1. Introduction
Memory and learning impairments are the most typical features in neuronal diseases, resulting from dysfunction and loss of neurons in the brain tissues, particularly the hippocampus [
1]. Within the brain, the hippocampus, basal forebrain, and amygdala are extremely vulnerable to oxidative damage due to high oxygen consumption and an abundance of unsaturated fatty acids and metal ions while maintaining a relatively low antioxidant capacity [
2,
3]. Cumulative oxidative stress can not only result in oxidized molecules but also impair key organelles, such as mitochondria, by reducing the production of ATP required for neuron survival and metabolism [
4]. Hippocampal oxidative stress is thereby acknowledged as a crucial causative factor of deterioration in the learning and memory performance.
It is well-documented that learning and memory are closely related to glutamatergic and cholinergic neurotransmission in the mammalian central nervous system (CNS) [
5]. Normally, glutamate, as a major endogenous excitatory neurotransmitter, is responsible for glutamatergic neurotransmission [
6]. However, an excess of extracellular glutamate can lead to neurotoxicity via ionotropic glutamate receptor-mediated excitotoxicity and cystine/glutamate antiporter-mediated oxidative stress, contributing to the development of many acute and chronic brain diseases [
7,
8,
9,
10]. Chronic exposure of neurons to glutamate results in persistent activation of postsynaptic glutamate receptors, destabilizing the intracellular Ca
2+ balance and eventually causing neuronal death [
11]. The mechanism of glutamate-induced oxidative toxicity has been extensively characterized, and extracellular glutamate accumulation has been speculated to inhibit cystine uptake by reversing the action of the cystine/glutamate antiporter, leading to glutathione (GSH) deprivation, which further promotes the accumulation of intracellular reactive oxygen species (ROS) [
12]. An increase in ROS activates 12-lipoxygenase and accelerates Ca
2+ influx [
13], promoting mitochondrial dysfunction-associated ATP deprivation, apoptosis-inducing factor (AIF) nuclear translocation, DNA fragmentation, and cell death [
14,
15,
16].
Scopolamine, a nonselective muscarinic acetylcholine receptor antagonist, is clinically suggested as an anti-muscarinic and anti-cholinergic drug. However, it was reported that scopolamine could promote glutamate release and rapidly increase extracellular glutamate levels, subsequently triggering a burst of glutamate transmission [
17], resulting in cognitive impairment [
18,
19], and the scopolamine-induced cognitive impairment presented strong positive correlation with changes in neuronal oxidative status and mitochondrial function [
20,
21,
22,
23]. In addition, consumption of a high-fat diet (HFD) has been shown to be a risk factor for glutamate-induced oxidative stress and mitochondrial dysfunction, contributing to metabolic dysfunction-associated neuronal death and cognitive decline [
24,
25]. Both scopolamine and HFD intervention have been widely used as animal models for the study of neuronal oxidative stress-associated cognitive impairment.
Chalcones, one of the subclasses of the flavonoid family, are precursors of flavones in the biosynthesis of flavonoids and have been reported to exhibit a broad spectrum of pharmacological properties [
26]. We previously designed and screened a novel chalcone analog, 1-(2,3,4-trimethoxyphenyl)-2-(3,4,5-trimethoxyphenyl)-acrylketone (Tak,
Supplemental Figure S1), which could significantly induce phase II enzyme expression in human retinal pigment epithelia (ARPE-19) cells via promoting nuclear factor-E2-related factor 2 (Nrf2) nuclear translocation due to its unique chemical structure [
27]. It is thus tempting to speculate that Tak, as an Nrf2 activator, may exhibit a neuroprotective effect through attenuating oxidative stress and recovering mitochondrial function. The present study was dedicated to investigating the neuronal protection of Tak and the possible underlying mechanisms in vitro and in vivo.
2. Materials and Methods
2.1. Chemicals and Reagents
Glutamate (G3291), LY294002 (L9908), JC-1 (T4069), ATP bioluminescent cell assay kit (MAK190), and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (M5655) were obtained from Sigma (St. Louis, MO, USA). Antibodies against p-Akt (9271), Akt (9272), p-Erk1/2 (9106), Erk1/2 (4695), p-p38 (4511), p38 (8690), p-JNK1/2 (9255), JNK1/2 (9252), BDNF (47808), caspase 3 (14220), and β-Actin (3700) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against Complex I (459130), Complex II (459230), Complex III (459140), Complex IV (459600), and Complex V (459240) were purchased from Thermo Fisher (Rockford, IL, USA). Antibodies against NQO-1 (sc-376023), HO-1 (sc-390991), SOD1 (sc-101523), and SDO2 (sc-133134) were purchased from Santa Cruz (Dallas, TX, USA). Antibodies against NGF (ab52918) and Nrf2 (ab92946) were purchased from Abcam (Cambridge, UK). Cell culture medium (11965092), H2DCF-DA (C369), bicinchoninic acid protein assay kit (23252), Mito-Tracker
® Green FM (M7514), and MitoSOX
TM Red Mitochondrial Superoxide Indicator (M36008) were purchased from Life Technologies (San Diego, CA, USA). Secondary antibodies including Goat Anti-Rabbit IgG (111-035-003), Goat Anti-Mouse IgG (115-035-003), and Rabbit Anti-Goat IgG (305-035-003) were purchased from Jackson ImmunoResearch (West Grove, PA, USA). A FITC Annexin V Apoptosis Detection Kit (556547) was obtained from BD Biosciences (San Jose, CA, USA). Hoechst staining kit (C1011) was purchased from Beyotime (Shanghai, China). TRIzol Reagent (9108), PrimeScript RT-PCR Kit (6210A), and TB Green Premix Ex Taq II kit (RR820B) were purchased from Takara (Shiga, Japan). Tak was synthesized from 1,2,3-trimethoxybenzene following a previously published study [
27], and Tak at the purity of 96.1% was used in the present study. The synthetic scheme and structure of Tak are shown in
Figure S1.
2.2. Cell Culture
HT22 mouse hippocampal neuronal cell line was purchased from Sigma (SCC129, St. Louis, MO, USA). SH-SY5Y human neuroblastoma cell line was purchased from ATCC (CRL-2266, Manassas, VA, USA). Cells were cultured in DMEM medium (11965092, Life Technology, Rockford, IL, USA) supplemented with 10% fetal calf serum (30-2030, ATCC), 100 U/mL penicillin G sodium (P3032, Sigma), and 100 μg/mL streptomycin sulfate (11860038, Thermo Fisher). The cells were maintained at 37 °C in a humidified atmosphere with 5% CO2. The medium was changed every two days. HT22 and SH-SY5Y cells were used within 8 generations.
2.3. Assessment of Cell Viability
Cell viability was analyzed using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. Both HT22 and SH-SY5Y cells were seeded at a density of 3.5 × 104 cells/mL in 96-well plates, allowing cells to attach and grow for 24 h, followed by indicated compound treatments. After the treatment, cultured medium was discarded and living cells were cultured with 200 μL of MTT–DMEM solution (0.5 mg/mL in serum free medium) for 4 h. The MTT solution was then discarded, and 200 μL DMSO was added to each well to dissolve the MTT formazan. The absorbance was measured at a wavelength of 590 nm using a microplate fluorometer (Fluoroskan Ascent; Thermo Fisher Scientific, Inc., Rockford, IL, USA).
