High-Content Screening for the Detection of Drug-Induced Oxidative Stress in Liver Cells


Abstract
1. Introduction
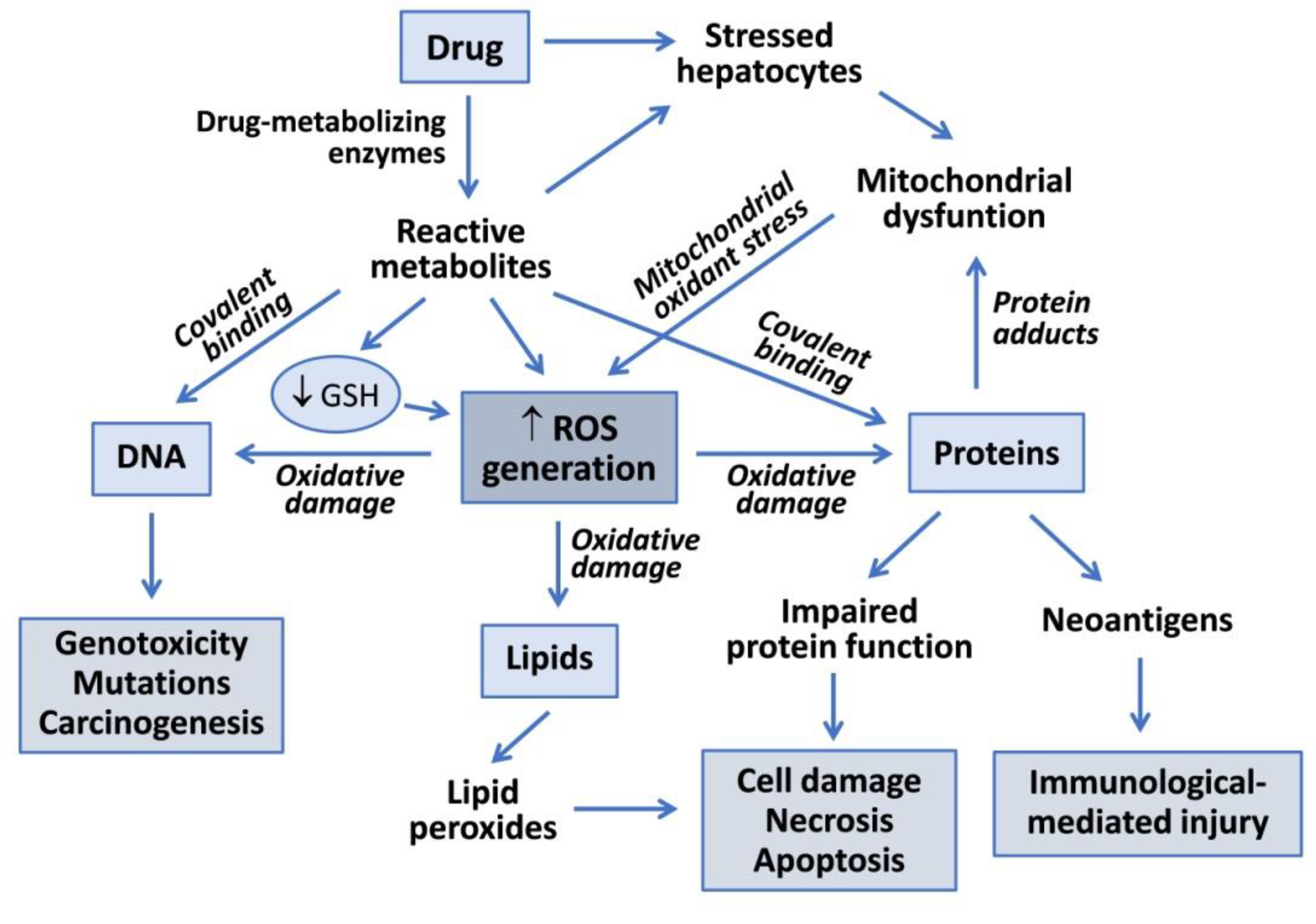
2. Mechanisms of Drug-Induced Hepatotoxicity
3. Drug-Induced ROS Generation
4. HCS Assays for the Detection of Oxidative Stress Induced by Drugs
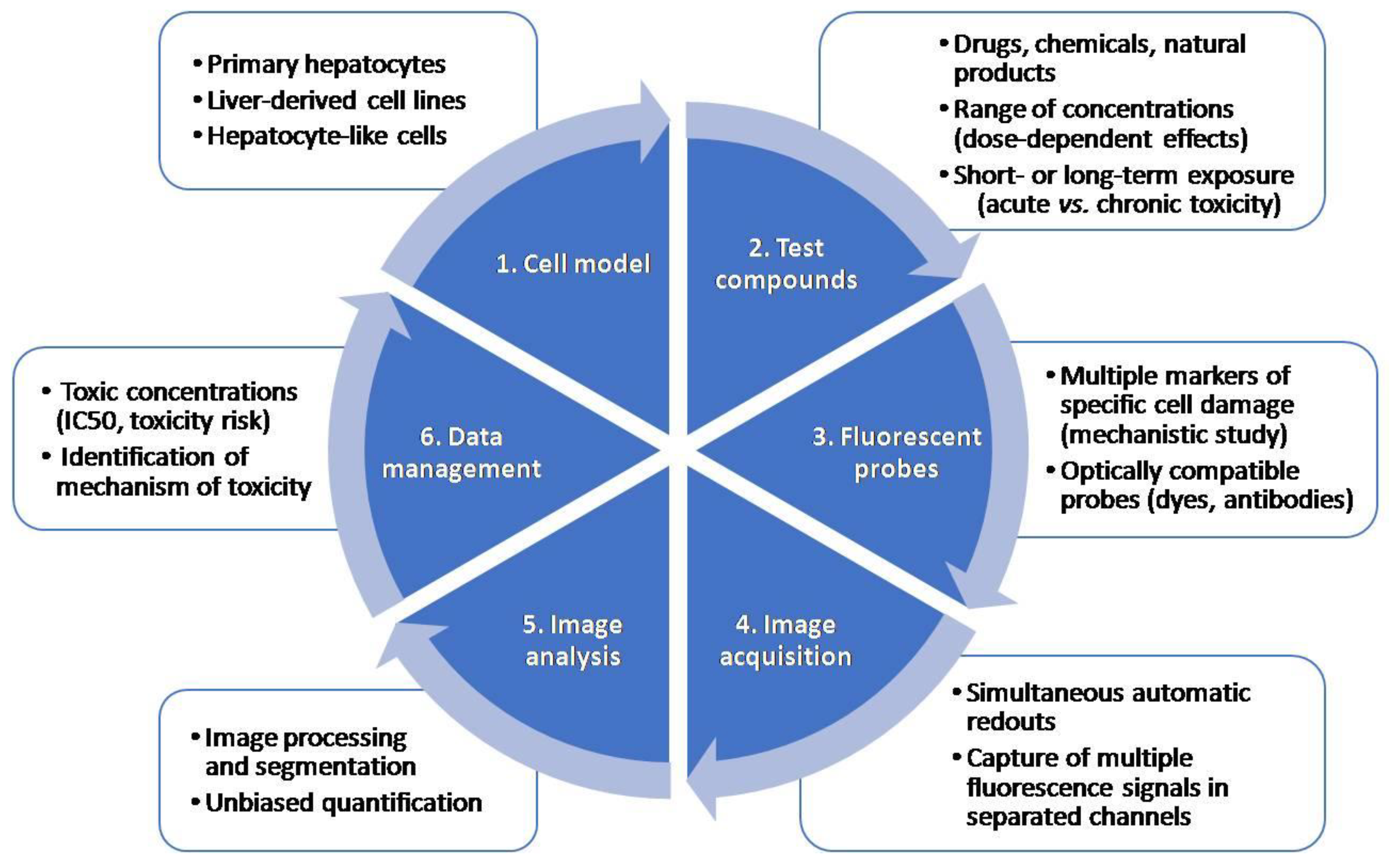
4.1. HCS Technology
4.2. HCS Probes for the Detection of Oxidative Stress
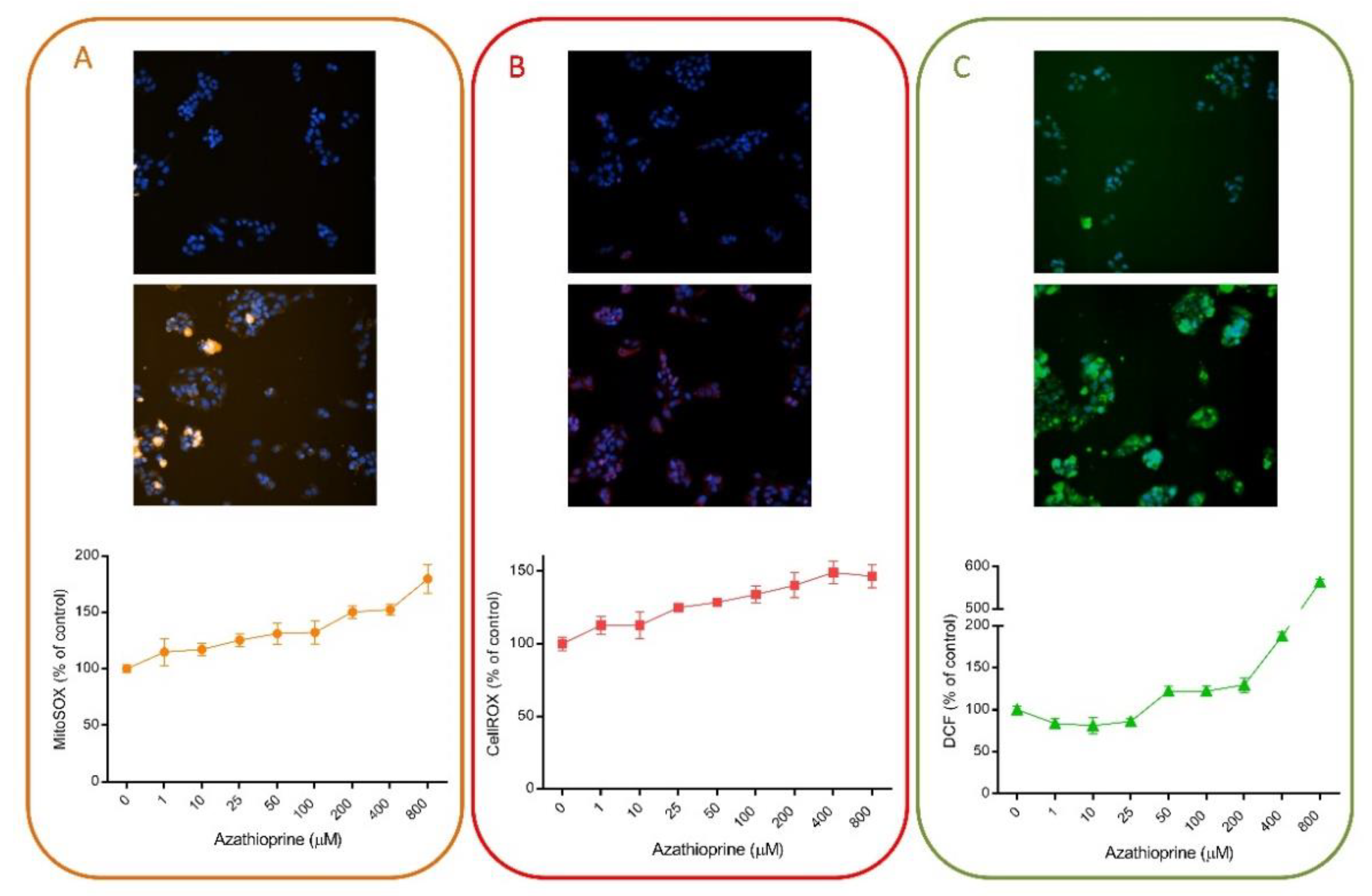
4.3. Examples of HCS Assays for the Detection of Oxidative Stress Induced by Drugs in Hepatic Cell Models
5. Conclusions and Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AZA | azathioprine |
| CYP | cytochrome P450 |
| DCF | 2′,7′-dichlorofluorescin |
| DILI | drug-induced liver injury |
| GSH | glutathione |
| HCS | high-content screening |
| HLCs | hepatocyte-like cells |
| iPSCs | induced pluripotent stem cells |
| mBCl | monochlorobimane |
| MRC | mitochondrial respiratory chain |
| NAPQI | N-acetyl-p-benzoquinone imine |
| PHH | primary human hepatocytes |
| ROS | reactive oxygen species |
| UHH | Upcyte human hepatocytes |
References
- Funk, C.; Roth, A. Current limitations and future opportunities for prediction of DILI from in vitro. Arch. Toxicol. 2017, 91, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cortes, M.; Robles-Diaz, M.; Stephens, C.; Ortega-Alonso, A.; Lucena, M.I.; Andrade, R.J. Drug induced liver injury: An update. Arch. Toxicol. 2020, 94, 3381–3407. [Google Scholar] [CrossRef]
- Lee, W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.L.; Baker, T.K. The future of drug safety testing: Expanding the view and narrowing the focus. Drug Discov. Today 2009, 14, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J. Pathogenesis of idiosyncratic drug-induced liver injury and clinical perspectives. Gastroenterology 2014, 146, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Tolosa, L.; Donato, M.T. Metabolic activation and drug-induced liver injury: In vitro approaches for the safety risk assessment of new drugs. J. Appl. Toxicol. 2016, 36, 752–768. [Google Scholar] [CrossRef]
