Microdose Lithium Protects against Pancreatic Islet Destruction and Renal Impairment in Streptozotocin-Elicited Diabetes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiment Design
2.2. Murine Model of STZ-Induced Diabetes
2.3. Blood Glucose Level Measurements
2.4. Isolation and Ex Vivo Treatment of Mouse Islets
2.5. Isolation of Glomeruli
2.6. Urine Analyses
2.7. Pancreas Morphology and Immunohistochemistry Analysis
2.8. Islet Immunofluorescence Analysis
2.9. Western Immunoblot Analysis
2.10. Statistical Analysis
3. Results
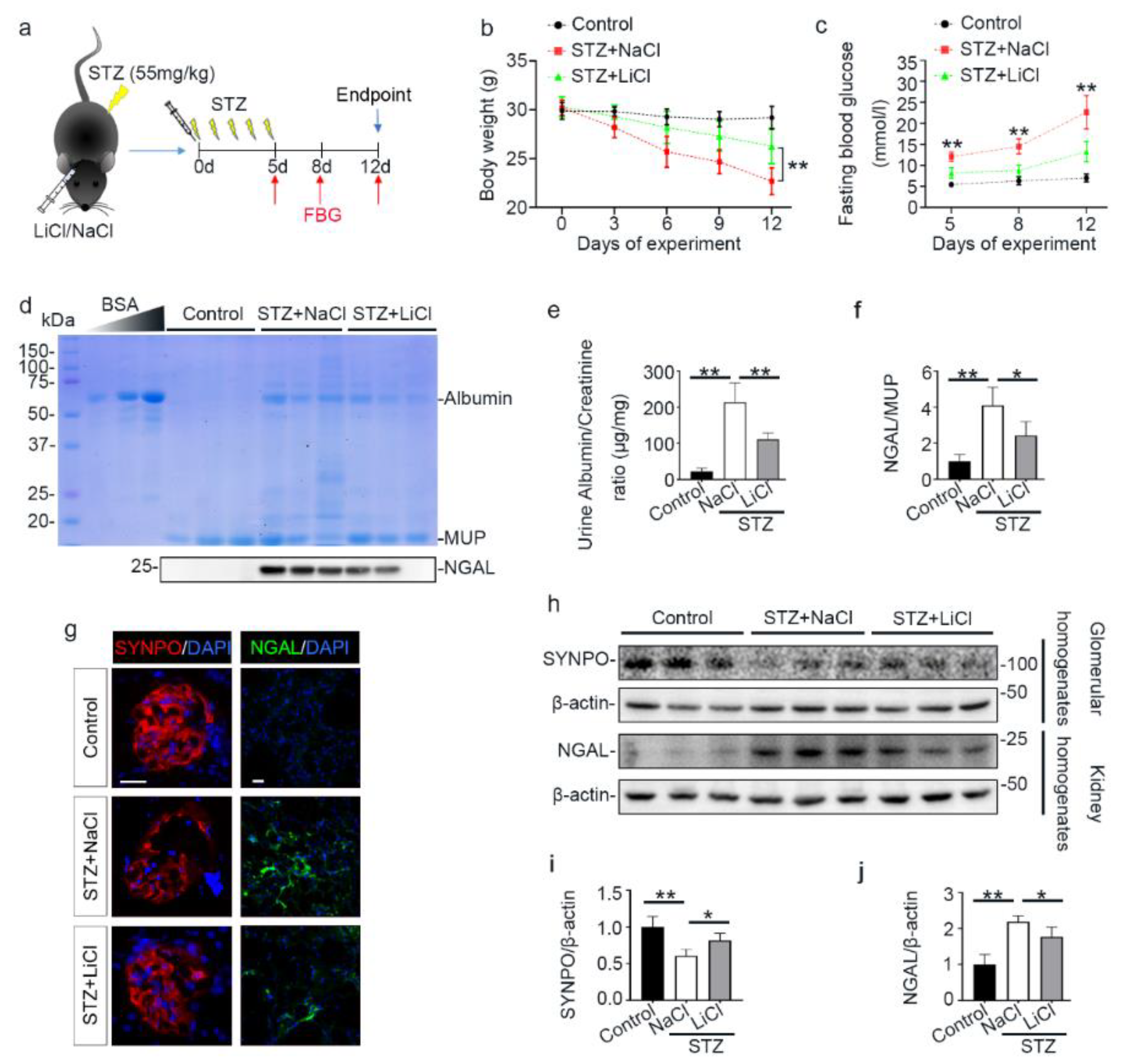
3.1. A Single Injection of Microdose Lithium Protects against STZ-Elicited Diabetes and Attenuates Early Signs of Diabetic Kidney Injury in Mice
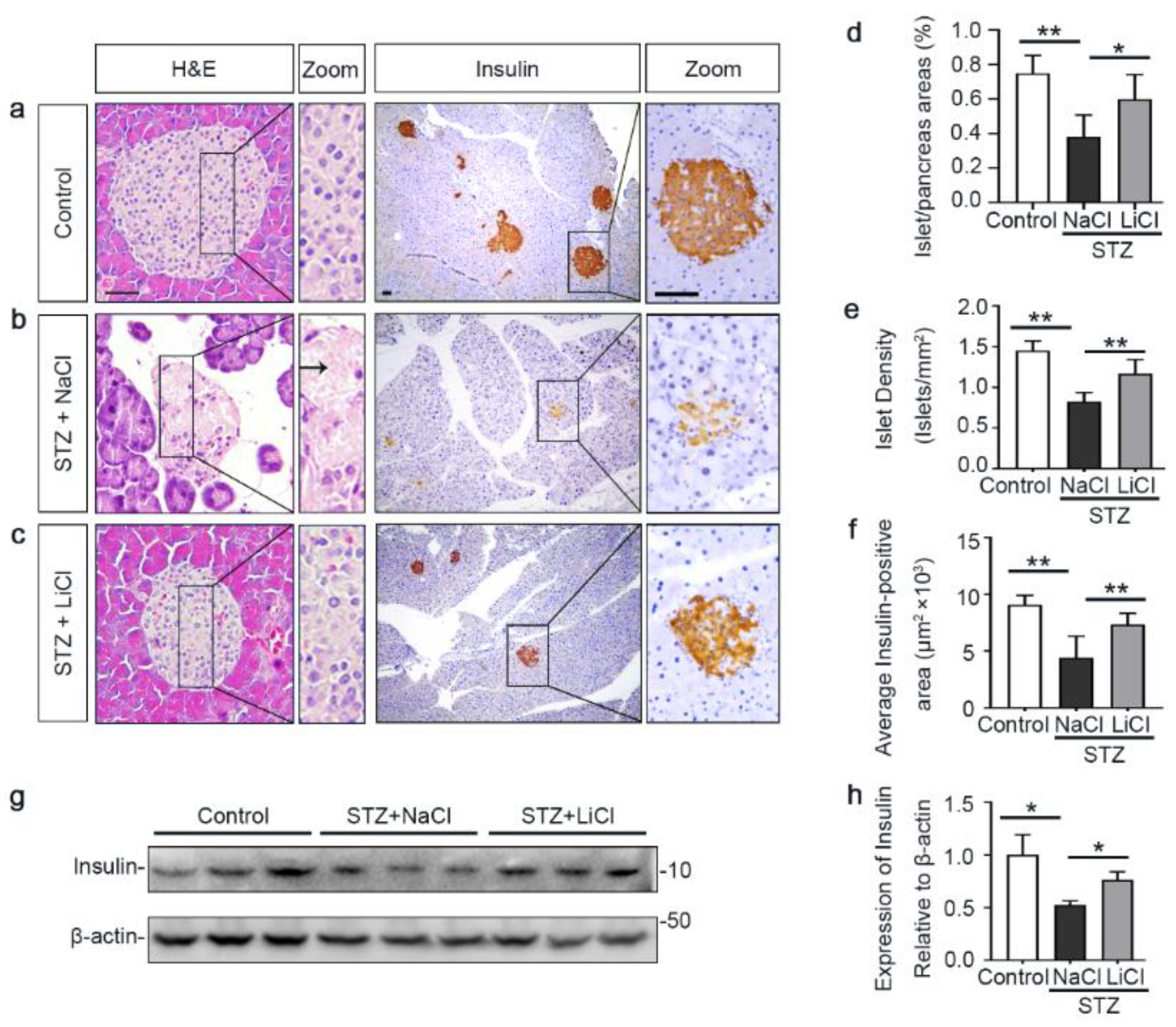
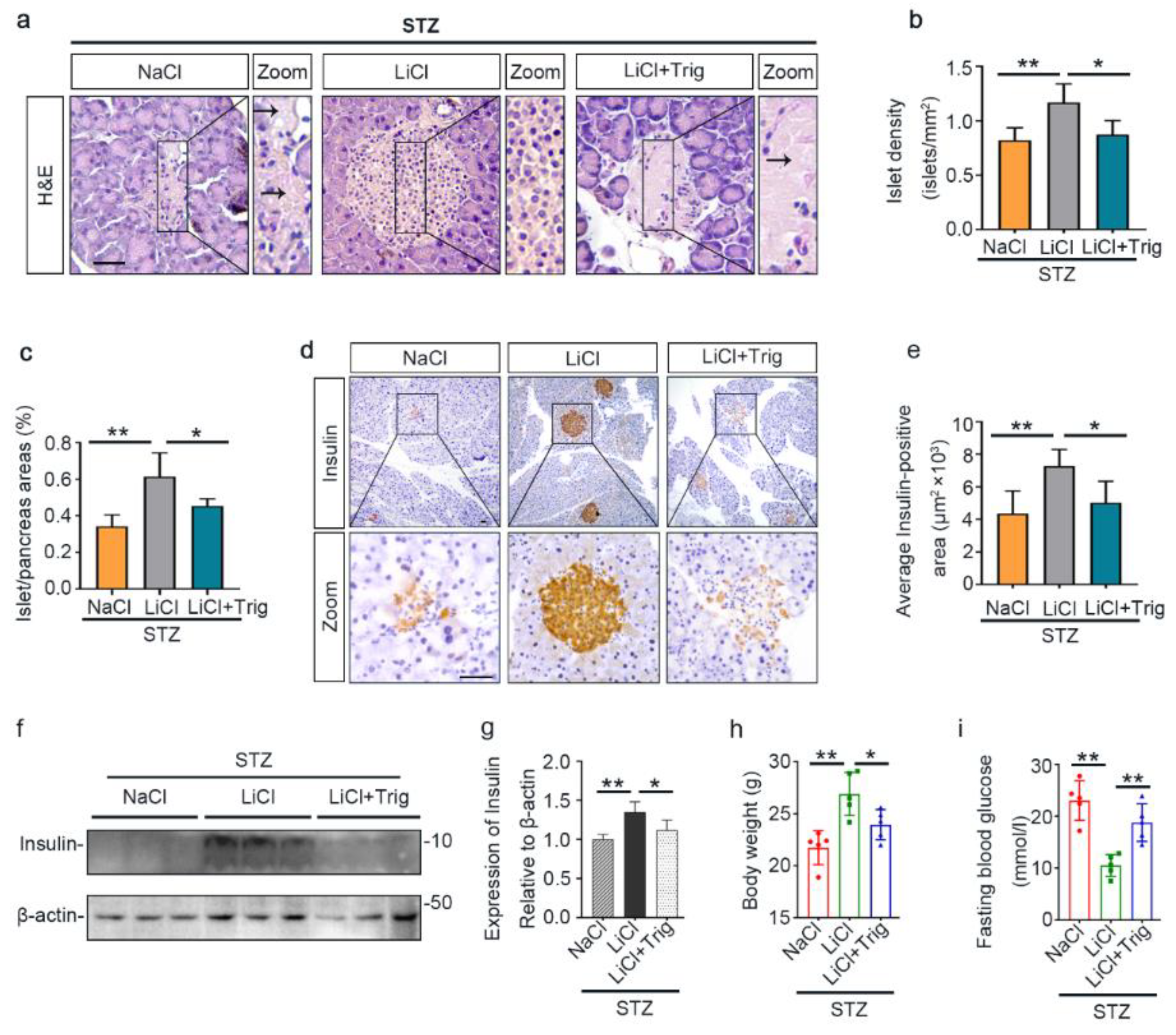
3.2. Lithium Preserves the Histology of Pancreatic Islets and Retains β-Cells Production of Insulin in STZ-Injured Mice
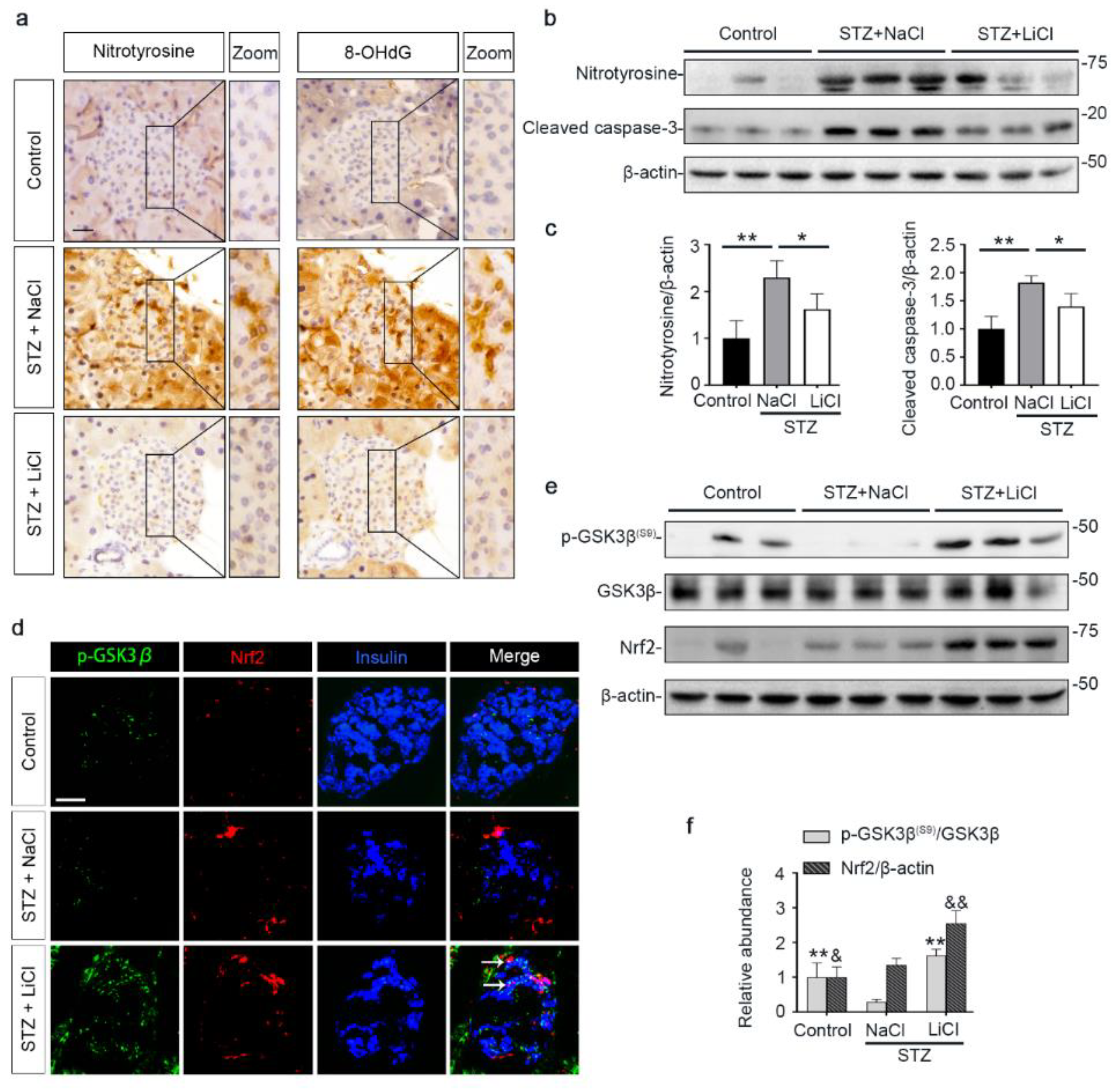
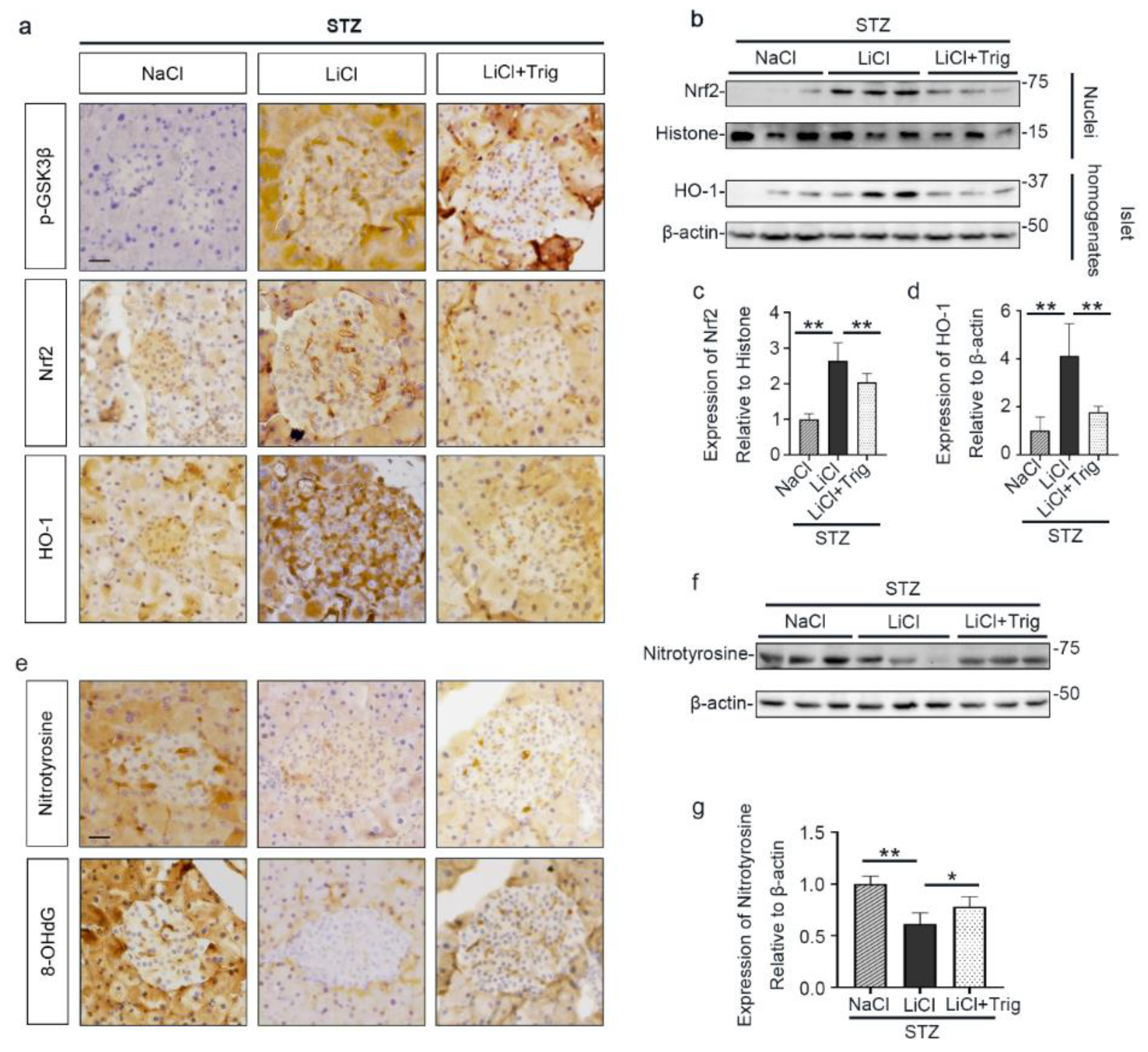
3.3. Lithium Therapy Attenuates STZ-Elicited Oxidative Stress in Pancreatic Islets, Concomitant with GSK3β Inhibition and Increased Nrf2 Expression
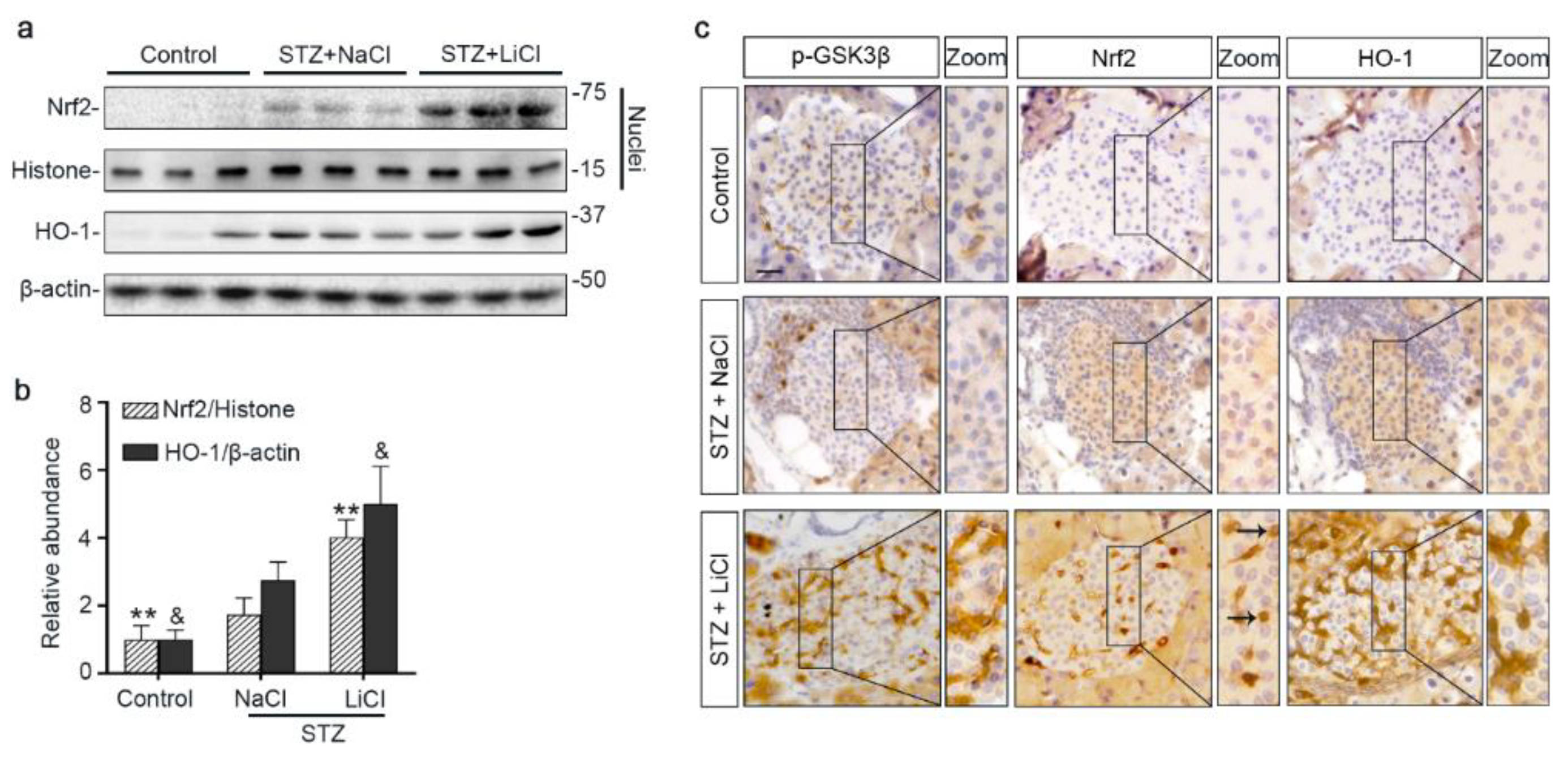
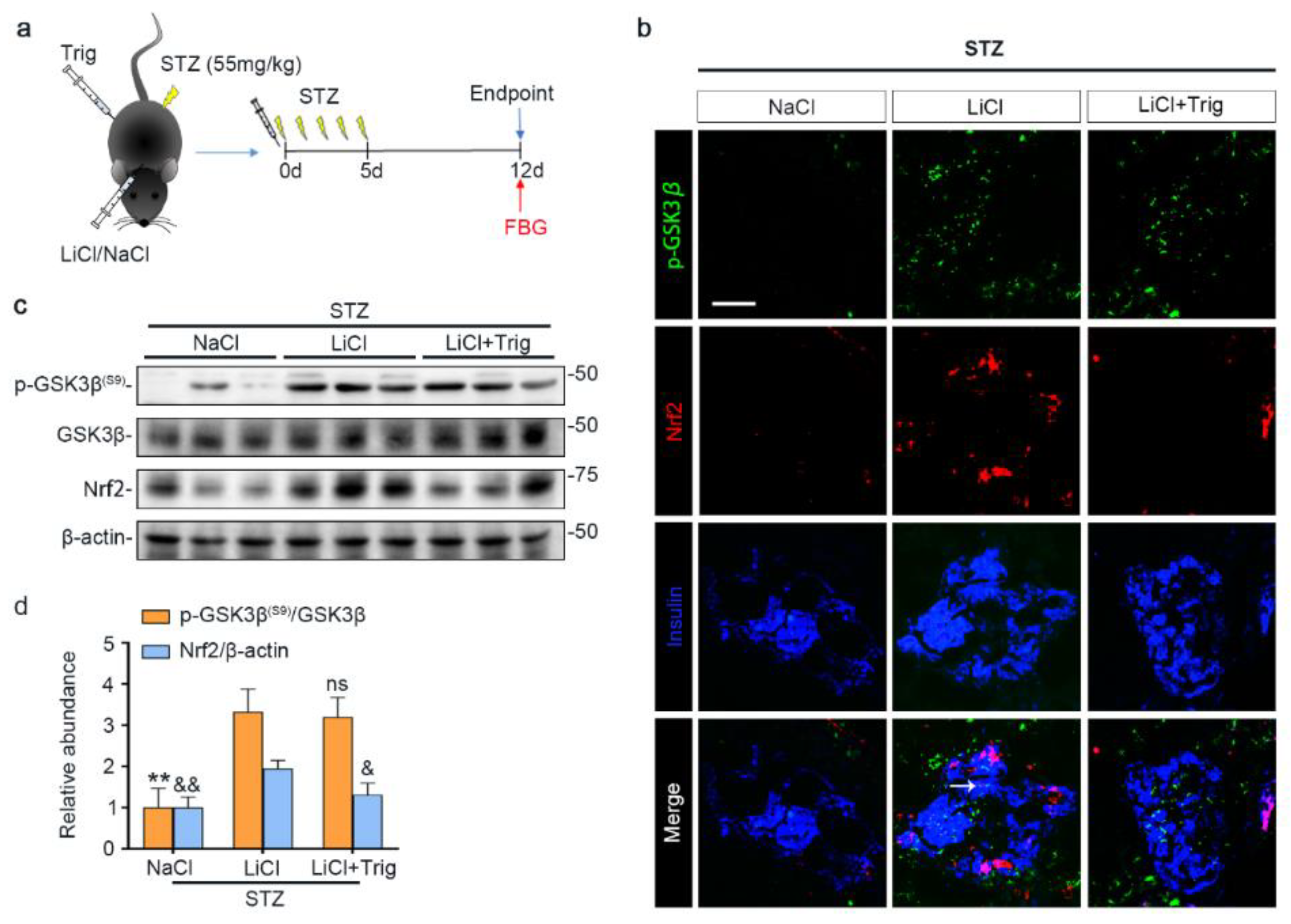
3.4. The Nrf2 Antioxidant Response to STZ Injury Is Reinforced in Pancreatic Islets by Lithium Therapy
3.5. Nrf2 Activity Is Required for the Lithium Reinforced Antioxidant Response to STZ Injury in Pancreatic Islets
3.6. The Potentiated Nrf2 Activity Is Essential for the Beneficial Effect of Lithium Therapy on Islet Injury, β-Cell Destruction and T1D in STZ-Injured Mice
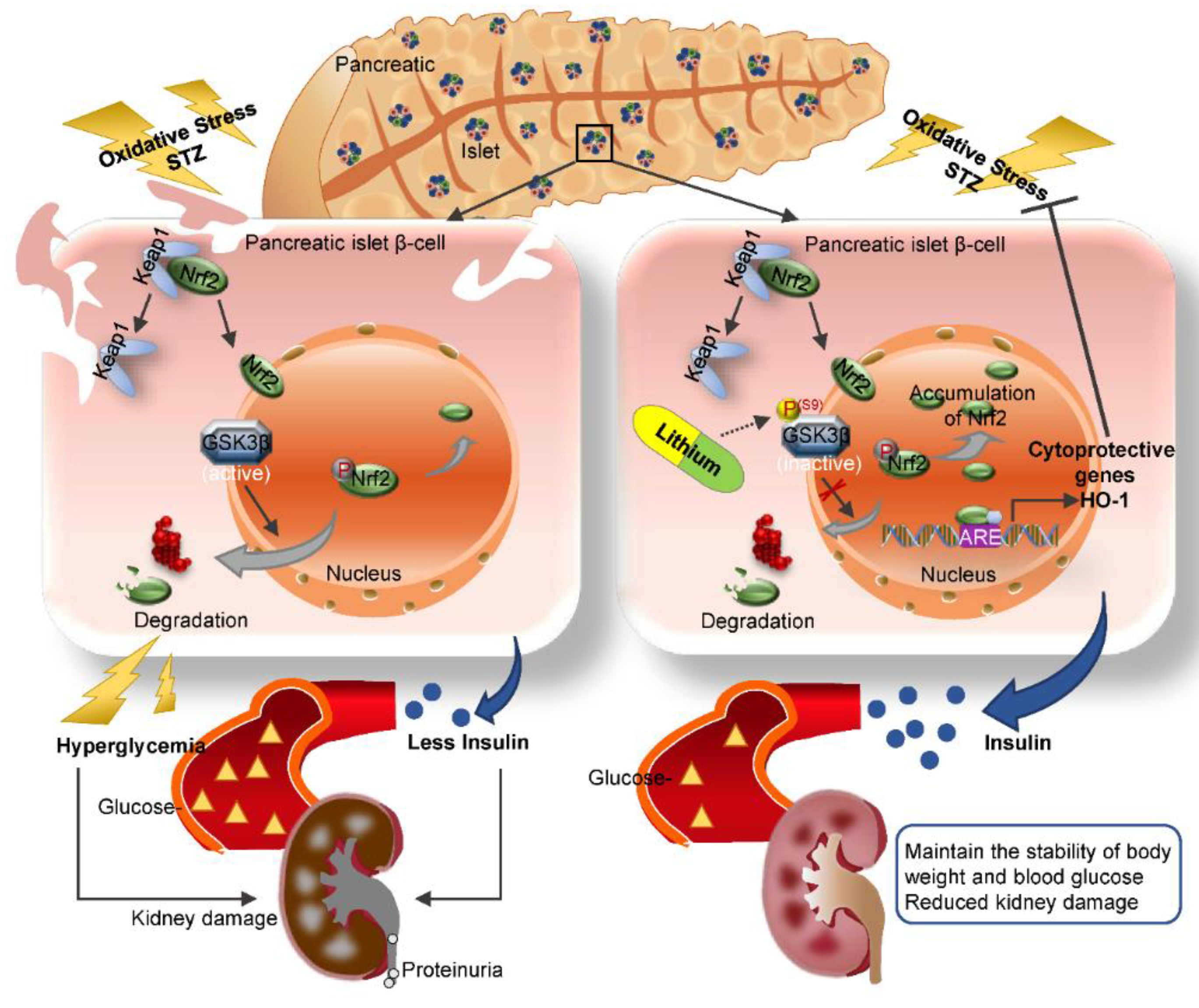
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef] [PubMed]
- Seuring, T.; Archangelidi, O.; Suhrcke, M. The Economic Costs of Type 2 Diabetes: A Global Systematic Review. Pharmacoeconomics 2015, 33, 811–831. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Salpea, P.; Karuranga, S.; Petersohn, I.; Malanda, B.; Gregg, E.W.; Unwin, N.; Wild, S.H.; Williams, R. Mortality attributable to diabetes in 20–79 years old adults, 2019 estimates: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2020, 162, 108086. [Google Scholar] [CrossRef] [PubMed]
- Rother, K.I. Diabetes treatment--bridging the divide. N. Engl. J. Med. 2007, 356, 1499–1501. [Google Scholar] [CrossRef]
- Mathis, D.; Vence, L.; Benoist, C. β-Cell death during progression to diabetes. Nature 2001, 414, 792–798. [Google Scholar] [CrossRef]
- Willcox, A.; Richardson, S.J.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Analysis of islet inflammation in human type 1 diabetes. Clin. Exp. Immunol. 2009, 155, 173–181. [Google Scholar] [CrossRef]
- DeWitt, D.E.; Hirsch, I.B. Outpatient insulin therapy in type 1 and type 2 diabetes mellitus: Scientific review. JAMA 2003, 289, 2254–2264. [Google Scholar] [CrossRef]
- Patel, D.K.; Prasad, S.K.; Kumar, R.; Hemalatha, S. An overview on antidiabetic medicinal plants having insulin mimetic property. Asian Pac. J. Trop. Biomed. 2012, 2, 320–330. [Google Scholar] [CrossRef]
- Saran, A.S. Antidiabetic effects of lithium. J. Clin. Psychiatry 1982, 43, 383–384. [Google Scholar]
- López-Muñoz, F.; Shen, W.W.; D’Ocon, P.; Romero, A.; Álamo, C. A History of the Pharmacological Treatment of Bipolar Disorder. Int. J. Mol. Sci. 2018, 19, 2143. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, C.D.; Gordon, M.W. Manic-depressive illness, diabetes mellitus, and lithium carbonate. Arch. Gen. Psychiatry 1969, 21, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Vendsborg, P.B.; Rafaelsen, O.J. Lithium in man: Effect on glucose tolerance and serum electrolytes. Acta Psychiatr. Scand. 1973, 49, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Männistö, P.; Koivisto, V. Antidiabetic effects of lithium. Lancet 1972, 2, 1031. [Google Scholar] [CrossRef]
- Altan, N.; Engin, A.; Hizel, N.; Ongun, O.; Cevik, C. The effect of short term lithium treatment on the leukocyte, liver and muscle carbohydrate metabolism of guinea-pigs. Gen. Pharmacol. 1993, 24, 1491–1493. [Google Scholar] [CrossRef]
- Rossetti, L. Normalization of insulin sensitivity with lithium in diabetic rats. Diabetes 1989, 38, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Okosieme, O.E.; Campbell, A.; Patton, K.; Evans, M.L. Transient diabetes associated with withdrawal of lithium therapy. Diabetes Care 2006, 29, 1181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sjöholm, A.; Welsh, N.; Hellerström, C. Lithium increases DNA replication, polyamine content, and insulin secretion by rat pancreatic beta-cells. Am. J. Physiol. 1992, 262, C391–C395. [Google Scholar] [CrossRef] [PubMed]
- Sjöholm, A. Lithium stimulation of rat pancreatic beta-cell replication is mediated through pertussis toxin-sensitive GTP-binding proteins and occurs independently of Ca2+ influx, cAMP, or protein kinase C activation. Diabetes 1996, 45, 1057–1062. [Google Scholar] [CrossRef]
- Yildirim, S.; Celikezen, F.C.; Oto, G.; Sengul, E.; Bulduk, M.; Tasdemir, M.; Ali Cinar, D. An Investigation of Protective Effects of Litium Borate on Blood and Histopathological Parameters in Acute Cadmium-Induced Rats. Biol. Trace Elem. Res. 2018, 182, 287–294. [Google Scholar] [CrossRef]