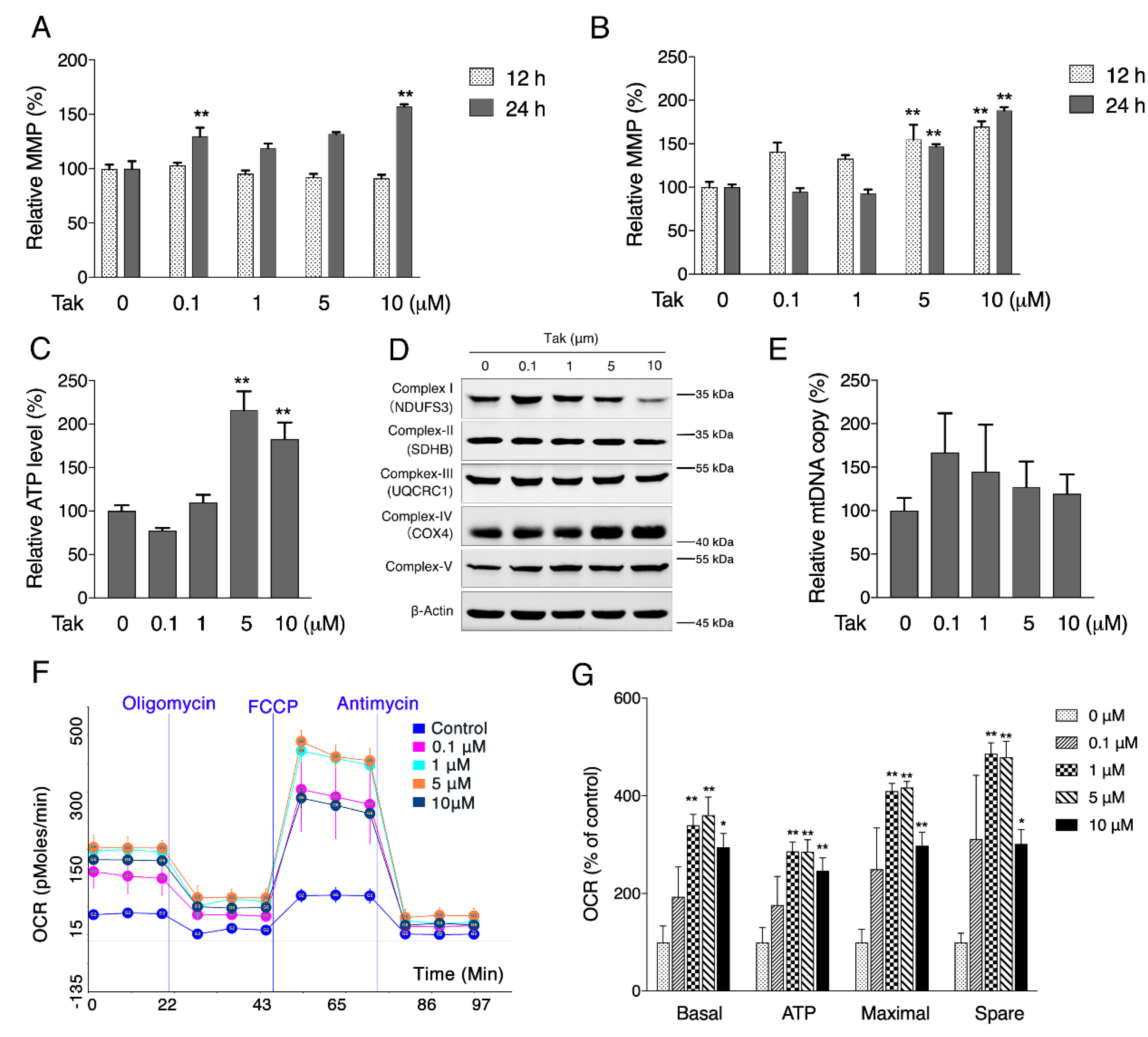
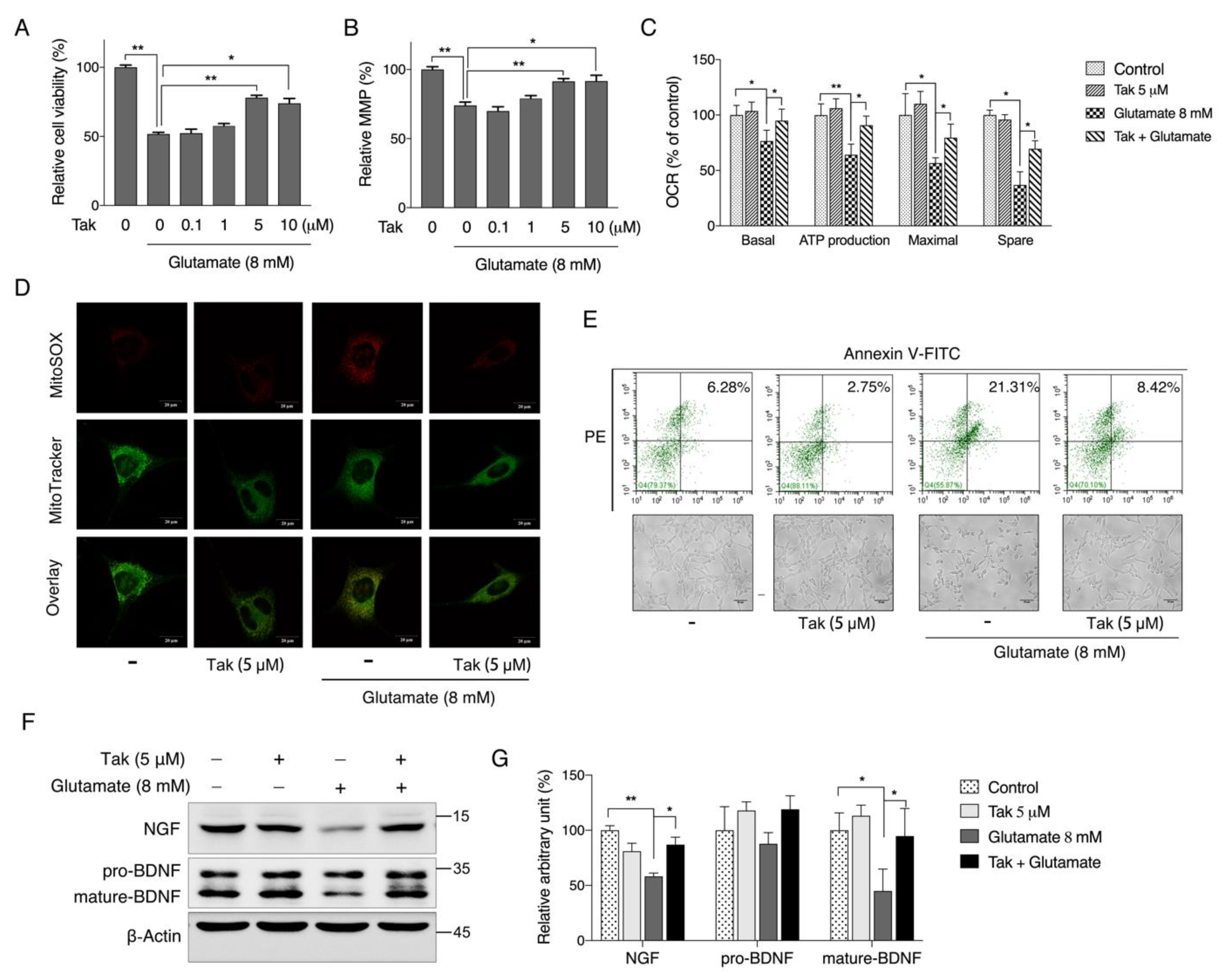
2.4. JC-1 Assay for MMP
Mitochondrial membrane potential (MMP) was analyzed, following a previously published study [
27]. Briefly, cells were cultured at a density of 3.5 × 10
4 cells/mL in 96-well plates. After treatment, MMP was analyzed using the lipophilic cationic probe 5,5′,6,6′-terachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1, 5 μg/mL in serum-free medium). The cells were washed with PBS once and scanned with a microplate fluorometer (Fluoroskan Ascent; Thermo Fisher) at a 488-nm excitation wavelength and 538- and 590-nm emission wavelengths to measure green and red JC-1 fluorescence, respectively. The red/green fluorescence intensity ratio reflects the MMP.
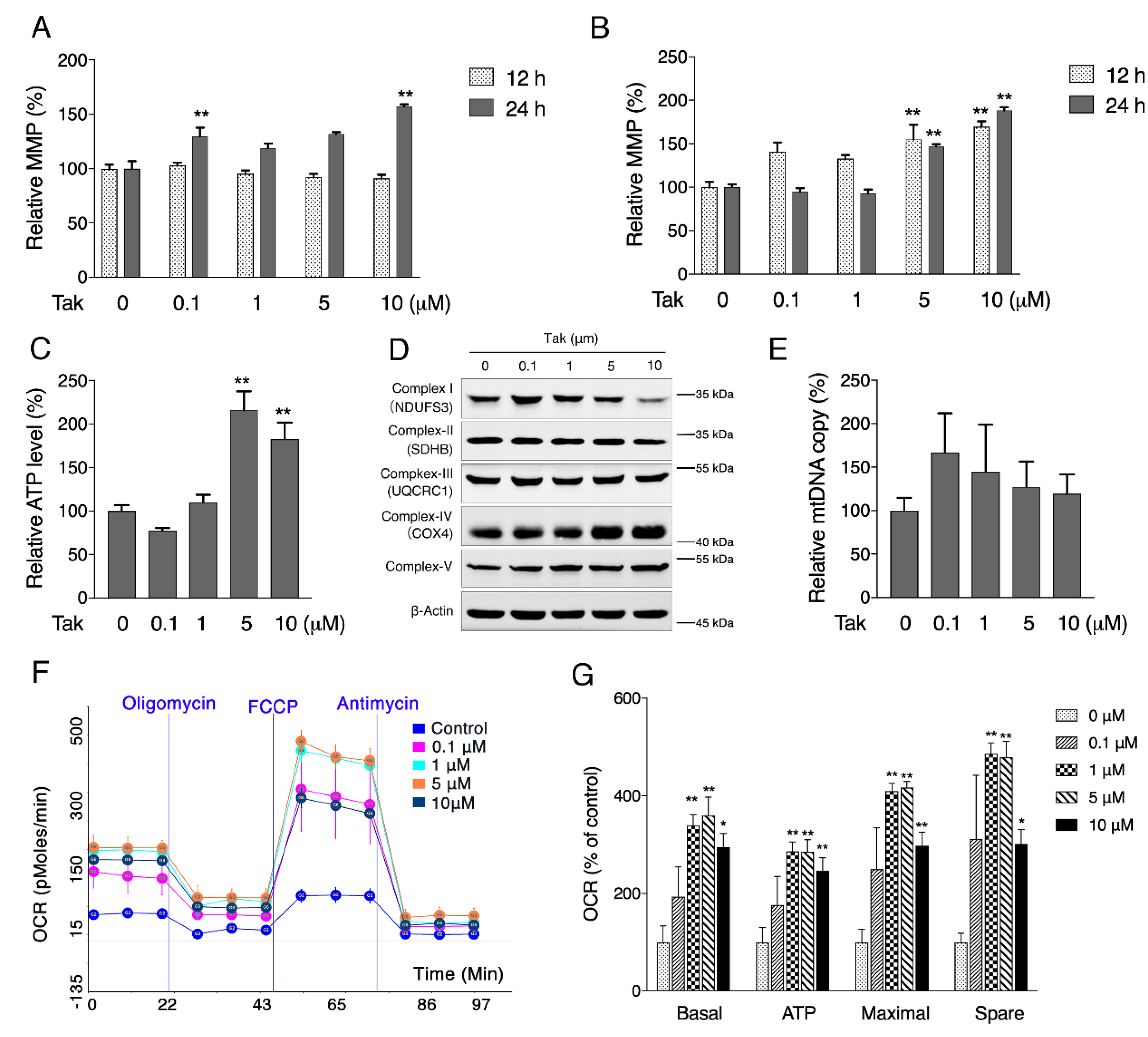
2.5. Oxygen Consumption Rate Analysis
Oxygen consumption rates of living cells were analyzed with the Seahorse method (Seahorse Bioscience, Santa Clara, CA, USA) [
28]. Cells were seeded in XF 24-well microplates. After treatment, oxygen consumption was measured using extracellular flux analysis. The final concentrations of the mitochondrial inhibitors were 1 μM antimycin A, 4 μM carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) and 4 μM oligomycin. Basal respiration was the baseline oxygen consumption reading before the compounds were injected. Basal, maximal, ATP-linked, and nonmitochondrial respiration and the reserve capacity were adjusted according to the protein concentration in each well, which was measured using the bicinchoninic acid method.