- Weaver, R.J.; Blomme, E.A.; Chadwick, A.E.; Copple, I.M.; Gerets, H.H.J.; Goldring, C.E.; Guillouzo, A.; Hewitt, P.G.; Ingelman-Sundberg, M.; Jensen, K.G.; et al. Managing the challenge of drug-induced liver injury: A roadmap for the development and deployment of preclinical predictive models. Nat. Rev. Drug Discov. 2020, 19, 131–148. [Google Scholar] [CrossRef]
- Kuijper, I.A.; Yang, H.; Van De Water, B.; Beltman, J.B. Unraveling cellular pathways contributing to drug-induced liver injury by dynamical modeling. Expert Opin. Drug Metab. Toxicol. 2017, 13, 5–17. [Google Scholar] [CrossRef]
- Jiang, J.; Pieterman, C.D.; Ertaylan, G.; Peeters, R.L.M.; de Kok, T. The application of omics-based human liver platforms for investigating the mechanism of drug-induced hepatotoxicity in vitro. Arch. Toxicol. 2019, 93, 3067–3098. [Google Scholar] [CrossRef]
- Gomez-Lechon, M.J.; Tolosa, L.; Conde, I.; Donato, M.T. Competency of different cell models to predict human hepatotoxic drugs. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1553–1568. [Google Scholar] [CrossRef]
- Joshi, P.; Lee, M.Y. High Content Imaging (HCI) on Miniaturized Three-Dimensional (3D) Cell Cultures. Biosensors 2015, 5, 768–790. [Google Scholar] [CrossRef] [PubMed]
- Dragovic, S.; Vermeulen, N.P.; Gerets, H.H.; Hewitt, P.G.; Ingelman-Sundberg, M.; Park, B.K.; Juhila, S.; Snoeys, J.; Weaver, R.J. Evidence-based selection of training compounds for use in the mechanism-based integrated prediction of drug-induced liver injury in man. Arch. Toxicol. 2016, 90, 2979–3003. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, L.; Pinto, S.; Donato, M.T.; Lahoz, A.; Castell, J.V.; O’Connor, J.E.; Gomez-Lechon, M.J. Development of a multiparametric cell-based protocol to screen and classify the hepatotoxicity potential of drugs. Toxicol. Sci. 2012, 127, 187–198. [Google Scholar] [CrossRef]
- Hewitt, M.; Enoch, S.J.; Madden, J.C.; Przybylak, K.R.; Cronin, M.T. Hepatotoxicity: A scheme for generating chemical categories for read-across, structural alerts and insights into mechanism(s) of action. Crit. Rev. Toxicol. 2013, 43, 537–558. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.A.; Isin, E.M.; Li, Y.; Weidolf, L.; Page, K.; Wilson, I.; Swallow, S.; Middleton, B.; Stahl, S.; Foster, A.J.; et al. In vitro approach to assess the potential for risk of idiosyncratic adverse reactions caused by candidate drugs. Chem. Res. Toxicol. 2012, 25, 1616–1632. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B.; Berson, A.; Robin, M.A.; Letteron, P.; Moreau, R.; Mansouri, A. Central role of mitochondria in drug-induced liver injury. Drug Metab. Rev. 2012, 44, 34–87. [Google Scholar] [CrossRef]
- Labbe, G.; Pessayre, D.; Fromenty, B. Drug-induced liver injury through mitochondrial dysfunction: Mechanisms and detection during preclinical safety studies. Fundam. Clin. Pharmacol. 2008, 22, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef]
- Pereira, C.V.; Nadanaciva, S.; Oliveira, P.J.; Will, Y. The contribution of oxidative stress to drug-induced organ toxicity and its detection in vitro and in vivo. Expert Opin. Drug Metab. Toxicol. 2012, 8, 219–237. [Google Scholar] [CrossRef]
- Grattagliano, I.; Bonfrate, L.; Diogo, C.V.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Biochemical mechanisms in drug-induced liver injury: Certainties and doubts. World J. Gastroenterol. 2009, 15, 4865–4876. [Google Scholar] [CrossRef]
- Thompson, R.A.; Isin, E.M.; Ogese, M.O.; Mettetal, J.T.; Williams, D.P. Reactive Metabolites: Current and Emerging Risk and Hazard Assessments. Chem. Res. Toxicol. 2016, 29, 505–533. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, F.; Alvarez-Suarez, J.M.; Gasparrini, M.; Forbes-Hernandez, T.Y.; Afrin, S.; Bompadre, S.; Rubini, C.; Zizzi, A.; Astolfi, P.; Santos-Buelga, C.; et al. Strawberry consumption alleviates doxorubicin-induced toxicity by suppressing oxidative stress. Food Chem. Toxicol. 2016, 94, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, R.; Kondeva-Burdina, M.; Vitcheva, V.; Mitcheva, M. Some in vitro/in vivo chemically-induced experimental models of liver oxidative stress in rats. Biomed. Res. Int. 2014, 2014, 706302. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Cho, W.C.; Upadhyay, G. Drug-Induced Liver Toxicity and Prevention by Herbal Antioxidants: An Overview. Front. Physiol. 2015, 6, 363. [Google Scholar] [CrossRef]
- Tong, V.; Teng, X.W.; Chang, T.K.; Abbott, F.S. Valproic acid II: Effects on oxidative stress, mitochondrial membrane potential, and cytotoxicity in glutathione-depleted rat hepatocytes. Toxicol. Sci. 2005, 86, 436–443. [Google Scholar] [CrossRef]
- Kang, P.; Dalvie, D.; Smith, E.; Zhou, S.; Deese, A.; Nieman, J.A. Bioactivation of flutamide metabolites by human liver microsomes. Drug Metab. Dispos. 2008, 36, 1425–1437. [Google Scholar] [CrossRef]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef]
- Teppner, M.; Boess, F.; Ernst, B.; Pahler, A. Biomarkers of Flutamide-Bioactivation and Oxidative Stress in Vitro and in Vivo. Drug Metab. Dispos. 2016, 44, 560–569. [Google Scholar] [CrossRef]
- Park, B.K.; Kitteringham, N.R.; Maggs, J.L.; Pirmohamed, M.; Williams, D.P. The role of metabolic activation in drug-induced hepatotoxicity. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 177–202. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Suda, C.; Horie, T. Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J. Hepatol. 2005, 42, 110–116. [Google Scholar] [CrossRef]
- Petit, E.; Langouet, S.; Akhdar, H.; Nicolas-Nicolaz, C.; Guillouzo, A.; Morel, F. Differential toxic effects of azathioprine, 6-mercaptopurine and 6-thioguanine on human hepatocytes. Toxicol. In Vitro 2008, 22, 632–642. [Google Scholar] [CrossRef]
- Ingawale, D.K.; Mandlik, S.K.; Naik, S.R. Models of hepatotoxicity and the underlying cellular, biochemical and immunological mechanism(s): A critical discussion. Environ. Toxicol. Pharmacol. 2014, 37, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Castell, J.V.; Donato, M.T.; Gomez-Lechon, M.J. Metabolism and bioactivation of toxicants in the lung. The in vitro cellular approach. Exp. Toxicol. Pathol. 2005, 57 (Suppl. S1), 189–204. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J.; Irwin, W.; Diaz, D.; Howard-Cofield, E.; Krejsa, C.M.; Slaughter, M.R.; Gao, B.; Kaludercic, N.; Angeline, A.; Bernardi, P.; et al. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch. Toxicol. 2006, 80, 580–604. [Google Scholar] [CrossRef]
- Donato, M.T.; Tolosa, L. Stem-cell derived hepatocyte-like cells for the assessment of drug-induced liver injury. Differentiation 2019, 106, 15–22. [Google Scholar] [CrossRef]
- Persson, M.; Loye, A.F.; Jacquet, M.; Mow, N.S.; Thougaard, A.V.; Mow, T.; Hornberg, J.J. High-content analysis/screening for predictive toxicology: Application to hepatotoxicity and genotoxicity. Basic Clin. Pharmacol. Toxicol. 2014, 115, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.C. Accuracy and precision in quantitative fluorescence microscopy. J. Cell Biol. 2009, 185, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D.; Ward, A.M.; Bard, F.; Calvert, M.E. Pushing the boundaries of high content imaging. Cytom. A 2017, 91, 113–114. [Google Scholar] [CrossRef]
- Bougen-Zhukov, N.; Loh, S.Y.; Lee, H.K.; Loo, L.H. Large-scale image-based screening and profiling of cellular phenotypes. Cytom. A 2017, 91, 115–125. [Google Scholar] [CrossRef]
- Liron, Y.; Paran, Y.; Zatorsky, N.G.; Geiger, B.; Kam, Z. Laser autofocusing system for high-resolution cell biological imaging. J. Microsc 2006, 221, 145–151. [Google Scholar] [CrossRef]
- Meijering, E.; Dzyubachyk, O.; Smal, I. Methods for cell and particle tracking. Methods Enzymol. 2012, 504, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Lee, H.K.; Hariharan, S.; Bu, W.; