
Impact of Silibinin A on Bioenergetics in PC12APPsw Cells and Mitochondrial Membrane Properties in Murine Brain Mitochondria


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Cell Lines
2.4. Measurement of Mitochondrial Membrane Potential (MMP)
2.5. Measurement of ATP Concentrations
2.6. High-Resolution Respirometry
2.7. Citrate Synthase Activity
2.8. Protein Content
2.9. Aβ1-40 Concentrations
2.10. ROS Concentrations
2.11. Lactate/Pyruvate Assay
2.12. Isolation of Mouse Brain Mitochondria for Mitochondrial Membrane Interactions
2.13. Mitochondrial Swelling (MS)
2.14. Mitochondrial Membrane Fluidity (MMF)
2.15. Statistics
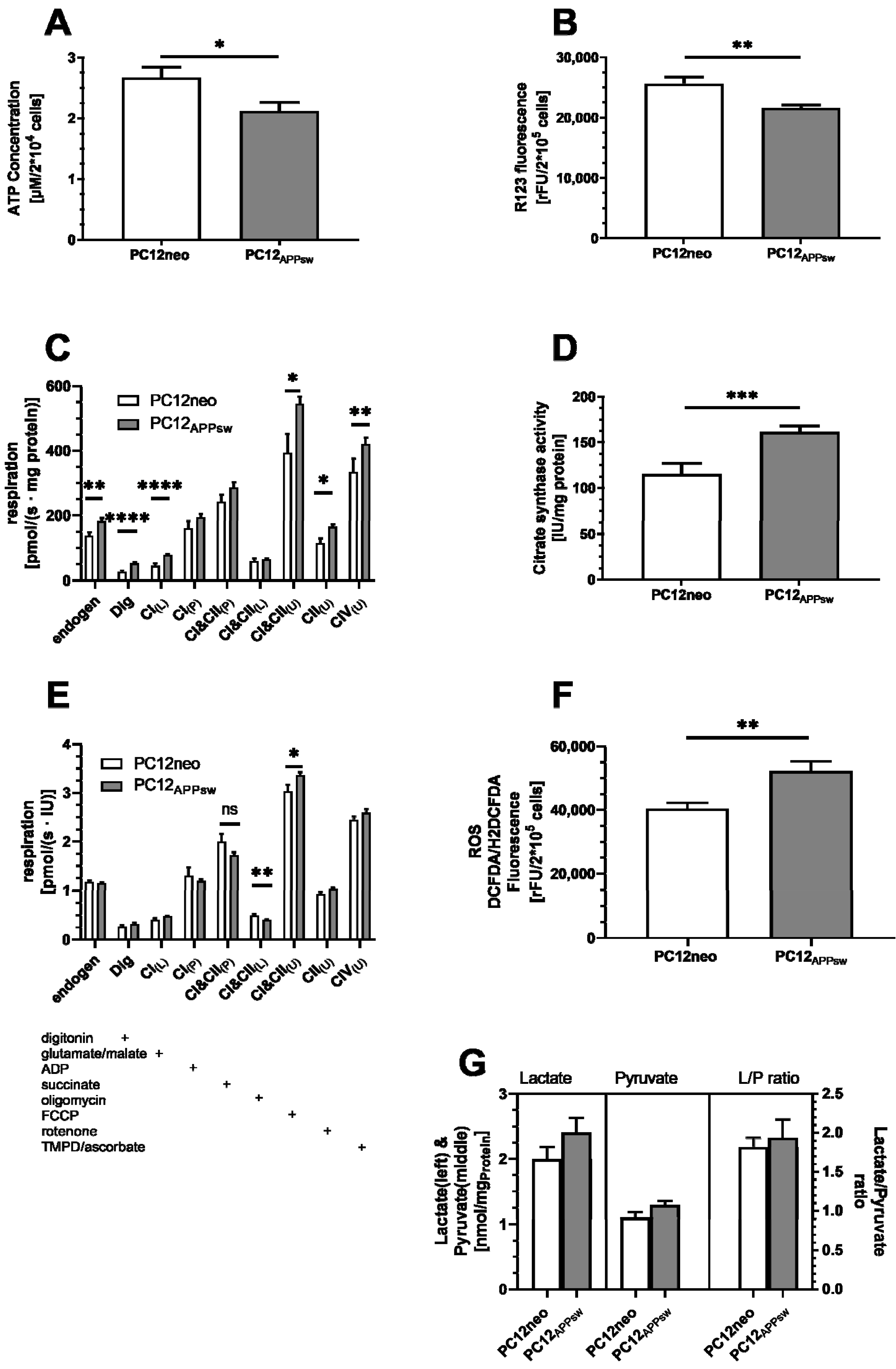
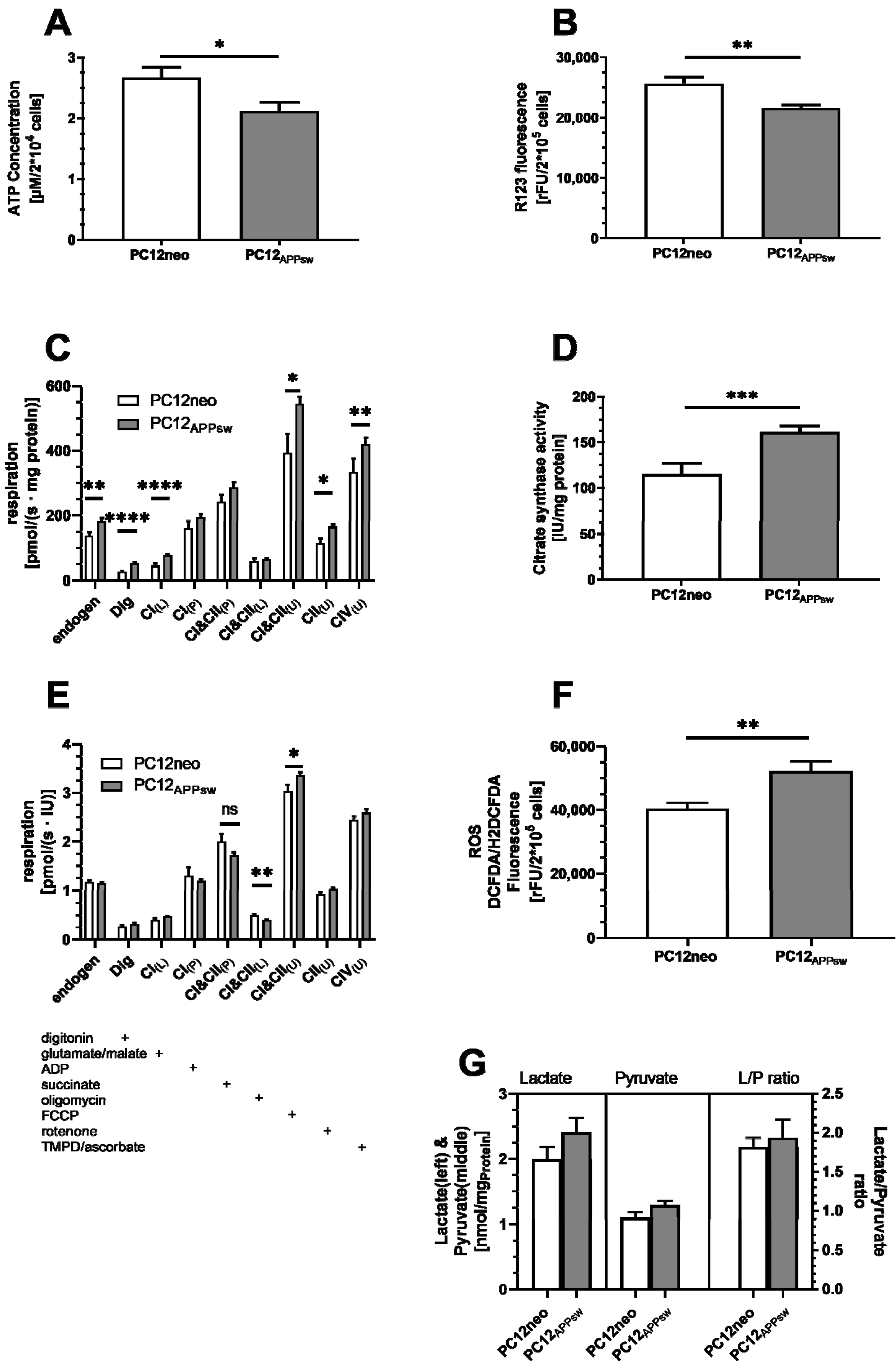
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vecchione, G.; Grasselli, E.; Cioffi, F.; Baldini, F.; Oliveira, P.J.; Sardão, V.A.; Cortese, K.; Lanni, A.; Voci, A.; Portincasa, P.; et al. The Nutraceutic Silybin Counteracts Excess Lipid Accumulation and Ongoing Oxidative Stress in an In Vitro Model of Non-Alcoholic Fatty Liver Disease Progression. Front. Nutr. 2017, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Wah Kheong, C.; Nik Mustapha, N.R.; Mahadeva, S. A Randomized Trial of Silymarin for the Treatment of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2017, 15, 1940–1949.e8. [Google Scholar] [CrossRef] [Green Version]
- Federico, A.; Dallio, M.; Loguercio, C. Silymarin/Silybin and Chronic Liver Disease: A Marriage of Many Years. Molecules 2017, 22, 191. [Google Scholar] [CrossRef] [Green Version]
- Fraschini, F.; DeMartini, G.; Esposti, D. Pharmacology of Silymarin. Clin. Drug Investig. 2002, 22, 51–65. [Google Scholar] [CrossRef]
- Esselun, C.; Bruns, B.; Hagl, S.; Grewal, R.; Eckert, G.P. Differential Effects of Silibinin A on Mitochondrial Function in Neuronal PC12 and HepG2 Liver Cells. Oxidative Med. Cell. Longev. 2019, 2019, 1652609. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Baulies, A.; Mari, M.; García-Rovés, P.M.; Fernandez-Checa, J.C. Mitochondrial dysfunction in non-alcoholic fatty liver disease and insulin resistance: Cause or consequence? Free Radic. Res. 2013, 47, 854–868. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.; Gerasimenko, O.V. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef] [Green Version]
- Elustondo, P.A.; Nichols, M.; Negoda, A.; Thirumaran, A.; Zakharian, E.; Robertson, G.S.; Pavlov, E.V. Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov. 2016, 2, 16070. [Google Scholar] [CrossRef]
- Moreno-Treviño, M.G.; Castillo-López, J.; Meester, I. Moving away from amyloid Beta to move on in Alzheimer research. Front. Aging Neurosci. 2015, 7, 2. [Google Scholar]
