
Fursultiamine Prevents Drug-Induced Ototoxicity by Reducing Accumulation of Reactive Oxygen Species in Mouse Cochlea


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Organotypic Cochlear Explants
2.2. Phalloidin Staining
2.3. Immunohistochemistry and Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
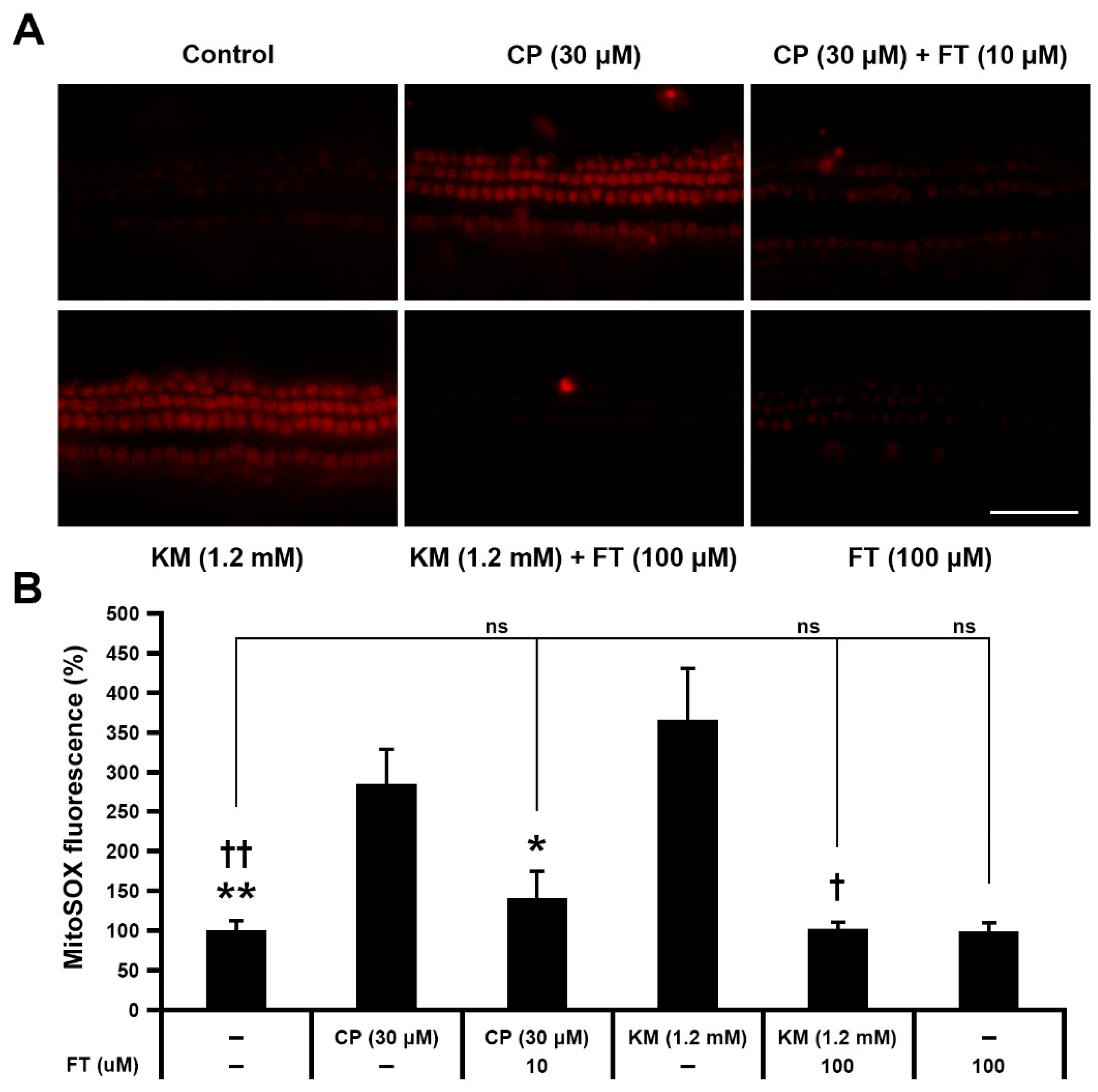
2.4. Examination of Mitochondrial ROS Levels
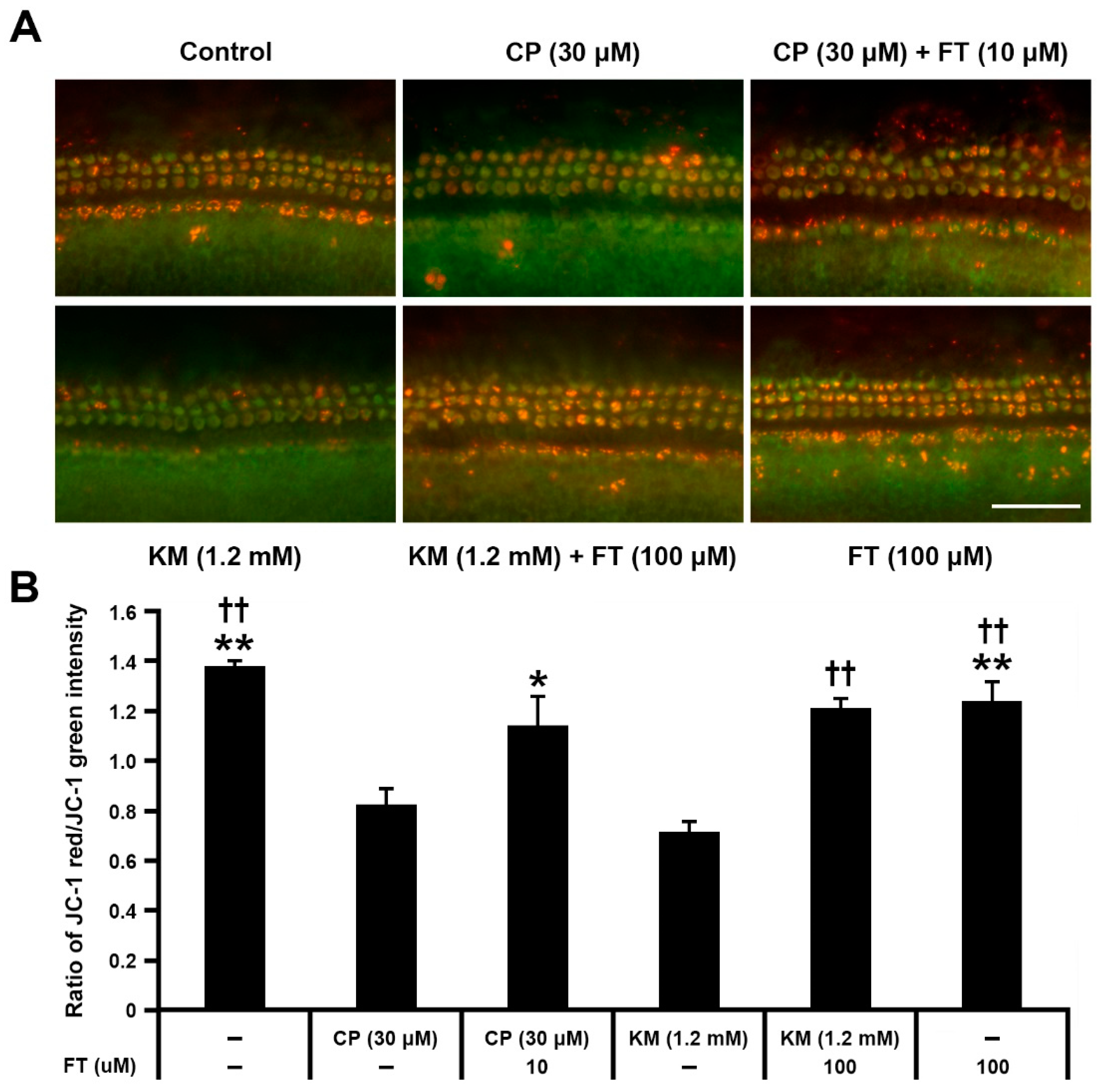
2.5. Analysis of Mitochondrial Membrane Potential (ΔYm)
2.6. Quantification of Hair Cell Survival and Statistical Analysis
3. Results
3.1. Fursultiamine Protects Hair Cells from Drug-Induced Damages
3.2. Fursultiamine Suppresses Activation of Apoptotic Hair Cell Death in Mouse Cochlear Explants
3.3. Fursultiamine Decreases Oxidative Stress and Prevents Disruption of Mitochondrial Membrane Potential in Hair Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Deafness and Hearing Loss 1 April 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 26 July 2021).
- Rybak, L.P.; Whitworth, C.A. Ototoxicity: Therapeutic opportunities. Drug Discov. Today 2005, 10, 1313–1321. [Google Scholar] [CrossRef]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.M.; Kim, H.K.; Shim, W.; Anwar, M.A.; Kwon, J.W.; Kwon, H.K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef] [Green Version]
- Marshak, T.; Steiner, M.; Kaminer, M.; Levy, L.; Shupak, A. Prevention of cisplatin-induced hearing loss by intratympanic dexamethasone: A randomized controlled study. Otolaryngol. Head Neck Surg. 2014, 150, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Rybak, L.P. Mechanisms of cisplatin ototoxicity and progress in otoprotection. Curr. Opin. Otolaryngol. Head Neck Surg. 2007, 15, 364–369. [Google Scholar] [CrossRef]
- Langer, T.; Zehnhoff-Dinnesen, A.; Radtke, S.; Meitert, J.; Zolk, O. Understanding platinum-induced ototoxicity. Trends Pharmacol. Sci. 2013, 34, 458–469. [Google Scholar] [CrossRef]
- Schacht, J.; Talaska, A.E.; Rybak, L.P. Cisplatin and aminoglycoside antibiotics: Hearing loss and its prevention. Anat. Rec. (Hoboken) 2012, 295, 1837–1850. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, M.E.; Perez, A.; Lin, R.; Sajjadi, A.; Ricci, A.J.; Cheng, A.G. Towards the prevention of aminoglycoside-related hearing loss. Front. Cell Neurosci. 2017, 11, 325. [Google Scholar] [CrossRef] [Green Version]
- Seligmann, H.; Podoshin, L.; Ben-David, J.; Fradis, M.; Goldsher, M. Drug-induced tinnitus and other hearing disorders. Drug Saf. 1996, 14, 198–212. [Google Scholar] [CrossRef]
- Lanvers-Kaminsky, C.; Ciarimboli, G. Pharmacogenetics of drug-induced ototoxicity caused by aminoglycosides and cisplatin. Pharmacogenomics 2017, 18, 1683–1695. [Google Scholar] [CrossRef]
- Kros, C.J.; Steyger, P.S. Aminoglycoside- and cisplatin-induced ototoxicity: Mechanisms and otoprotective strategies. Cold Spring Harb. Perspect. Med. 2019, 9, a033548. [Google Scholar] [CrossRef]
- Lonsdale, D. A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. Evid. Based Complement. Alternat. Med. 2006, 3, 49–59. [Google Scholar] [CrossRef]
- Said, H.M.; Ortiz, A.; Subramanian, V.S.; Neufeld, E.J.; Moyer, M.P.; Dudeja, P.K. Mechanism of thiamine uptake by human colonocytes: Studies with cultured colonic epithelial cell line NCM460. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G144–G150. [Google Scholar] [CrossRef]
- Nabokina, S.M.; Said, H.M. A high-affinity and specific carrier-mediated mechanism for uptake of thiamine pyrophosphate by human colonic epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G389–G395. [Google Scholar] [CrossRef] [Green Version]
- Winkler, B.S.; DeSantis, N.; Solomon, F. Multiple NADPH-producing pathways control glutathione (GSH) content in retina. Exp. Eye Res. 1986, 43, 829–847. [Google Scholar] [CrossRef]
- Martin, P.R.; Singleton, C.K.; Hiller-Sturmhofel, S. The role of thiamine deficiency in alcoholic brain disease. Alcohol Res. Health 2003, 27, 134–142. [Google Scholar]
- Lindqvist, Y.; Schneider, G.; Ermler, U.; Sundstrom, M. Three-dimensional structure of transketolase, a thiamine diphosphate dependent enzyme, at 2.5. A resolution. EMBO J. 1992, 11, 2373–2379. [Google Scholar] [CrossRef] [PubMed]
- Portari, G.V.; Marchini, J.S.; Vannucchi, H.; Jordao, A.A. Antioxidant effect of thiamine on acutely alcoholized rats and lack of efficacy using thiamine or glucose to reduce blood alcohol content. Basic Clin. Pharmacol. Toxicol. 2008, 103, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Turan, M.I.; Siltelioglu Turan, I.; Mammadov, R.; Altinkaynak, K.; Kisaoglu, A. The effect of thiamine and thiamine pyrophosphate on oxidative liver damage induced in rats with cisplatin. Biomed. Res. Int. 2013, 2013, 783809. [Google Scholar] [CrossRef] [PubMed]
- Kamogashira, T.; Fujimoto, C.; Yamasoba, T. Reactive oxygen species, apoptosis, and mitochondrial dysfunction in hearing loss. Biomed. Res. Int. 2015, 2015, 617207. [Google Scholar] [CrossRef] [Green Version]
- Esterberg, R.; Linbo, T.; Pickett, S.B.; Wu, P.; Ou, H.C.; Rubel, E.W.; Raible, D.W. Mitochondrial calcium uptake underlies ROS generation during aminoglycoside-induced hair cell death. J. Clin. Investig. 2016, 126, 3556–3566. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Nakai, Y.; Konishi, K.; Chang, K.C.; Ohashi, K.; Morisaki, N.; Minowa, Y.; Morimoto, A. Ototoxicity of the anticancer drug cisplatin. An experimental study. Acta Otolaryngol. 1982, 93, 227–232. [Google Scholar] [CrossRef]
- Hill, G.W.; Morest, D.K.; Parham, K. Cisplatin-induced ototoxicity: Effect of intratympanic dexamethasone injections. Otol. Neurotol. 2008, 29, 1005–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Pecka, J.L.; Zhang, Q.; Soukup, G.A.; Beisel, K.W.; He, D.Z. Characterization of transcriptomes of cochlear inner and outer hair cells. J. Neurosci. 2014, 34, 11085–11095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, H.; Barta, C.L.; Judge, P.D.; Zhao, L.; Zhang, W.J.; Gong, S.; Beisel, K.W.; He, D.Z. Transcription factors expressed in mouse cochlear inner and outer hair cells. PLoS ONE 2016, 11, e0151291. [Google Scholar] [CrossRef] [PubMed]
- Petros, A.M.; Medek, A.; Nettesheim, D.G.; Kim, D.H.; Yoon, H.S.; Swift, K.; Matayoshi, E.D.; Oltersdorf, T.; Fesik, S.W. Solution structure of the antiapoptotic protein bcl-2. Proc. Natl. Acad. Sci. USA 2001, 98, 3012–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrill, S.; He, D.Z.Z. Apoptosis in inner ear sensory hair cells. J. Otol. 2017, 12, 151–164. [Google Scholar] [CrossRef]
- Rybak, L.P.; Husain, K.; Morris, C.; Whitworth, C.; Somani, S. Effect of protective agents against cisplatin ototoxicity. Am. J. Otol. 2000, 21, 513–520. [Google Scholar]
- Brenner, C.; Grimm, S. The permeability transition pore complex in cancer cell death. Oncogene 2006, 25, 4744–4756. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, A.; Wu, J.; He, Y.; Yu, H.; Chai, R.; Li, H. MiR-182-5p protects inner ear hair cells from cisplatin-induced apoptosis by inhibiting FOXO3a. Cell Death Dis. 2016, 7, e2362. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.A.; Ikeda, K.; Oshima, T.; Suzuki, M.; Kawase, T.; Kikuchi, T.; Takasaka, T. Cisplatin-induced apoptotic cell death in Mongolian gerbil cochlea. Hear. Res. 2000, 141, 28–38. [Google Scholar] [CrossRef]
- Liu, Y.H.; Ke, X.M.; Qin, Y.; Gu, Z.P.; Xiao, S.F. Adeno-associated virus-mediated Bcl-xL prevents aminoglycoside-induced hearing loss in mice. Chin. Med. J. 2007, 120, 1236–1240. [Google Scholar] [CrossRef]
- Lee, J.S.; Kang, S.U.; Hwang, H.S.; Pyun, J.H.; Choung, Y.H.; Kim, C.H. Epicatechin protects the auditory organ by attenuating cisplatin-induced ototoxicity through inhibition of ERK. Toxicol. Lett. 2010, 199, 308–316. [Google Scholar] [CrossRef]
- Orzaez, M.; Sancho, M.; Marchan, S.; Mondragon, L.; Montava, R.; Valero, J.G.; Landeta, O.; Basanez, G.; Carbajo, R.J.; Pineda-Lucena, A.; et al. Apaf-1 inhibitors protect from unwanted cell death in in vivo models of kidney ischemia and chemotherapy induced ototoxicity. PLoS ONE 2014, 9, e110979. [Google Scholar] [CrossRef] [PubMed]
- Van Blerkom, J. Mitochondrial function in the human oocyte and embryo and their role in developmental competence. Mitochondrion 2011, 11, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Sun, G.; Yin, H.; Li, H.; Cao, Z.; Wang, J.; Zhou, M.; Wang, H.; Li, J. PINK1 Protects auditory hair cells and spiral ganglion neurons from cisplatin-induced ototoxicity via inducing autophagy and inhibiting JNK signaling pathway. Free Radic. Biol. Med. 2018, 120, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; Lee, M.Y.; Chung, P.S.; Kim, S.; Choi, B.; Suh, M.W.; Rhee, C.K.; Jung, J.Y. Enhanced mitochondrial membrane potential and ATP synthesis by photobiomodulation increases viability of the auditory cell line after gentamicin-induced intrinsic apoptosis. Sci. Rep. 2019, 9, 19248. [Google Scholar] [CrossRef]
- McLure, K.G.; Takagi, M.; Kastan, M.B. NAD+ modulates p53 DNA binding specificity and function. Mol. Cell Biol. 2004, 24, 9958–9967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheline, C.T.; Wei, L. Free radical-mediated neurotoxicity may be caused by inhibition of mitochondrial dehydrogenases in vitro and in vivo. Neuroscience 2006, 140, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Babaei-Jadidi, R.; Karachalias, N.; Ahmed, N.; Battah, S.; Thornalley, P.J. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes 2003, 52, 2110–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor, M.; Adams, R.D.; Collins, G.H. The Wernicke-Korsakoff syndrome. A clinical and pathological study of 245 patients, 82 with post-mortem examinations. Contemp. Neurol. Ser. 1971, 7, 1–206. [Google Scholar] [PubMed]
- Park, W.S.; Lee, J.; Hong, T.; Park, G.; Youn, S.; Seo, Y.; Lee, S.; Han, S. Comparative pharmacokinetic analysis of thiamine and its phosphorylated metabolites administered as multivitamin preparations. Clin. Ther. 2016, 38, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Harisa, G.I. Benfotiamine enhances antioxidant defenses and protects against cisplatin-induced DNA damage in nephrotoxic rats. J. Biochem. Mol. Toxicol. 2013, 27, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Cheng, Z.; Li, S.; Liu, X.; Guo, X.; Yu, P.; Gu, Z. Pharmacokinetic study of benfotiamine and the bioavailability assessment compared to thiamine hydrochloride. J. Clin. Pharmacol. 2014, 54, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Volvert, M.L.; Seyen, S.; Piette, M.; Evrard, B.; Gangolf, M.; Plumier, J.C.; Bettendorff, L. Benfotiamine, a synthetic S-acyl thiamine derivative, has different mechanisms of action and a different pharmacological profile than lipid-soluble thiamine disulfide derivatives. BMC Pharmacol. 2008, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Ziems, M.; Netzel, M.; Bitsch, I. Biokinetic parameters and metabolism of S-benzoylthiamine-O-monophosphate. Biofactors 2000, 11, 109–110. [Google Scholar] [CrossRef]
- Lonsdale, D. Thiamine tetrahydrofurfuryl disulfide: A little known therapeutic agent. Med. Sci. Monit. 2004, 10, RA199–RA203. [Google Scholar]
- Shimazono, N.K.E. Beriberi and Thiammine; Igaku Shoin Ltd.: Tokyo, Japan, 1965; pp. 1–295. [Google Scholar]
- Kitzushima, Y.J.V. Anti-inflammatory effect of thiamine derivatives. Vitamins 1967, 36, 305–310. [Google Scholar]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Oh, G.S.; Kim, H.J.; Choi, J.H.; Shen, A.; Choe, S.K.; Karna, A.; Lee, S.H.; Jo, H.J.; Yang, S.H.; Kwak, T.H.; et al. Pharmacological activation of NQO1 increases NAD(+) levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 2014, 85, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Kim, Y.R.; Lee, I.K.; Kim, U.K.; Baek, J.I.; Lee, K.Y. KL1333, a derivative of beta-lapachone, protects against cisplatin-induced ototoxicity in mouse cochlear cultures. Biomed. Pharmacother. 2020, 126, 110068. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, A.; Clendening, J.W.; Goard, C.A.; Penn, L.Z. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: Potential therapeutic relevance. BMC Cancer 2010, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altwairgi, A.K. Statins are potential anticancerous agents (review). Oncol. Rep. 2015, 33, 1019–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.-R.; Kwon, T.-J.; Kim, U.-K.; Lee, I.-K.; Lee, K.-Y.; Baek, J.-I. Fursultiamine Prevents Drug-Induced Ototoxicity by Reducing Accumulation of Reactive Oxygen Species in Mouse Cochlea. Antioxidants 2021, 10, 1526. https://doi.org/10.3390/antiox10101526
Kim Y-R, Kwon T-J, Kim U-K, Lee I-K, Lee K-Y, Baek J-I. Fursultiamine Prevents Drug-Induced Ototoxicity by Reducing Accumulation of Reactive Oxygen Species in Mouse Cochlea. Antioxidants. 2021; 10(10):1526. https://doi.org/10.3390/antiox10101526
Chicago/Turabian StyleKim, Ye-Ri, Tae-Jun Kwon, Un-Kyung Kim, In-Kyu Lee, Kyu-Yup Lee, and Jeong-In Baek. 2021. "Fursultiamine Prevents Drug-Induced Ototoxicity by Reducing Accumulation of Reactive Oxygen Species in Mouse Cochlea" Antioxidants 10, no. 10: 1526. https://doi.org/10.3390/antiox10101526
APA StyleKim, Y.-R., Kwon, T.-J., Kim, U.-K., Lee, I.-K., Lee, K.-Y., & Baek, J.-I. (2021). Fursultiamine Prevents Drug-Induced Ototoxicity by Reducing Accumulation of Reactive Oxygen Species in Mouse Cochlea. Antioxidants, 10(10), 1526. https://doi.org/10.3390/antiox10101526