2.6. Mitochondrial Superoxide Analysis
Cells were cultured at a density of 5 × 10
4 cells/mL in 24-well plates. After treatment, the generation of mitochondrial superoxides was observed using a mitochondrial superoxide indicator (MitoSOX
TM Red), and the mitochondria were assessed using Mito-Tracker
® Green FM, following a previous study [
27]. Briefly, the cells were stained with 500 nM Mito-Tracker
® Green FM in serum-free medium for 30 min, followed by incubation with 10 μM MitoSOX
TM Red in serum-free medium for 10 min. After washing with PBS, the cells were observed by laser scanning confocal microscopy (Zeiss, Jena, Germany).
2.7. ATP Analysis
The ATP content was determined using an ATP bioluminescent cell assay kit according to the manufacturer’s instructions. Cells were lysed with 0.5% Triton X-100 in 100 mM glycine buffer (pH 7.4). The supernatant was collected after centrifugation at 21,000× g for 10 min at 4 °C. Then, 40 μL of the supernatant was transferred to an appropriate bioluminescence plate. The mixture was agitated for 5 min before the addition of 160 μL reaction solution. The luminescence was measured and normalized to the protein content.
2.8. Hoechst Staining
Hoechst staining was employed to identify apoptotic cells by condensed and fragmented nuclei [
29]. Briefly, cells were plated onto coverslips at a density of 9 × 10
4 cells/mL in 6-well plates. After treatment, the cells were fixed with 4% formaldehyde for 30 min at room temperature, followed by washes with PBS. The cells were incubated with the membrane-permeable fluorescent dye Hoechst for 30 min and then observed using fluorescence microscopy (Olympus, Tokyo, Japan) with a peak excitation wavelength of 340 nm.
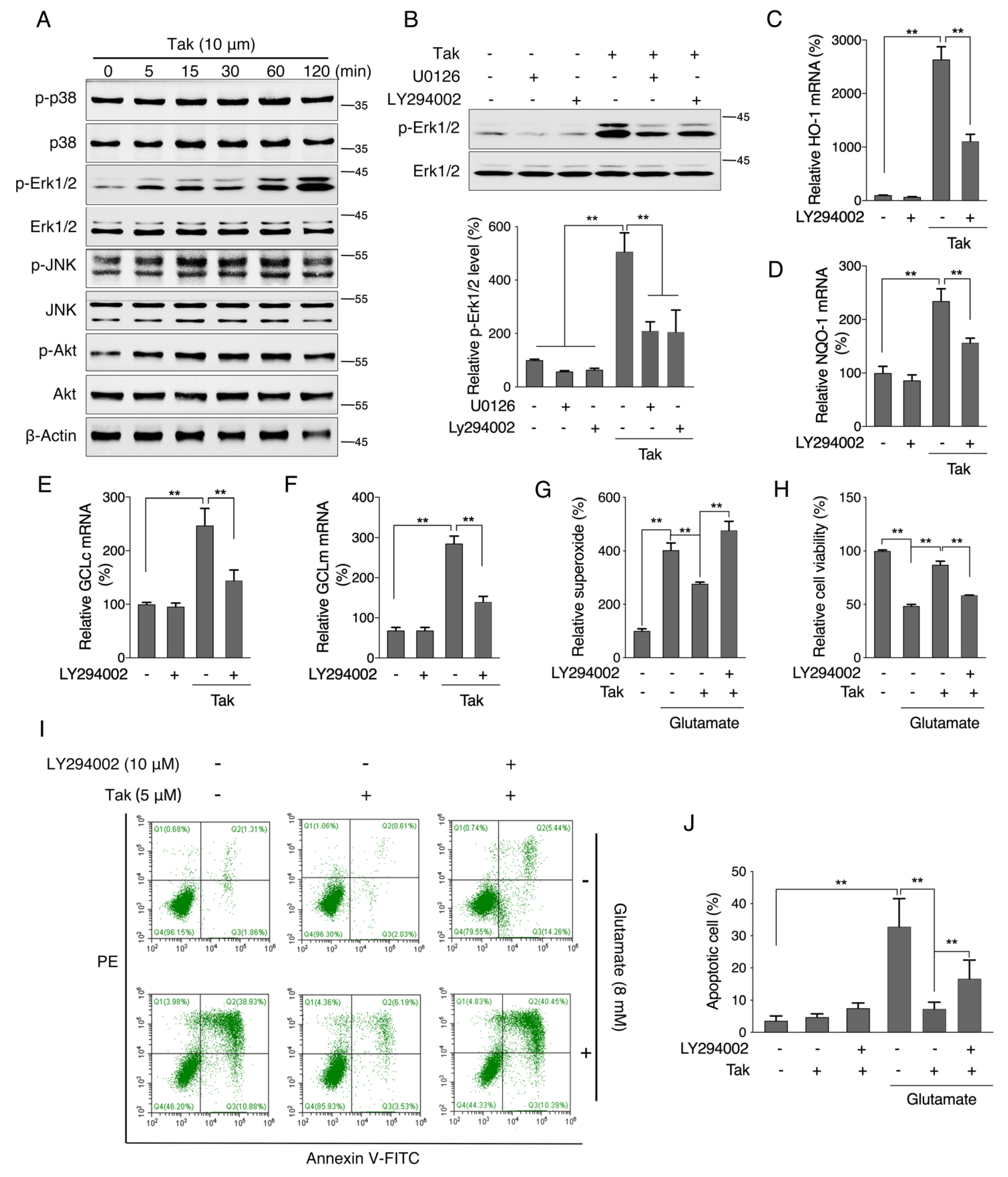
2.9. Annexin V-FITC/PI Double Staining Assay
Cells in the early and late apoptotic stages were analyzed by an Annexin V-FITC/PI double staining assay. Early apoptotic cells showed the externalization of phosphatidylserine (PS) on the outer leaflets of the plasma membrane. Annexin V had a high affinity for PS. Cells were plated at a density of 9 × 104 cells/mL in 6-well plates. When reaching confluence of 80–90%, cells were exposed to Tak or glutamate treatment for the indicated time, then both floating and attached cells were all collected and washed twice with PBS. Cells were resuspended in 500 μL binding buffer, followed by staining with 10 μL Annexin V and 5 μL propidium iodide (PI) in the dark at room temperature for 15 min. The stained cells were analyzed immediately using a FACS flow cytometry analyzer (Beckman Coulter, Indianapolis, IN, USA) with emission filters at wavelengths of 488–530 nm for green fluorescence of Annexin V (FL1) and 488–630 nm for red fluorescence of PI (FL2). A total of at least 10,000 cells per sample were acquired to ensure adequate data. The lower left quadrant indicates the normal cells (Annexin V−, PI−), the lower right quadrant indicates early apoptotic cells (Annexin V+, PI−), and the upper right quadrant indicates late apoptotic cells (Annexin V+, PI+).
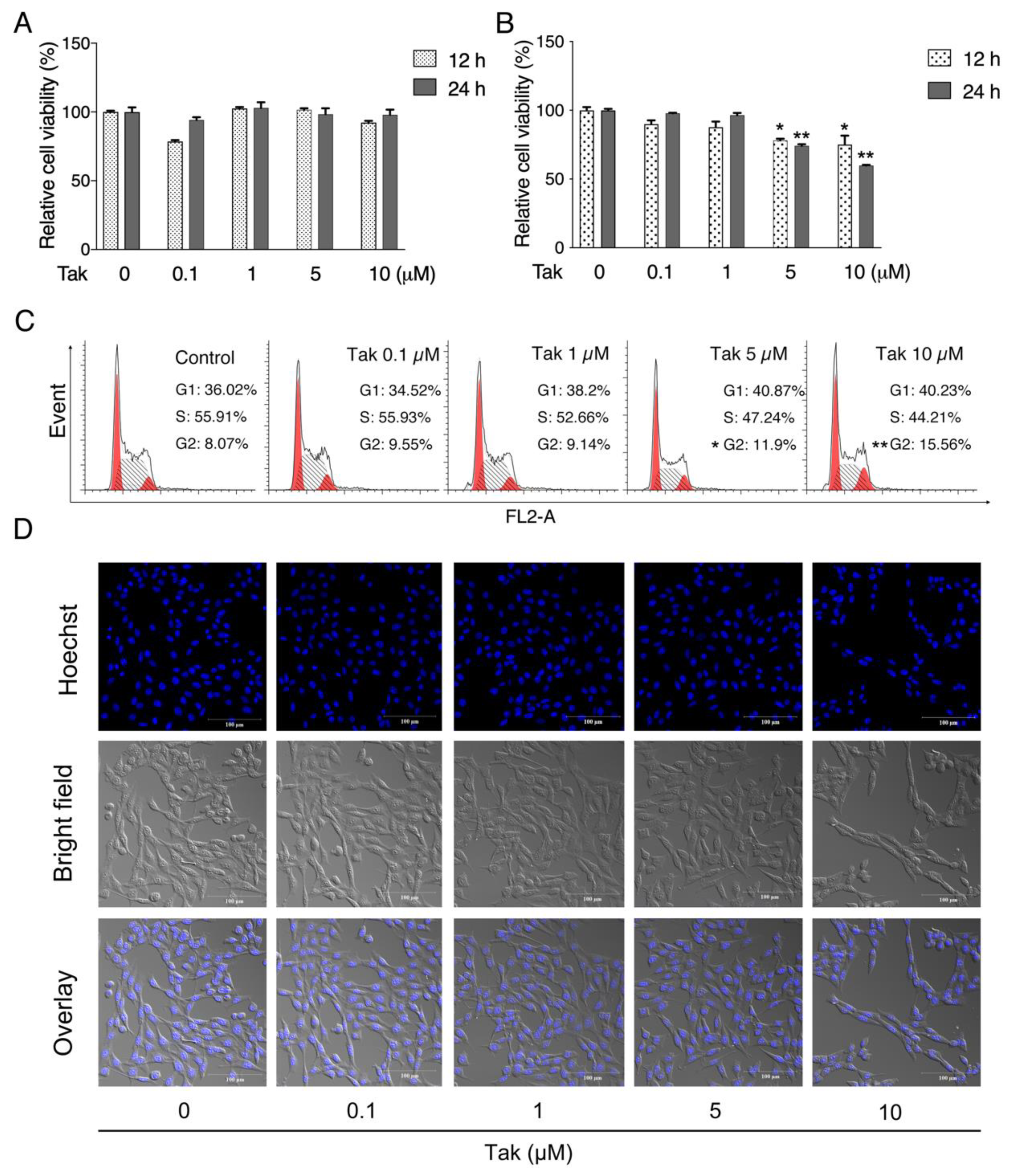
2.10. Cell Cycle Analysis
Cells were plated at a density of 9 × 104 cells/mL in 6-well plates. When cells were grown to 80–90% confluence, cells were exposed to Tak treatment for 24 h. At the end of the treatment, the cells were serum starved for 3 h. After synchronization, the floating and attached cells were collected and centrifugated at 4 °C 1000× g for 5 min, and then cell pellets were washed twice with PBS and resuspended in 500 μL PBS with the addition of 4.5 mL 70% ethanol, followed by incubation at −20 °C for at least 2 h. Afterwards, the cells were collected and centrifugated at 1000× g for 5 min. Cell pellets were resuspended in 500 μL 1 mg/mL PI solution (0.1% Triton X-100, 100 μg/mL Rnase A, and 40 μg/mL PI) for 30 min on ice. A total of at least 10,000 cells per sample were recorded and monitored by a FACS flow cytometry analyzer (Beckman Coulter, Brea, CA, USA).
2.11. Measurements of Superoxide Dismutase (SOD) Activity, Catalase (CAT) Activity, Glutathione (GSH) Level and Malondialdehyde (MDA) Level
SOD activity, CAT activity, GSH level, and MDA level were measured using commercially available kits (Jiancheng, Nanjing, China) according to the manufacturer’s instructions. The cell or hippocampus tissue samples were dissected rapidly and homogenized in ice-cold 0.9% saline (pH 7.4). After centrifugation, supernatants were collected for the determination of SOD activity, CAT activity, GSH level, and MDA level. SOD and CAT activities were expressed as units (U) per milligram of protein. GSH and MDA levels were calculated by means of a calibration curve and normalized to the protein concentration.
2.12. Western Blot Analysis
Samples were lysed with western and immunoprecipitation (IP) lysis buffer (P10013, Beyotime, China). The lysates were homogenized, and the homogenates were centrifuged at 13,000× g for 12 min at 4 °C. The supernatants were collected, and the protein concentrations were determined with a bicinchoninic acid (BCA) protein assay kit. Equal aliquots (20 μg) of the protein were separated by 10% SDS-PAGE, transferred to pure nitrocellulose membranes, and blocked with 5% nonfat milk in TBST buffer (8 g/L NaCl, 2.42 g/L Tris, 0.1% Tween20, PH 7.6). The membranes were incubated with primary antibodies at 4 °C overnight. Then, the membranes were incubated with secondary antibodies at room temperature for 1 h. Chemiluminescent detection was performed using an ECL western blotting detection kit (Thermo Fisher, Rockford, IL, USA). The results were analyzed by Quantity One software (Bio-Rad, Shanghai, China) to obtain the optical density ratio of the target proteins relative to β-actin. The antibodies used in the present study were: p-Akt (1:1000), Akt (1:1000), p-Erk1/2 (1:1000), Erk1/2 (1:1000), p-p38 (1:1000), p38 (1:1000), p-JNK1/2 (1:1000), JNK1/2 (1:1000), β-Actin (1:10000), Complex I (1:1000), Complex II (1:1000), Complex III (1:1000), Complex IV (1:1000), Complex V (1:1000), NQO-1 (1:700), HO-1 (1:700), SOD1 (1:700), SOD2 (1:700), BDNF (1:1000), NGF (1:1000), caspase 3 (1:1000), Goat Anti-Rabbit IgG (1:3000), Goat Anti-Mouse IgG (1:3000), and Rabbit Anti-Goat IgG (1:3000).
2.13. Real-Time Quantitative PCR
Total RNA was isolated using TRIzol Reagent followed by treatment with chloroform and precipitation with 2-propanol. The total RNA pellet was then washed with 75% ethanol and resuspended in water. The RNA was subjected to reverse transcription using a PrimeScript RT-PCR Kit, and quantitative real-time PCR analysis (CFX96, Bio-Rad, Hercules, CA, USA) of the genes of interest was conducted using a TB Green Premix Ex Taq II kit. Relative gene expression was calculated using the 2
−ΔΔCt method. The data were normalized to the expression level of β-actin in percentage, and control in each group was normalized to 100%. Primers are shown in
Supplementary Materials (Accession gene IDs: 15368, 18104, 14629, 14630, 12359, 20655, 20656, 11461) (
Table S1).