- Morris, G.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef] [Green Version]
- Demarest, T.G.; Varma, V.R.; Estrada, D.; Babbar, M.; Basu, S.; Mahajan, U.V.; Moaddel, R.; Croteau, D.L.; Thambisetty, M.; Mattson, M.P.; et al. Biological sex and DNA repair deficiency drive Alzheimer’s disease via systemic metabolic remodeling and brain mitochondrial dysfunction. Acta Neuropathol. 2020, 140, 25–47. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Alzheimer’s disease pathologic cascades: Who comes first, what drives what. Neurotox. Res. 2012, 22, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Et Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [Green Version]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Payne, B.A.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1347–1353. [Google Scholar] [CrossRef] [Green Version]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef]
- Larsen, S.B.; Hanss, Z.; Krüger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Hervias, I.; Beal, M.F.; Manfredi, G. Mitochondrial dysfunction and amyotrophic lateral sclerosis. Muscle Nerve 2006, 33, 598–608. [Google Scholar] [CrossRef]
- Lee, Y.; Park, H.R.; Chun, H.J.; Lee, J. Silibinin prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease via mitochondrial stabilization. J. Neurosci. Res. 2015, 93, 755–765. [Google Scholar] [CrossRef]
- Liu, B.; Yang, P.; Ye, Y.; Zhou, Y.; Li, L.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Role of ROS in the protective effect of silibinin on sodium nitroprusside-induced apoptosis in rat pheochromocytoma PC12 cells. Free. Radic. Res. 2011, 45, 835–847. [Google Scholar] [CrossRef]
- Jiang, H.-H.; Yan, F.-S.; Shen, L.; Ji, H.-F. Silymarin versus Silibinin: Differential Antioxidant and Neuroprotective Effects against H2O2-induced Oxidative Stress in PC12 Cells. Nat. Prod. Commun. 2016, 11, 633–636. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Guan, X.; Lin, R.; Liu, X.; Yan, Y.; Lin, R.; Zhang, T.; Chen, X.; Huang, J.; Sun, X.; et al. Silibinin inhibits acetylcholinesterase activity and amyloid β peptide aggregation: A dual-target drug for the treatment of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 1792–1807. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Liu, J.; Ji, X.; Wang, Y.; Zidichouski, J.; Zhang, J. Silibinin: A novel inhibitor of Aβ aggregation. Neurochem. Int. 2011, 58, 399–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagl, S.; Grewal, R.; Ciobanu, I.; Helal, A.; Khayyal, M.T.; Muller, W.E.; Eckert, G.P. Rice bran extract compensates mitochondrial dysfunction in a cellular model of early Alzheimer’s disease. J. Alzheimers Dis. JAD 2015, 43, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Steiner, B.; Marques, C.; Leutz, S.; Romig, H.; Haass, C.; Müller, W.E. Elevated vulnerability to oxidative stress-induced cell death and activation of caspase-3 by the Swedish amyloid precursor protein mutation. J. Neurosci. Res. 2001, 64, 183–192. [Google Scholar] [CrossRef]