- Ostrovskaya, R.U.; Ivanov, S.V.; Durnev, A.D. Neuroprotective Lithium Salts Protect Pancreatic β-Cells from Damage. Bull. Exp. Biol. Med. 2018, 165, 758–762. [Google Scholar] [CrossRef]
- Chuang, D.M.; Wang, Z.; Chiu, C.T. GSK-3 as a Target for Lithium-Induced Neuroprotection against Excitotoxicity in Neuronal Cultures and Animal Models of Ischemic Stroke. Front. Mol. Neurosci. 2011, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, A.; Dehpour, A.R.; Safa, M.; Mirzamohammadi, S.; Sharifi, A.M. Investigating the protective effect of lithium against high glucose-induced neurotoxicity in PC12 cells: Involvements of ROS, JNK and P38 MAPKs, and apoptotic mitochondria pathway. Cell. Mol. Neurobiol. 2014, 34, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA. 1996, 93, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Ruel, L.; Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. CB 1996, 6, 1664–1668. [Google Scholar] [CrossRef]
- Rayasam, G.V.; Tulasi, V.K.; Sodhi, R.; Davis, J.A.; Ray, A. Glycogen synthase kinase 3: More than a namesake. Br. J. Pharmacol. 2009, 156, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef]
- Rojo, A.I.; Sagarra, M.R.; Cuadrado, A. GSK-3beta down-regulates the transcription factor Nrf2 after oxidant damage: Relevance to exposure of neuronal cells to oxidative stress. J. Neurochem. 2008, 105, 192–202. [Google Scholar] [CrossRef]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef]
- Lu, M.; Wang, P.; Qiao, Y.; Jiang, C.; Ge, Y.; Flickinger, B.; Malhotra, D.K.; Dworkin, L.D.; Liu, Z.; Gong, R. GSK3β-mediated Keap1-independent regulation of Nrf2 antioxidant response: A molecular rheostat of acute kidney injury to chronic kidney disease transition. Redox Biol. 2019, 26, 101275. [Google Scholar] [CrossRef]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K.; Huan, Y. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. Pharmacol. 2008, 40, 5.47.1–5.47.14. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, H.; Chen, F.; Fu, J.; Xu, Y.; Hou, Y.; Kou, H.H.; Zhai, C.; Nelson, M.B.; Zhang, Q.; et al. An overview of chemical inhibitors of the Nrf2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic. Biol. Med. 2016, 99, 544–556. [Google Scholar] [CrossRef] [PubMed]
- García-Ocaña, A.; Vasavada, R.C.; Cebrian, A.; Reddy, V.; Takane, K.K.; López-Talavera, J.C.; Stewart, A.F. Transgenic overexpression of hepatocyte growth factor in the beta-cell markedly improves islet function and islet transplant outcomes in mice. Diabetes 2001, 50, 2752–2762. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, P.; Qiao, Y.; Ge, Y.; Wang, Y.; Quan, S.; Yao, R.; Zhuang, S.; Wang, L.J.; Du, Y.; et al. Genetic and Pharmacologic Targeting of Glycogen Synthase Kinase 3β Reinforces the Nrf2 Antioxidant Defense against Podocytopathy. J. Am. Soc. Nephrol. JASN 2016, 27, 2289–2308. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.B.; Gao, L.; Wang, J.; Wang, Y.G.; Dong, Z.; Zhao, J.; Mi, Q.S.; Zhou, L. Conditional ablation of HDAC3 in islet beta cells results in glucose intolerance and enhanced susceptibility to STZ-induced diabetes. Oncotarget 2016, 7, 57485–57497. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Ge, Y.; Wang, Z.; Zhuang, S.; Dworkin, L.; Peng, A.; Gong, R. Delayed administration of a single dose of lithium promotes recovery from AKI. J. Am. Soc. Nephrol. JASN 2014, 25, 488–500. [Google Scholar] [CrossRef]
- Bao, H.; Zhang, Q.; Liu, X.; Song, Y.; Li, X.; Wang, Z.; Li, C.; Peng, A.; Gong, R. Lithium targeting of AMPK protects against cisplatin-induced acute kidney injury by enhancing autophagy in renal proximal tubular epithelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 14370–14381. [Google Scholar] [CrossRef]