2.14. Animal Experiments
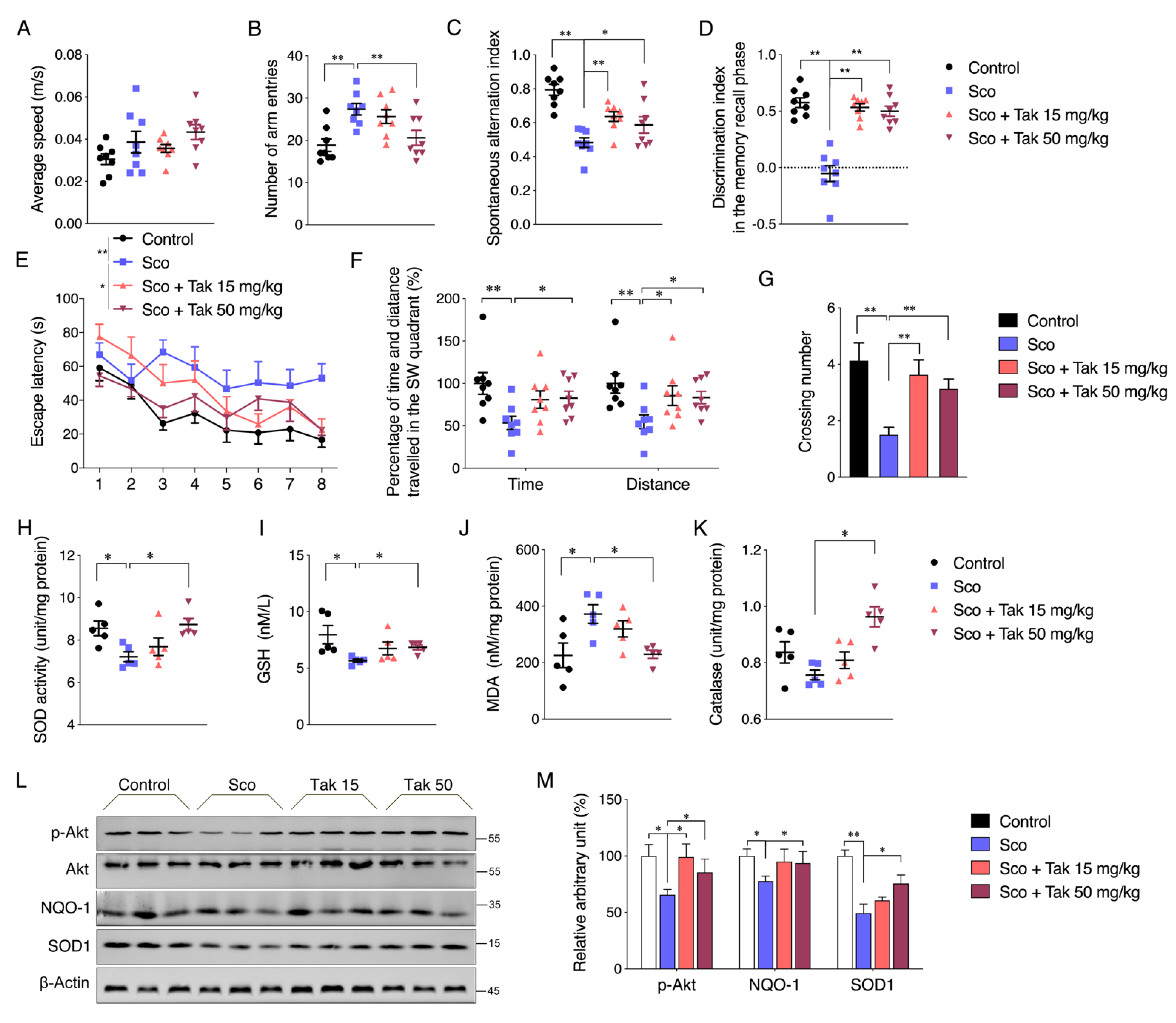
For the scopolamine-induced dementia mouse model, 12-week-old male C57BL/6J mice weighing 25–30 g were purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). After acclimatization for 1 week, the mice were randomly assigned to following four groups (
n = 8 in each group): (1) mice were administered with 0.9% saline (Control), (2) mice were administered with the vehicle followed by intraperitoneal (i.p.) injection of 1 mg/kg scopolamine (Sco), (3) mice were intraperitoneally administered with 15 mg/kg Tak prior to intraperitoneal injection of 1 mg/kg scopolamine (Sco + Tak 15 mg/kg), and (4) mice were intraperitoneally administered with 50 mg/kg Tak prior to intraperitoneal injection of 1 mg/kg scopolamine (Sco + Tak 50 mg/kg). The doses of 15 and 50 mg/kg were determined following a previous study showing the protective effect of another chalcone derivative in mice at the dose of 30 mg/kg [
19]. Tak was dissolved in 0.4 % DMSO in 0.9% saline and intraperitoneally administered once daily for 9 consecutive days. Hereafter, scopolamine was dissolved in 0.9% saline and intraperitoneally injected to induce cognitive deficit 30 min before each behavioral task.
For the HFD-induced diabetic mouse model, 4-week-old male C57BL/6J mice were purchased from the SLAC Laboratory Animal Co., Ltd. (Shanghai, China). After 1 week of acclimatization, the mice were randomly divided into four groups (n = 8 in each group) as follows: (1) mice fed a normal diet (control, 10% kcal fat content, D12492, Research Diets, New Brunswick, NJ, USA), (2) mice fed a high-fat diet (HFD, 60% kcal fat content, D12450, Research Diets), (3) mice fed a HFD with 10 mg/kg/day Tak (taken the long-term feeding into consideration, we changed the dose of Tak from 15 mg/kg/day to 10 mg/kg/day), and (4) mice fed a HFD with 50 mg/kg/day Tak. The HFD feeding proceeded for a total of 8 weeks, and intraperitoneal injections of the vehicle and Tak (0.4% DMSO in PBS) were administered from week 5 to week 8. All animals were housed in a temperature (25–28 °C)- and humidity (60%)-controlled animal room and maintained on a 12-h light/12-h dark cycle with food and water provided during the experiments. Animal procedures were approved by the Xi’an Jiaotong University Animal Care and Use Committee (XJTU-2018-033 for HFD model, XJTU-2019-021 for scopolamine model). All the methods were performed in accordance with approved guidelines, and all efforts were made to minimize the suffering and the number of animals used in this study.
2.15. Open Field Test
The open field test is the behavioral test to evaluate locomotion, based on the natural tendency of rodents to explore new environments and to avoid open and brightly lit spaces [
30]. The open field chamber is an open area, and a video camera is positioned at the top of chamber. The mice were habituated to the testing room for at least 30 min prior to testing. In the trial, each mouse was placed into the chamber center and allowed to explore freely for 3 min. In the next 15 min, the total distance and average speed were automatically recorded by an animal tracking program (AnyMaze, Shanghai, China). After each trial, the chamber was cleaned and dried in order to avoid any olfactory cues.
2.16. Y Maze Test
The Y maze test is based on the natural tendency of rodents to prefer entering a new arm rather than returning to an arm that has just been explored [
31]. The Y maze apparatus consists of three identical arms placed at 120° around an equilateral triangular platform in the center, and a video camera is fixed at the top of Y maze apparatus. The mice were habituated to the testing room for at least 30 min prior to testing. In the trial, the mouse was placed at one arm end, facing the short wall, and allowed to move freely through the maze during a 5 min session. After each trial, the Y maze apparatus was cleaned and dried. The sequence and number of arm entries were recorded by AnyMaze (Shanghai, China). An arm entry was completed when the hind paws of the mouse were completely placed in the arm. A triplet was defined as the mouse’s respective alternations between the arms, including spontaneous alternation (every arm was mentioned in a triplet), alternate arm return (the first and third arms were the same), or same arm return (the first and second arms were the same). A spontaneous alternation was defined as consecutive entries into each arm of the Y maze without any repeats. The percentage of spontaneous alternations was calculated based on the following formula: spontaneous alternation percentage = number of spontaneous alternation/number of triplets. The number of arm entries per trial is used as an indicator of locomotor activity.
2.17. Novel Object Recognition Test
This test is used to assess short-term spatial recognition memory, based on the natural tendency of rodents to spend more time exploring an unfamiliar stimulus than a familiar one [
31]. The general procedure consists of three sessions: the habituation, memory acquisition, and memory recall phases. In habituation phase (2 days), the mice were allowed to freely explore the arena without objects. In the training trial of the memory acquisition phase, the mouse was allowed to explore the arena with two identical objects placed on opposite sides of the cage for 10 min. The cage and objects were cleaned at the end of each trial. During a retention interval of 3 h, the mice were kept individually in a cage until the retention trial. In the retention trial of the memory recall phase, one of the previously familiar objects was substituted for a novel, unfamiliar one. The mouse was allowed to explore the area with two differential objects placed in opposite sides of cage for 10 min. Exploration was defined as sniffing or touching the object with the nose and/or forepaws at a distance of no more than 2 cm. Sitting and standing or leaning on the objects were not considered as an exploration. A discrimination index in the memory recall phase was calculated as time spent exploring the “novel” object compared with the “familiar” object relative to the total time spent exploring the two objects, according to the formula:
ETNO = exploration time of “novel” object; ETFO = exploration time of “familiar” object.
2.18. Morris Water Maze (MWM) Test
MWM test is used to assess hippocampal-dependent spatial learning and reference memory [
32]. The MWM is a white circular pool with a diameter of 122 cm and a height of 51 cm, located in a room with ample surrounding visual cues. The pool is filled with water containing white dye to a depth of 30 cm (24 ± 1 °C). The pool is conceptually divided into four quadrants. A white platform is centered in one of the quadrants and submerged 1 cm below the water surface so that it is invisible at water level. One day before the acquisition trial, the mice were given a pre-training trial for 60 s in the absence of the platform to acclimatize to the situation. Thereafter, in the acquisition trial, each mouse was subjected to four trials each day for four subsequent days. In each acquisition trial, the mouse was gently placed into the water facing the edge of the pool in a randomly selected quadrant which was changed in each trial per day. If the mouse reached the platform within 60 s, it was permitted to stay on the platform for 5 s to remember the position of the goal in relation to surrounding cues. If the mouse did not find the platform within 60 s, it was gently guided to the platform and allowed to stay there for 15 s. To assess memory consolidation, a probe trial was conducted 24 h after the last acquisition trial. The platform was removed from the pool. The mouse was placed into the pool side opposite to the target quadrant and allowed to freely swim for 60 s. A video camera connected to corresponding software monitored the behavior of the mice in the pool, including the escape latency to the platform, cumulative path length, average speed in each acquisition trial, crossing numbers over the target quadrant, and the time spent and distance traveled in the target quadrant.
2.19. Mitochondrial Complex Activity Determination
Hippocampus tissues were collected, resuspended in 1 mL of hypotonic lysis buffer (10 mmol/L NaCl, 2.5 mmol/L MgCl
2, and 10 mmol/L Tris base (pH 7.5)), and homogenized on ice with a glass homogenizer (Fisher Scientific, Pittsburgh, PA, USA). The homogenates were then centrifugated at 1300×
g for 5 min at 4 °C. The supernatant was centrifugated at 17,000×
g for 15 min at 4 °C, and the mitochondrial pellet was resuspended in 100 μL of isotonic buffer (210 mmol/L mannitol, 70 mmol/L sucrose, 5 mmol/L Tris base, and 1 mmol/L EDTA-Na
2 (pH 7.5)). Assays for reduced nicotinamide adenine dinucleotide (NADH)–ubiquinone reductase (complex I), ubiquinol–cytochrome c reductase (complex III), and Mg
2+-ATPase (complex V) activities were performed using the activity kits according to our previous method [
28].
2.20. Statistical Analysis
All cell experiments were repeated at least three times. The data are presented as the mean ± S.E.M. All statistical analyses were performed using SPSS 23 software. The significance of differences was assessed by unpaired Student’s t-test or one-way ANOVA followed by Tukey’s multiple comparisons test. For all comparisons, the level of significance was set at p < 0.05.
4. Discussion
Synaptic plasticity is the key cellular mechanism involved in learning and memory, which requires glutamatergic neurons to form a neural circuit in the hippocampus. However, excessive excitation of glutamatergic neurons leads to glutamate accumulation in the extracellular compartment, followed by a number of deleterious consequences, including the generation of free radicals and the activation of mitochondrial permeability transition, contributing to neuronal death [
34]. In addition, clinical use of the anti-cholinergic drug scopolamine and long-term HFD intake were suggested to disrupt glutamate homeostasis, resulting in increased oxidative stress, mitochondrial dysfunction, and cognitive impairment [
18,
19,
24,
25]. Targeting oxidative stress and mitochondrial protection is thereby considered a popular strategy for neuronal survival and cognitive improvement [
12,
35,
36].
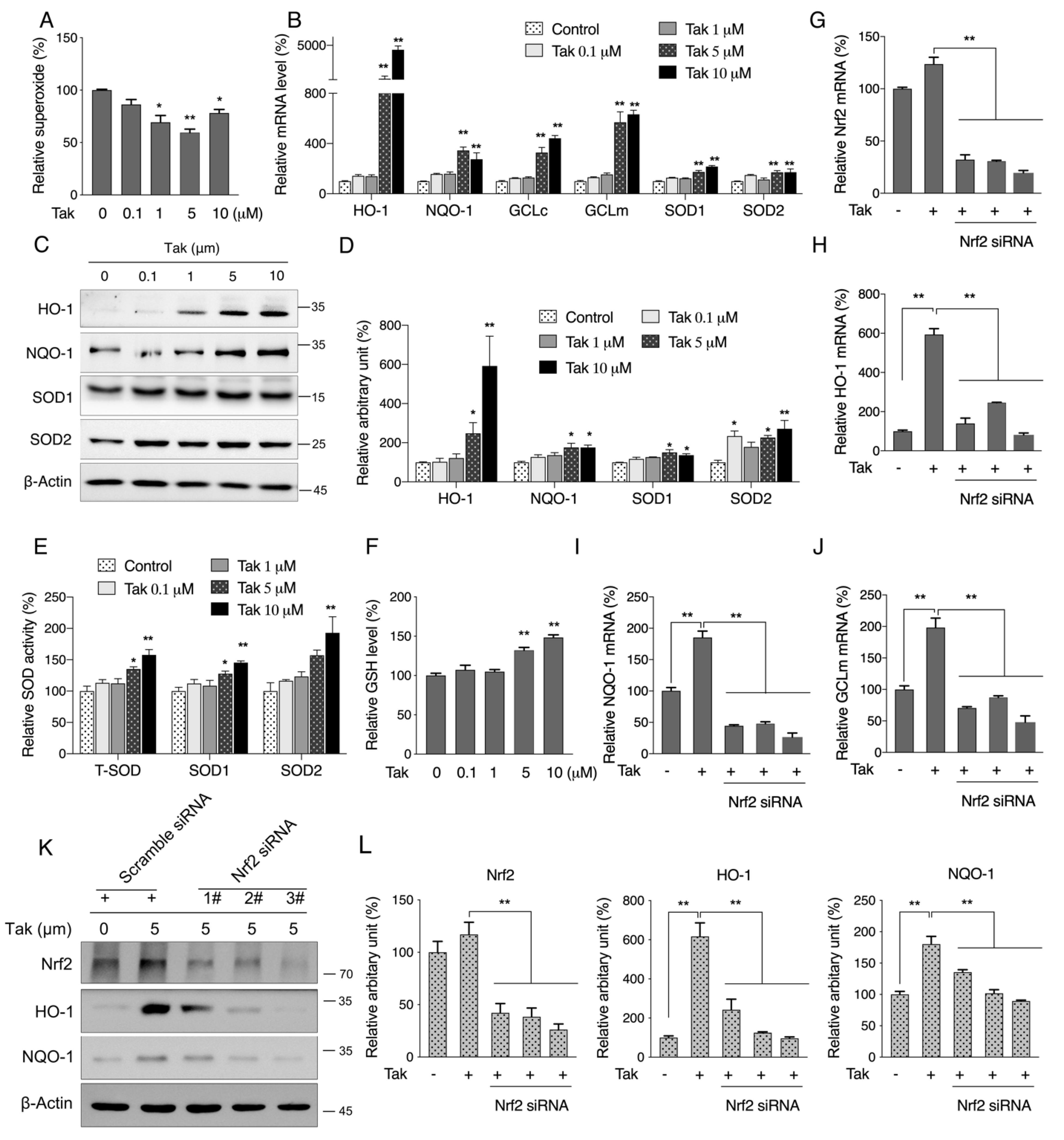
The Nrf2 signaling pathway, which orchestrates key cellular antioxidant response mechanisms, has great potential for protecting cells against oxidative stress-mediated brain diseases associated with cognitive impairment [
37]. Studies have indicated that overexpression of Nrf2 induced memory improvement in a mouse model of Alzheimer’s disease [
38], while impaired Nrf2 signaling and increased cerebral oxidative stress exacerbated deleterious effects of obesity on cognitive performance [
24]. Chalcones, biosynthetic precursors of flavonoids, have gained increasing attention for their derivates which could regulate Nrf2 activity [
39]. The ability of chalcone derivatives activating Nrf2 was markedly improved with one or two electron-withdrawing groups on phenyl ring B and up to three methoxyl and/or hydroxyl groups on phenyl ring A [
40]. We recently synthesized and screened a new chalcone analog that contains three methoxyl groups on both phenyl rings A and B, which effectively activated Nrf2 in vitro [
27]. In the current study, both HT22 and SH-SY5Y cell lines were cultured to evaluate the potential toxicity of Tak. Notably, Tak had no significant toxic effect except for slight cell cycle arrest in SH-SY5Y cells. Since the HT22 cell line is an immortalized mouse hippocampal cell line that lacks functional ionotropic glutamate receptors, similar to the undifferentiated neuronal stem cell line, it expresses neuron-specific enolase and neurofilament proteins [
41], making it an excellent candidate for a glutamate-induced neuronal oxidative stress model for compound screening.