- Liu, W.; Kong, S.; Xie, Q.; Su, J.; Li, W.; Guo, H.; Li, S.; Feng, X.; Su, Z.; Xu, Y.; et al. Protective effects of apigenin against 1-methyl-4-phenylpyridinium ion-induced neurotoxicity in PC12 cells. Int. J. Mol. Med. 2014, 35, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.-C.; Juan, C.-W.; Lin, C.-S.; Chen, C.-C.; Chang, C.-L. Neuroprotective effects of terminalia chebubal extracts and ellagic acid in PC12 cells. Afr. J. Tradit. Complementary Altern. Med. AJTCAM 2017, 14, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-Y.; Chen, X.-X.; Chen, H.-Y.; Shi, J.; Leung, G.P.-H.; Tang, S.C.-W.; Lao, L.-X.; Yip, H.K.-F.; Lee, K.-F.; Sze, S.C.-W.; et al. Downregulation of Aquaporin 9 Exacerbates Beta-amyloid-induced Neurotoxicity in Alzheimer’s Disease Models In vitro and In vivo. Neuroscience 2018, 394, 72–82. [Google Scholar] [CrossRef]
- Eckert, A.; Marques, C.A.; Keil, U.; Schüssel, K.; Müller, W.E. Increased apoptotic cell death in sporadic and genetic Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2003, 1010, 604–609. [Google Scholar] [CrossRef]
- Czubowicz, K.; Wójtowicz, S.; Wencel, P.L.; Strosznajder, R.P. The role of ceramide and SEW 2871 in the transcription of enzymes involved in amyloid b precursor protein metabolism in an experimental model of Alzheimer’s disease. Folia Neuropathol. 2018, 56, 196–205. [Google Scholar] [CrossRef]
- Chalimoniuk, M.; Stolecka, A.; Cakała, M.; Hauptmann, S.; Schulz, K.; Lipka, U.; Leuner, K.; Eckert, A.; Muller, W.E.; Strosznajder, J. Amyloid beta enhances cytosolic phospholipase A2 level and arachidonic acid release via nitric oxide in APP-transfected PC12 cells. Acta Biochim. Pol. 2007, 54, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Hagl, S.; Kocher, A.; Schiborr, C.; Kolesova, N.; Frank, J.; Eckert, G.P. Curcumin micelles improve mitochondrial function in neuronal PC12 cells and brains of NMRI mice–Impact on bioavailability. Neurochem. Int. 2015, 89, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Strosznajder, J.; Cieslik, M.; Cakala, M.; Jęśko, H.; Eckert, A.; Strosznajder, R. Lipoxygenases and Poly(ADP-Ribose) Polymerase in Amyloid Beta Cytotoxicity. Neurochem. Res. 2011, 36, 839–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagl, S.; Berressem, D.; Bruns, B.; Sus, N.; Frank, J.; Eckert, G.P. Beneficial Effects of Ethanolic and Hexanic Rice Bran Extract on Mitochondrial Function in PC12 Cells and the Search for Bioactive Components. Molecules 2015, 20, 16524–16539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control: An Introduction to OXPHOS Analysis. In Mitochondr Physiol Network 19.12; OROBOROS MiPNet Publications: Innsbruck, Austria, 2014. [Google Scholar]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schrøder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Lenkiewicz, A.M.; Czapski, G.A.; Jęsko, H.; Wilkaniec, A.; Szypuła, W.; Pietrosiuk, A.; Uszyńska, A.M.; Adamczyk, A. Potent effects of alkaloid-rich extract from Huperzia selago against sodium nitroprusside-evoked PC12 cells damage via attenuation of oxidative stress and apoptosis. Folia Neuropathol. 2016, 2, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Lozinsky, O.V.; Lushchak, O.V.; Storey, J.M.; Storey, K.B.; Lushchak, V.I. The mitochondrial uncoupler 2,4-dinitrophenol attenuates sodium nitroprusside-induced toxicity in Drosophila melanogaster: Potential involvement of free radicals. Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 2013, 158, 244–252. [Google Scholar] [CrossRef]
- Camello-Almaraz, C.; Gomez-Pinilla, P.J.; Pozo, M.J.; Camello, P.J. Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. Cell Physiol. 2006, 291, C1082–C1088. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Vianello, A.; Casolo, V.; Petrussa, E.; Peresson, C.; Patui, S.; Bertolini, A.; Passamonti, S.; Braidot, E.; Zancani, M. The mitochondrial permeability transition pore (PTP)—An example of multiple molecular exaptation? Biochim. et Biophys. Acta (BBA)—Bioenerg. 2012, 1817, 2072–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brustovetsky, N.; Brustovetsky, T.; Purl, K.J.; Capano, M.; Crompton, M.; Dubinsky, J.M. Increased Susceptibility of Striatal Mitochondria to Calcium-Induced Permeability Transition. J. Neurosci. 2003, 23, 4858–4867. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.; Alonso-Navarro, H.; Herrero, M.; García-Martín, E.; Agúndez, J. An Update on the Role of Nitric Oxide in the Neurodegenerative Processes of Parkinson’s Disease. CMC 2016, 23, 2666–2679. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.; Leeuwenburgh, C. Aging and the role of reactive nitrogen species. Ann. N. Y. Acad. Sci. 2002, 959, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Pourova, J.; Kottova, M.; Voprsalova, M.; Pour, M. Reactive oxygen and nitrogen species in normal physiological processes. Acta Physiol. 2010, 198, 15–35. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Um, J.-H.; Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress1,2. Free. Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Yatin, S.M.; Varadarajan, S.; Butterfield, D.A. Vitamin E Prevents Alzheimer’s Amyloid beta-Peptide (1-42)-Induced Neuronal Protein Oxidation and Reactive Oxygen Species Production. J. Alzheimers Dis. JAD 2000, 2, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.X.; Du Yan, S. Amyloid-beta-induced mitochondrial dysfunction. J. Alzheimers Dis. JAD 2007, 12, 177–184. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Harrison, D.G. Methods for Detection of Mitochondrial and Cellular Reactive Oxygen Species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Liemburg-Apers, D.; Willems, P.H.; Koopman, W.J.H.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [Green Version]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Eckert, G.P.; Eckert, S.H.; Eckmann, J.; Hagl, S.; Muller, W.E.; Friedland, K. Olesoxime improves cerebral mitochondrial dysfunction and enhances Aβ levels in preclinical models of Alzheimer’s disease. Exp. Neurol. 2020, 329, 113286. [Google Scholar] [CrossRef]
- Matsuo, M.; Sasaki, N.; Saga, K.; Kaneko, T. Cytotoxicity of Flavonoids toward Cultured Normal Human Cells. Biol. Pharm. Bull. 2005, 28, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Procházková, D.; Boušová, I.; Wilhelmová, N. Antioxidant and prooxidant properties of flavonoids. Fitoterapia 2011, 82, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Dorta, D.J.; Pigoso, A.A.; Mingatto, F.E.; Rodrigues, T.; Pestana, C.R.; Uyemura, S.A.; Santos, A.C.; Curti, C. Antioxidant activity of flavonoids in isolated mitochondria. Phytother. Res. 2008, 22, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Detaille, D.; Sanchez, C.; Sanz, N.; Lopez-Novoa, J.M.; Leverve, X.; El-Mir, M.-Y. Interrelation between the inhibition of glycolytic flux by silibinin and the lowering of mitochondrial ROS production in perifused rat hepatocytes. Life Sci. 2008, 82, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Grewal, R.; Reutzel, M.; Dilberger, B.; Hein, H.; Zotzel, J.; Marx, S.; Tretzel, J.; Sarafeddinov, A.; Fuchs, C.; Eckert, G.P. Purified oleocanthal and ligstroside protect against mitochondrial dysfunction in models of early Alzheimer’s disease and brain ageing. Exp. Neurol. 2020, 328, 113248. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Kushnareva, Y.E.; Sokolove, P.M. Prooxidants Open Both the Mitochondrial Permeability Transition Pore and a Low-Conductance Channel in the Inner Mitochondrial Membrane. Arch. Biochem. Biophys. 2000, 376, 377–388. [Google Scholar] [CrossRef]
- Muriel, P.; Mourelle, M. Prevention by silymarin of membrane alterations in acute CCI4 liver damage. J. Appl. Toxicol. 1990, 10, 275–279. [Google Scholar] [CrossRef]
- Meiß, R.; Heinrich, U.; Robenek, H.; Themann, H. Effect of Silybin on hepatic cell membranes after damage by polycyclic aromatic hydrocarbons (PAH). Inflamm. Res. 1982, 12, 254–257. [Google Scholar] [CrossRef]
- Lettéron, P.; Labbe, G.; Degott, C.; Berson, A.; Fromenty, B.; Delaforge, M.; Larrey, D.; Pessayre, D. Mechanism for the protective effects of silymarin against carbon tetrachloride-induced lipid peroxidation and hepatotoxicity in mice: Evidence that silymarin acts both as an inhibitor of metabolic activation and as a chain-breaking antioxidant. Biochem. Pharmacol. 1990, 39, 2027–2034. [Google Scholar] [CrossRef]
- Basiglio, C.L.; Pozzi, E.J.S.; Mottino, A.D.; Roma, M.G. Differential effects of silymarin and its active component silibinin on plasma membrane stability and hepatocellular lysis. Chem. Interact. 2009, 179, 297–303. [Google Scholar] [CrossRef]
- Farghali, H.; Kameniková, L.; Hynie, S.; Kmonicková, E. Silymarin effects on intracellular calcium and cytotoxicity: A study in perfused rat hepatocytes after oxidative stress injury. Pharmacol. Res. 2000, 41, 231–237. [Google Scholar] [CrossRef]
- Yao, J.; Zhi, M.; Minhu, C. Effect of silybin on high-fat-induced fatty liver in rats. Braz. J. Med. Biol. Res. 2011, 44, 652–659. [Google Scholar] [CrossRef] [Green Version]
- Parasassi, T.; Martellucci, A.; Conti, F.; Messina, B. Drug—membrane interactions: Silymarin, silibyn and microsomal membranes. Cell Biochem. Funct. 1984, 2, 85–88. [Google Scholar] [CrossRef]
- Kidd, P.; Head, K. A review of the bioavailability and clinical efficacy of milk thistle phytosome: A silybin-phosphatidylcholine complex (Siliphos). Altern. Med. Rev. A J. Clin. Ther. 2005, 10, 193–203. [Google Scholar]
- Pais, P.; D’Amato, M. In vivo efficacy study of milk thistle extract (ETHIS-094™) in STAM™ model of nonalcoholic steatohepatitis. Drugs R&D 2014, 14, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Sánchez, A.; Cuyàs, E.; Ruiz-Torres, V.; Agulló-Chazarra, L.; Verdura, S.; González-Álvarez, I.; Bermejo, M.; Joven, J.; Micol, V.; Bosch-Barrera, J.; et al. Intestinal Permeability Study of Clinically Relevant Formulations of Silibinin in Caco-2 Cell Monolayers. Int. J. Mol. Sci. 2019, 20, 1606. [Google Scholar] [CrossRef] [Green Version]
- Bai, D.; Jin, G.; Yin, S.; Zou, D.; Zhu, Q.; Yang, Z.; Liu, X.; Ren, L.; Sun, Y.; Gan, S. Antioxidative and Anti-Apoptotic Roles of Silibinin in Reversing Learning and Memory Deficits in APP/PS1 Mice. Neurochem. Res. 2017, 42, 3439–3445. [Google Scholar] [CrossRef]
- Bai, D.; Jin, G.; Zhang, D.; Zhao, L.; Wang, M.; Zhu, Q.; Zhu, L.; Sun, Y.; Liu, X.; Chen, X.; et al. Natural silibinin modulates amyloid precursor protein processing and amyloid-β protein clearance in APP/PS1 mice. J. Physiol. Sci. 2019, 69, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Liu, W.; Liu, P.; Liu, X.; Song, X.; Hayashi, T.; Onodera, S.; Ikejima, T. Silibinin Alleviates the Learning and Memory Defects in Overtrained Rats Accompanying Reduced Neuronal Apoptosis and Senescence. Neurochem. Res. 2019, 44, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Bai, D.; Yin, S.; Yang, Z.; Zou, D.; Zhang, Z.; Li, X.; Sun, Y.; Zhu, Q. Silibinin rescues learning and memory deficits by attenuating microglia activation and preventing neuroinflammatory reactions in SAMP8 mice. Neurosci. Lett. 2016, 629, 256–261. [Google Scholar] [CrossRef]
- Kim, S.; Jung, U.J.; Oh, Y.S.; Jeon, M.T.; Kim, H.J.; Shin, W.H.; Hong, J.; Kim, S.R. Beneficial Effects of Silibinin Against Kainic Acid-induced Neurotoxicity in the Hippocampus in vivo. Exp. Neurobiol. 2017, 26, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, U.J.; Jeon, M.-T.; Choi, M.-S.; Kim, S.R. Silibinin Attenuates MPP+-Induced Neurotoxicity in the Substantia Nigra In Vivo. J. Med. Food 2014, 17, 599–605. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cells | State 3 Respiration (CI(P)) | State 4 Respiration (CI(L)) | RCR |
|---|---|---|---|
| PC12neo | 1.30 ± 0.17 | 0.407 ± 0.036 | 3.11 ± 0.60 |
| PC12APPsw | 1.19 ± 0.040 | 0.478 ± 0.014 | 2.52 ± 0.14 pn.s. = 0.69 |
| Ctrl | SIL50 | p | |
|---|---|---|---|
| Aβ1-40 [pg/mgProtein] | 82.22 ± 7.98 | 99.26 ± 10.08 | 0.20, n.s. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esselun, C.; Bruns, B.; Hagl, S.; Grewal, R.; Eckert, G.P. Impact of Silibinin A on Bioenergetics in PC12APPsw Cells and Mitochondrial Membrane Properties in Murine Brain Mitochondria. Antioxidants 2021, 10, 1520. https://doi.org/10.3390/antiox10101520
Esselun C, Bruns B, Hagl S, Grewal R, Eckert GP. Impact of Silibinin A on Bioenergetics in PC12APPsw Cells and Mitochondrial Membrane Properties in Murine Brain Mitochondria. Antioxidants. 2021; 10(10):1520. https://doi.org/10.3390/antiox10101520
Chicago/Turabian StyleEsselun, Carsten, Bastian Bruns, Stephanie Hagl, Rekha Grewal, and Gunter P. Eckert. 2021. "Impact of Silibinin A on Bioenergetics in PC12APPsw Cells and Mitochondrial Membrane Properties in Murine Brain Mitochondria" Antioxidants 10, no. 10: 1520. https://doi.org/10.3390/antiox10101520
APA StyleEsselun, C., Bruns, B., Hagl, S., Grewal, R., & Eckert, G. P. (2021). Impact of Silibinin A on Bioenergetics in PC12APPsw Cells and Mitochondrial Membrane Properties in Murine Brain Mitochondria. Antioxidants, 10(10), 1520. https://doi.org/10.3390/antiox10101520