- Yu, F.; Wang, Z.; Tchantchou, F.; Chiu, C.T.; Zhang, Y.; Chuang, D.M. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J. Neurotrauma 2012, 29, 362–374. [Google Scholar] [CrossRef]
- O’Donnell, K.C.; Gould, T.D. The behavioral actions of lithium in rodent models: Leads to develop novel therapeutics. Neurosci. Biobehav. Rev. 2007, 31, 932–962. [Google Scholar] [CrossRef]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Jörns, A.; Günther, A.; Hedrich, H.J.; Wedekind, D.; Tiedge, M.; Lenzen, S. Immune cell infiltration, cytokine expression, and beta-cell apoptosis during the development of type 1 diabetes in the spontaneously diabetic LEW.1AR1/Ztm-iddm rat. Diabetes 2005, 54, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Osorio, A.S.; González-Reyes, S.; Pedraza-Chaverri, J. Natural Nrf2 activators in diabetes. Clin. Chim. Acta 2015, 448, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Park, K.-G. A systematic review of oxidative stress and safety of antioxidants in diabetes: Focus on islets and their defense. Diabetes Metab. J. 2013, 37, 106–112. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Grankvist, K.; Marklund, S.L.; Täljedal, I.B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J. 1981, 199, 393–398. [Google Scholar] [CrossRef]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53 (Suppl. S1), S119–S124. [Google Scholar] [CrossRef]
- Yoh, K.; Hirayama, A.; Ishizaki, K.; Yamada, A.; Takeuchi, M.; Yamagishi, S.; Morito, N.; Nakano, T.; Ojima, M.; Shimohata, H.; et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells Devoted Mol. Cell. Mech. 2008, 13, 1159–1170. [Google Scholar] [CrossRef]
- National Clinical Guideline Centre. National Institute for Health and Care Excellence: Clinical Guidelines. In Type 1 Diabetes in Adults: Diagnosis and Management; National Institute for Health and Care Excellence (UK). Copyright © 2021 National Clinical Guideline Centre: London, UK, 2015. [Google Scholar]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef]
- Yagishita, Y.; Fukutomi, T.; Sugawara, A.; Kawamura, H.; Takahashi, T.; Pi, J.; Uruno, A.; Yamamoto, M. Nrf2 Protects Pancreatic β-Cells From Oxidative and Nitrosative Stress in Diabetic Model Mice. Diabetes 2014, 63, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef]
- Shelton, P.M. Glycogen Synthase Kinase 3-Beta (GSK3beta) Is a Central Regulator in the Non-Canonical NF-E2 Related Factor (Nrf2) Antioxidant Response. Ph.D. Thesis, University of Maryland, College Park, MD, USA, 2013. [Google Scholar]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed]
- Jauhar, S.; Young, A.H. Controversies in bipolar disorder; role of second-generation antipsychotic for maintenance therapy. Int. J. Bipolar Disord. 2019, 7, 10. [Google Scholar] [CrossRef]
- Malhi, G.S.; Tanious, M.; Gershon, S. The lithiumeter: A measured approach. Bipolar Disord. 2011, 13, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, M. Lithium side effects and toxicity: Prevalence and management strategies. Int. J. Bipolar Disord. 2016, 4, 27. [Google Scholar] [CrossRef]
- Shine, B.; McKnight, R.F.; Leaver, L.; Geddes, J.R. Long-term effects of lithium on renal, thyroid, and parathyroid function: A retrospective analysis of laboratory data. Lancet 2015, 386, 461–468. [Google Scholar] [CrossRef]
- Razzak, M. Basic research: The long and the short of it-the temporal effects of renal lithium exposure are beginning to be unravelled. Nat. Rev. Nephrol. 2014, 10, 123. [Google Scholar] [CrossRef]
- Gong, R.; Wang, P.; Dworkin, L. What we need to know about the effect of lithium on the kidney. Am. J. Physiol. Ren. Physiol. 2016, 311, F1168–F1171. [Google Scholar] [CrossRef]
- Lenox, R.H.; Wang, L. Molecular basis of lithium action: Integration of lithium-responsive signaling and gene expression networks. Mol. Psychiatry 2003, 8, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Harwood, A.J. Lithium therapy and signal transduction. Trends Pharmacol. Sci. 2000, 21, 61–64. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Anshul, F.; Malhotra, D.K.; Jaume, J.; Dworkin, L.D.; Gong, R. Microdose Lithium Protects against Pancreatic Islet Destruction and Renal Impairment in Streptozotocin-Elicited Diabetes. Antioxidants 2021, 10, 138. https://doi.org/10.3390/antiox10010138
Zhang J, Anshul F, Malhotra DK, Jaume J, Dworkin LD, Gong R. Microdose Lithium Protects against Pancreatic Islet Destruction and Renal Impairment in Streptozotocin-Elicited Diabetes. Antioxidants. 2021; 10(1):138. https://doi.org/10.3390/antiox10010138
Chicago/Turabian StyleZhang, Jiahui, Fnu Anshul, Deepak K. Malhotra, Juan Jaume, Lance D. Dworkin, and Rujun Gong. 2021. "Microdose Lithium Protects against Pancreatic Islet Destruction and Renal Impairment in Streptozotocin-Elicited Diabetes" Antioxidants 10, no. 1: 138. https://doi.org/10.3390/antiox10010138
APA StyleZhang, J., Anshul, F., Malhotra, D. K., Jaume, J., Dworkin, L. D., & Gong, R. (2021). Microdose Lithium Protects against Pancreatic Islet Destruction and Renal Impairment in Streptozotocin-Elicited Diabetes. Antioxidants, 10(1), 138. https://doi.org/10.3390/antiox10010138