Google Scholar]
- Look, M.P.; Gerard, A.; Rao, G.S.; Sudhop, T.; Fischer, H.P.; Sauerbruch, T.; Spengler, U. Interferon/antioxidant combination therapy for chronic hepatitis C—A controlled pilot trial. Antivir. Res. 1999, 43, 113–122. [Google Scholar] [CrossRef]
- Wagoner, J.; Negash, A.; Kane, O.J.; Martinez, L.E.; Nahmias, Y.; Bourne, N.; Owen, D.M.; Grove, J.; Brimacombe, C.; McKeating, J.A.; et al. Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology 2010, 51, 1912–1921. [Google Scholar] [CrossRef] [PubMed]
- Umetsu, T.; Inoue, J.; Kogure, T.; Kakazu, E.; Ninomiya, M.; Iwata, T.; Takai, S.; Nakamura, T.; Sano, A.; Shimosegawa, T. Inhibitory effect of silibinin on hepatitis B virus entry. Biochem. Biophys. Rep. 2018, 14, 20–25. [Google Scholar] [CrossRef]
- Wu, Y.F.; Fu, S.L.; Kao, C.H.; Yang, C.W.; Lin, C.H.; Hsu, M.T.; Tsai, T.F. Chemopreventive effect of silymarin on liver pathology in HBV X protein transgenic mice. Cancer Res. 2008, 68, 2033–2042. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Polyak, S.J.; Ferenci, P.; Pawlotsky, J.M. Hepatoprotective and antiviral functions of silymarin components in hepatitis C virus infection. Hepatology 2013, 57, 1262–1271. [Google Scholar] [CrossRef]
- Gordon, A.; Hobbs, D.A.; Bowden, D.S.; Bailey, M.J.; Mitchell, J.; Francis, A.J.P.; Roberts, S.K. Effects of Silybum marianum on serum hepatitis C virus RNA, alanine aminotransferase levels and well-being in patients with chronic hepatitis C. J. Gastroenterol. Hepatol. 2006, 21, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Hawke, R.L.; Schrieber, S.J.; Soule, T.A.; Wen, Z.; Smith, P.C.; Reddy, K.R.; Wahed, A.S.; Belle, S.H.; Afdhal, N.H.; Navarro, V.J.; et al. Silymarin ascending multiple oral dosing phase I study in noncirrhotic patients with chronic hepatitis C. J. Clin. Pharmacol. 2010, 50, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Fried, M.W.; Navarro, V.J.; Afdhal, N.; Belle, S.H.; Wahed, A.S.; Hawke, R.L.; Doo, E.; Meyers, C.M.; Reddy, K.R. Effect of silymarin (milk thistle) on liver disease in patients with chronic hepatitis C unsuccessfully treated with interferon therapy: A randomized controlled trial. JAMA 2012, 308, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Liu, S.K.; Liu, X.Y.; Li, Z.J.; Li, B.; Zhou, Y.L.; Zhang, H.Y.; Li, Y.W. Meta-analysis: Silymarin and its combination therapy for the treatment of chronic hepatitis B. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 657–669. [Google Scholar] [CrossRef]
- Mohamadkhani, A. On the potential role of intestinal microbial community in hepatocarcinogenesis in chronic hepatitis B. Cancer Med. 2018, 7, 3095–3100. [Google Scholar] [CrossRef]
- Fooladi, A.A.I.; Hosseini, H.M.; Nourani, M.R.; Khani, S.; Alavian, S.M. Probiotic as a novel treatment strategy against liver disease. Hepat. Mon. 2013, 13, e7521. [Google Scholar] [CrossRef]
- Chen, C.; Yu, X.; Lu, H.; Xiao, D.; Mao, W.; Li, L. Antioxidant protective effects of lactitol against endotoxemia in patients with chronic viral hepatitis. Mol. Med. Rep. 2013, 7, 401–405. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, S.; Fu, Y.; Wu, W.; Chen, T.; Chen, J.; Yang, B.; Ou, Q. Gut microbiota dysbiosis in patients with hepatitis B virus–induced chronic liver disease covering chronic hepatitis, liver cirrhosis and hepatocellular carcinoma. J. Viral Hepat. 2020, 27, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, R.; Bedi, O.; Trehanpati, N. Role of Microbiota in Pathogenesis and Management of Viral Hepatitis. Front. Cell. Infect. Microbiol. 2020, 10, 341. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Marra, M.; Sordelli, I.M.; Lombardi, A.; Lamberti, M.; Tarantino, L.; Giudice, A.; Stiuso, P.; Abbruzzese, A.; Sperlongano, R.; Accardo, M.; et al. Molecular targets and oxidative stress biomarkers in hepatocellular carcinoma: An overview. J. Transl. Med. 2011, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Janevska, D.; Chaloska-ivanova, V.; Janevski, V. Hepatocellular Treatment Carcinoma: Risk Diagnosis and Treatment. Open Access Maced. J. Med. Sci. 2015, 3, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Haque, E.; Rezaul Karim, M.; Teeli, A.S.; Śmiech, M.; Leszczynski, P.; Winiarczyk, D.; Parvanov, E.D.; Atanasov, A.G.; Taniguchi, H. Molecular mechanisms underlying hepatocellular carcinoma induction by aberrant nrf2 activation-mediated transcription networks: Interaction of nrf2-keap1 controls the fate of hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 21, 5378. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, C.; Ma, Q.; Chen, W.; Atyah, M.; Yin, Y.; Fu, P.; Liu, S.; Hu, B.; Ren, N.; et al. High GCLC level in tumor tissues is associated with poor prognosis of hepatocellular carcinoma after curative resection. J. Cancer 2019, 10, 3333–3343. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Shao, S.; Duan, W.; Xu, Q.; Li, X.; Han, L.; Li, W.; Zhang, D.; Wang, Z.; Lei, J. Curcumin suppresses hepatic stellate cell-induced hepatocarcinoma angiogenesis and invasion through downregulating CTGF. Oxid. Med. Cell. Longev. 2019, 2019, 8148510. [Google Scholar] [CrossRef]
- Cheng, C.-S.; Wang, N.; Feng, Y. Selenium in the Prevention and Treatment of Hepatocellular Carcinoma: From Biomedical Investigation to Clinical Application. In Importance of Selenium in the Environment and Human Health; IntechOpen: London, UK, 2020. [Google Scholar]
- Brigelius-Flohé, R.; Kipp, A. Glutathione peroxidases in different stages of carcinogenesis. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1555–1568. [Google Scholar] [CrossRef]
- Liu, T.; Kan, X.F.; Ma, C.; Chen, L.L.; Cheng, T.T.; Zou, Z.W.; Li, Y.; Cao, F.J.; Zhang, W.J.; Yao, J.; et al. GPX2 overexpression indicates poor prognosis in patients with hepatocellular carcinoma. Tumor Biol. 2017, 39, 28635398. [Google Scholar] [CrossRef]
- Guerriero, E.; Capone, F.; Accardo, M.; Sorice, A.; Costantini, M.; Colonna, G.; Castello, G.; Costantini, S. GPX4 and GPX7 over-expression in human hepatocellular carcinoma tissues. Eur. J. Histochem. 2015, 59, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Ghalia, A.H.A.; Fouad, I.M. Glutathione and Its Metabolizing Enzymes in Patients with Different Benign and Malignant Diseases. Clin. Biochem. 2001, 33, 657–662. [Google Scholar] [CrossRef]
- Czeczot, H.; Ścibior, D.; Skrzycki, M.; Podsiad, M. Glutathione and GSH-dependent enzymes in patients with liver cirrhosis and hepatocellular carcinoma. Acta Biochim. Pol. 2006, 53, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhao, X.P.; Wang, L.Y.; Gao, S.; Zhao, J.; Fan, Y.C.; Wang, K. Glutathione S-transferase P1 correlated with oxidative stress in hepatocellular carcinoma. Int. J. Med. Sci. 2013, 10, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cai, Y.; Chen, H.; Mao, L. Clinical significance and association of GSTP1 hypermethylation with hepatocellular carcinoma: A meta-analysis. J. Cancer Res. Ther. 2018, 14, S486–S489. [Google Scholar] [CrossRef]
- Huang, Z.Z.; Chen, C.; Zeng, Z.; Yang, H.; Oh, J.; Chen, L.; Lu, S.C. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2011, 15, 19–21. [Google Scholar] [CrossRef]
- Diaz Vivancos, P.; Wolff, T.; Markovic, J.; Pallardò, F.V.; Foyer, C.H. A nuclear glutathione cycle within the cell cycle. Biochem. J. 2010, 178, 169–178. [Google Scholar] [CrossRef]
- Cheng, S.; Liu, H.; Chen, S.; Lin, P.; Lai, C.; Huang, C. Changes of Oxidative Stress, Glutathione, and Its Dependent Antioxidant Enzyme Activities in Patients with Hepatocellular Carcinoma before and after Tumor Resection. PLoS ONE 2017, 12, e0170016. [Google Scholar] [CrossRef] [PubMed]
- Baulies, A.; Montero, J.; Matías, N.; Insausti, N.; Terrones, O.; Basañez, G.; Vallejo, C.; de La Rosa, L.C.; Martinez, L.; Robles, D.; et al. The 2-oxoglutarate carrier promotes liver cancer by sustaining mitochondrial GSH despite cholesterol loading. Redox Biol. 2018, 14, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Miyanishi, K.; Hoki, T.; Tanaka, S.; Kato, J. Prevention of hepatocellular carcinoma: Focusing on antioxidant therapy. World J. Hepatol. 2015, 7, 593–599. [Google Scholar] [CrossRef]