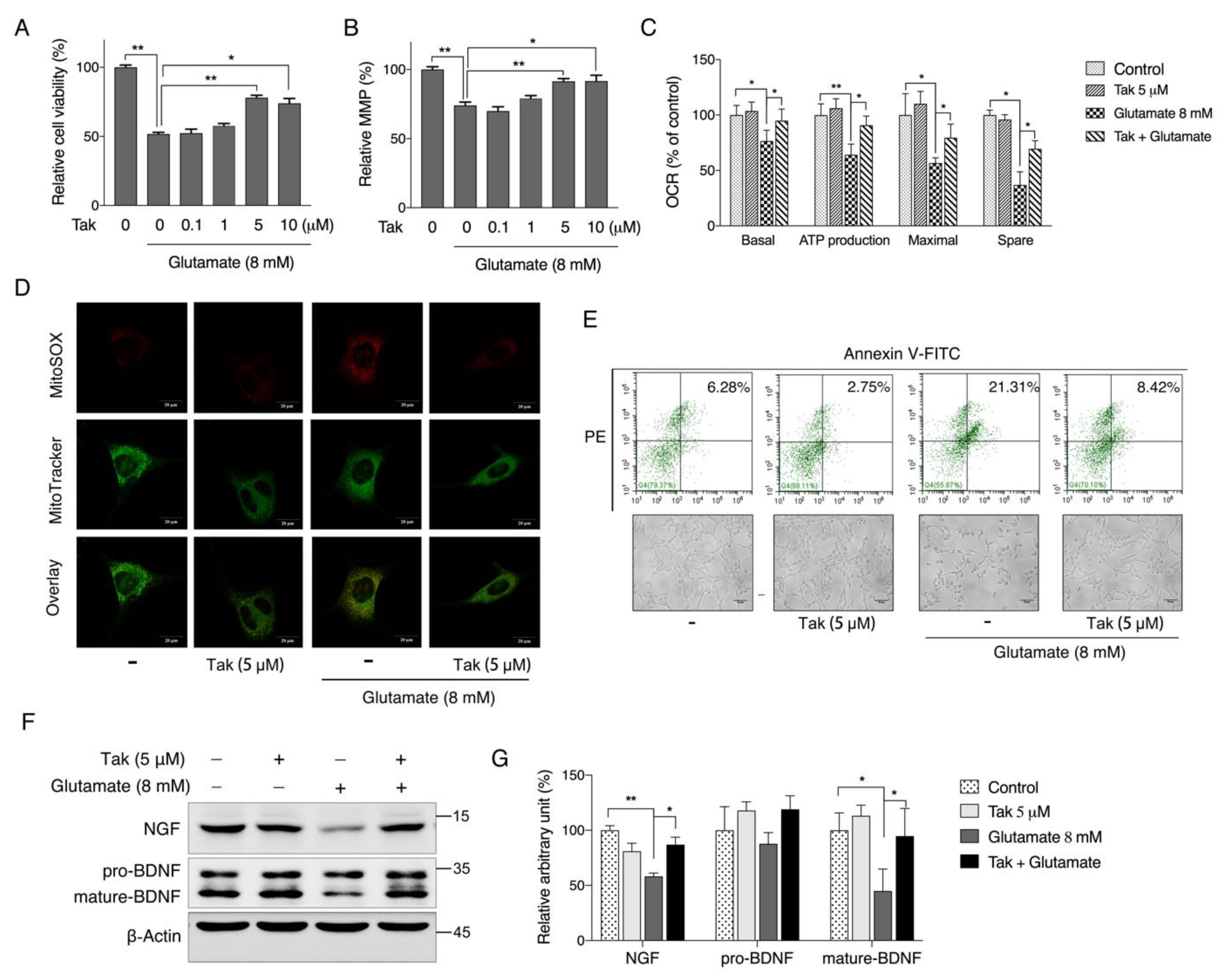
Considering that mitochondria are the key organelles for neuronal function and survival, we evaluated the effect of Tak on mitochondrial function and found that Tak significantly increased the MMP, cellular ATP level, and oxygen respiration capacity. However, these notable benefits were not attributed to mitochondrial biogenesis. It is estimated that 1–3% of mitochondrial oxygen consumed during the process of oxidative phosphorylation is incompletely reduced, resulting in the formation of superoxide anions, which are a predominant form of ROS produced in mitochondria, and their accumulation could induce oxidative stress to suppress mitochondrial activity [
41]. We hypothesized that the improvement in mitochondrial function by Tak was due to its enhanced redox status, which was confirmed by measuring mitochondrial ROS. Phase II enzymes are known as the endogenous antioxidative system that is regulated by Nrf2, which forms heterodimers with small oncogene family proteins for the selective recognition of the antioxidant responsive element (ARE) on target genes, encoding a set of phase II detoxifying enzymes, including HO-1, NQO-1, and GCL [
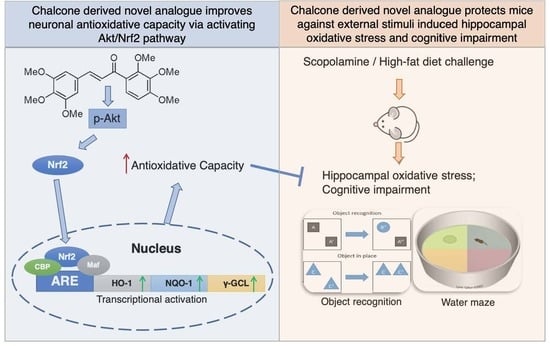
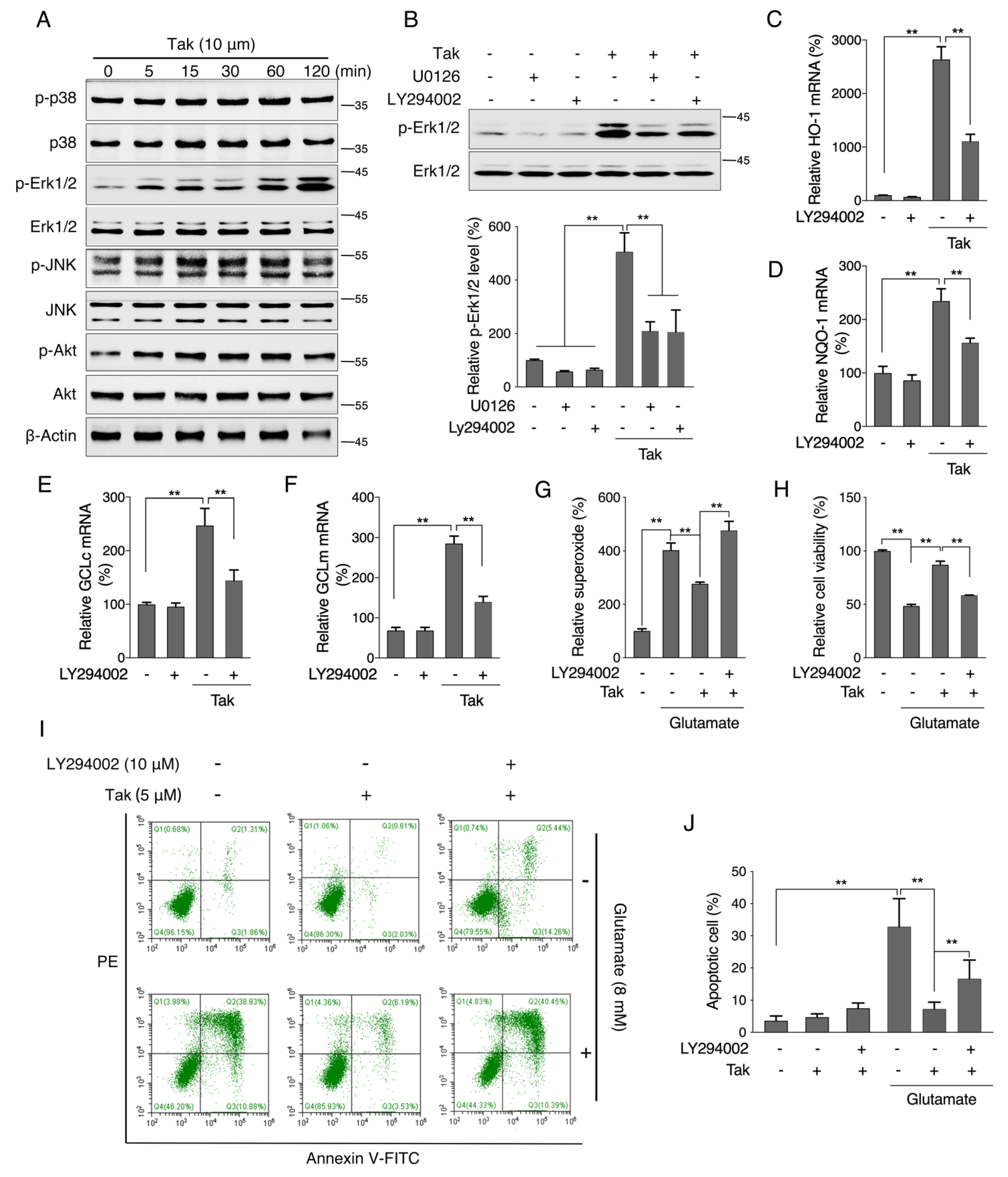
42]. Consistently, we showed that Tak markedly increased the mRNA expression levels of phase II enzymes via Nrf2, and we confirmed that Tak activated Nrf2 through Akt signaling. We thereby concluded that Tak activated phase II enzymes and enhanced cellular redox status, contributing to the improvement of mitochondrial function via Akt-mediated Nrf2 activation.
Abnormal glutamate homeostasis has been reported to be involved in multiple risk factors that contribute to declined cognitive function, such as utilization of clinical drug scopolamine or a lifestyle of long-term HFD intake [
18,
19,
24,
25,
43,
44,
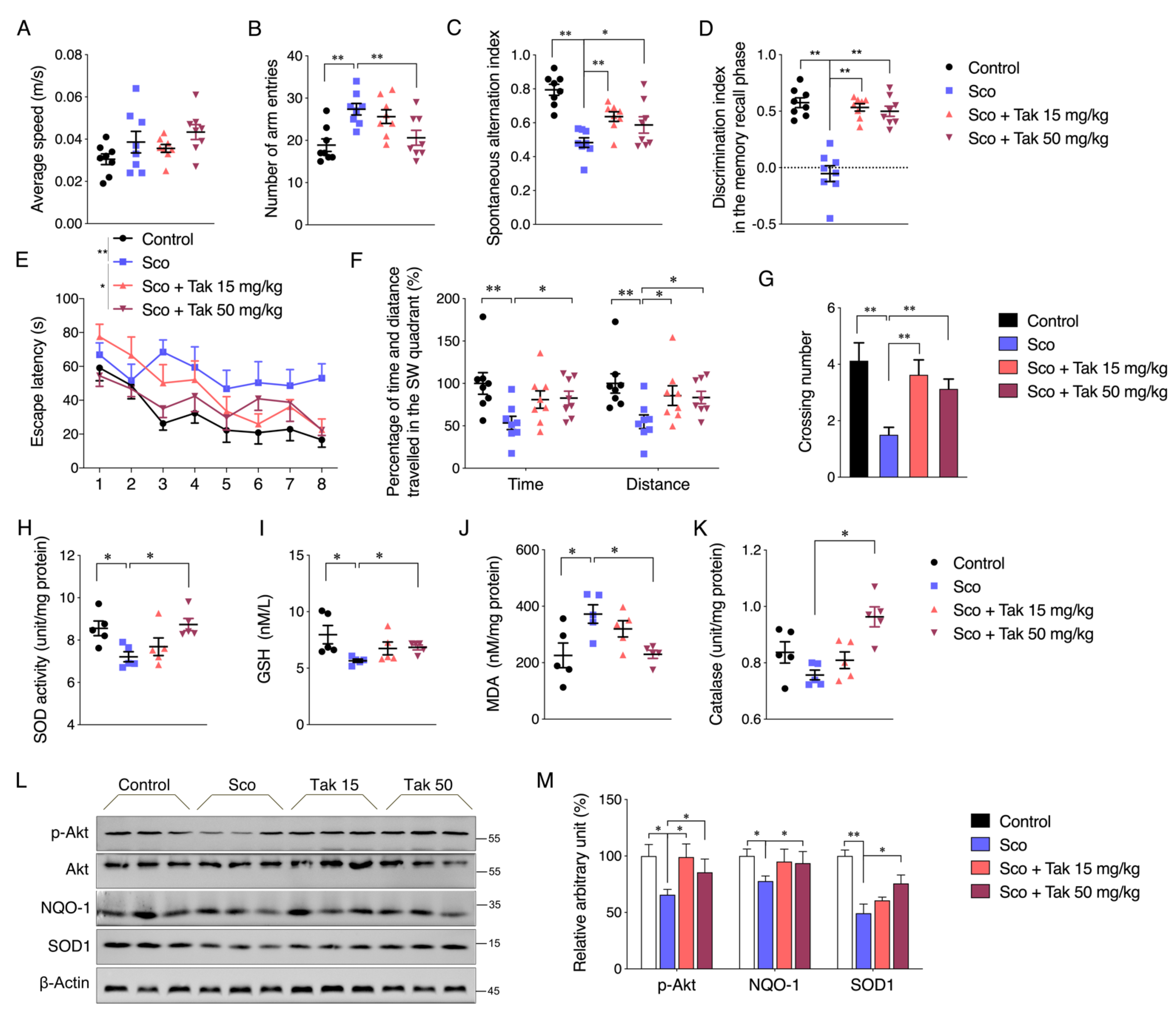
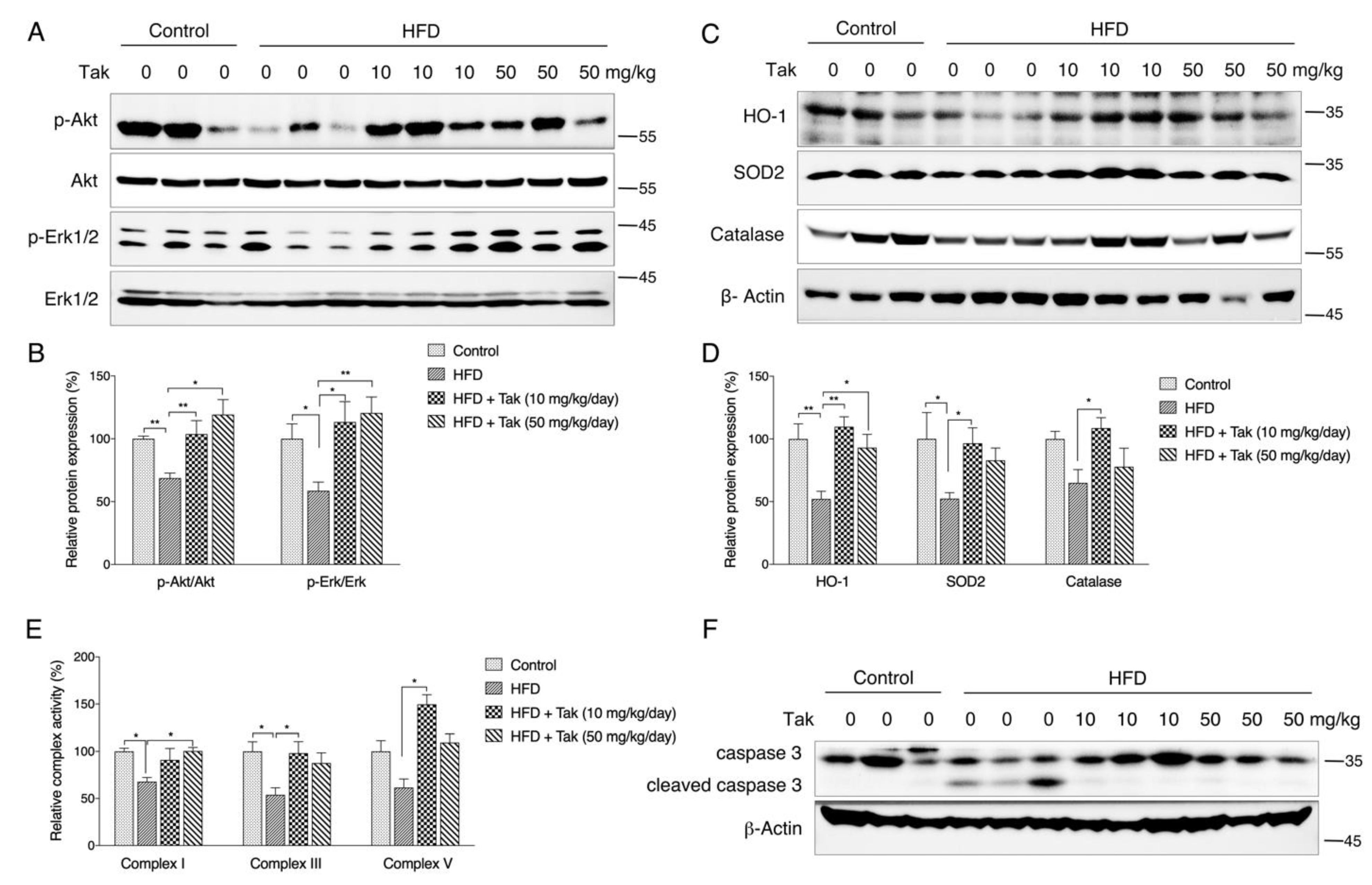
45]. Therefore, an in vitro glutamate-challenged HT22 cell model and in vivo scopolamine- and HFD-challenged mice models were used to thoroughly investigate the protective effects of Tak. We found that 5 μM Tak sufficiently prevented glutamate-induced mitochondrial superoxide overproduction and mitochondrial dysfunction, and it inhibited the activation of apoptosis in HT22 cells. Furthermore, we found that the PI3K/Akt pathway was the major upstream kinase that regulated the effects of Tak on phase II enzyme activation and HT22 cell protection against the glutamate challenge. In scopolamine-treated mice, significant cognitive impairment was observed; while mouse locomotor activity was not affected, three behavior tests, including Y-Maze, novel object recognition, and MWM test, all consistently supported the improvement of learning and memory in mice by Tak supplement, accompanied by significantly increased neuronal Akt signaling and redox status. On the other hand, in HFD-treated mice, Tak had no significant effects on mouse metabolic features, such as body weight gain and hyperlipidemia. Interestingly, Tak intervention significantly activated the Akt pathway, induced phase II enzymatic expression, improved mitochondrial function, and inhibited the activation of apoptosis, consistent with the in vitro observations. These data suggest that Tak could specifically work on neurons to exert protective effects via activating Akt-mediated antioxidative systems, and the underlying mechanism of this hippocampus-specific effect requires further study.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}