- Brandon-Warner, E.; Sugg, J.A.; Schrum, L.W.; McKillop, I.H. Silibinin inhibits ethanol metabolism and ethanol-dependent cell proliferation in an in vitro model of hepatocellular carcinoma. Cancer Lett. 2010, 291, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, G.; Lo Muzio, L.; Elinos-Báez, C.M.; Jagan, S.; Augustine, T.A.; Kamaraj, S.; Anandakumar, P.; Devaki, T. Silymarin inhibited proliferation and induced apoptosis in hepatic cancer cells. Cell Prolif. 2009, 42, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, G.; Raghavendran, H.R.B.; Vinodhkumar, R.; Devaki, T. Suppression of N-nitrosodiethylamine induced hepatocarcinogenesis by silymarin in rats. Chem. Biol. Interact. 2006, 161, 104–114. [Google Scholar] [CrossRef]
- Stagos, D.; Amoutzias, G.D.; Matakos, A.; Spyrou, A.; Tsatsakis, A.M.; Kouretas, D. Chemoprevention of liver cancer by plant polyphenols. Food Chem. Toxicol. 2012, 50, 2155–2170. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Garzón, V.; Arellanes-Robledo, J.; García-Román, R.; Aparicio-Rautista, D.; Villa-Treviño, S. Inhibition of reactive oxygen species and pre-neoplastic lesions by quercetin through an antioxidant defense mechanism. Free Radic. Res. 2009, 43, 128–137. [Google Scholar] [CrossRef]
- Wang, L.; Zhan, J.; Huang, W. Grape seed proanthocyanidins induce apoptosis and cell cycle arrest of hepg2 cells accompanied by induction of the MAPK pathway and NAG-1. Antioxidants 2020, 9, 1200. [Google Scholar] [CrossRef]
- Fatima, N.; Akhtar, T.; Sheikh, N. Prebiotics: A Novel Approach to Treat Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2017, 2017, 6238106. [Google Scholar] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef]
- Duan, B.; Huang, C.; Bai, J.; Zhang, Y.L.; Wang, X.; Yang, J.; Li, J. Multidrug Resistance in Hepatocellular Carcinoma. In Hepatocellular Carcinoma; Codon Publications: Brisbane, Australia, 2019; pp. 141–158. [Google Scholar]
- Lohitesh, K.; Chowdhury, R.; Mukherjee, S. Resistance a major hindrance to chemotherapy in hepatocellular carcinoma: An insight. Cancer Cell Int. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, S.C.; Sheldon, R.D.; Rauckhorst, A.J.; Noterman, M.F.; Solst, S.R.; Buchanan, J.L.; Mapuskar, K.A.; Pewa, A.D.; Gray, L.R.; Oonthonpan, L.; et al. Disrupting Mitochondrial Pyruvate Uptake Directs Glutamine into the TCA Cycle away from Glutathione Synthesis and Impairs Hepatocellular Tumorigenesis. Cell Rep. 2019, 28, 2608–2619.e6. [Google Scholar] [CrossRef] [PubMed]
- Duval, A.P.; Troquier, L.; Silva, O.D.S.; Demartines, N.; Dormond, O. Diclofenac potentiates sorafenib-based treatments of hepatocellular carcinoma by enhancing oxidative stress. Cancers 2019, 11, 1453. [Google Scholar] [CrossRef] [PubMed]
- Van der Vijgh, W.J.F. Protection of Normal Tissues from the Cytotoxic Effects of Chemotherapy and Radiation by Amifostine (WR-2721): Preclinical Aspects. Eur. J. Cancer Part A 1995, 31, S1–S7. [Google Scholar] [CrossRef]
- Sidi, V.; Arsos, G.; Papakonstantinou, E.; Hatzipantelis, E.; Fragandrea, I.; Gombakis, N.; Koliouskas, E. Use of amifostine in the treatment of recurrent solid tumours in children. Hippokratia 2007, 11, 25–29. [Google Scholar] [PubMed]
- Feng, M.; Smith, D.E.; Normolle, D.P.; Knol, J.A.; Pan, C.C.; Ben-Josef, E.; Lu, Z.; Feng, M.R.; Chen, J.; Ensminger, W.; et al. A phase i clinical and pharmacology study using amifostine as a radioprotector in dose-escalated whole liver radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Symon, Z.; Levi, M.; Ensminger, W.D.; Smith, D.E.; Lawrence, T.S. Selective radioprotection of hepatocytes by systemic and portal vein infusions of amifostine in a rat liver tumor model. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 473–478. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).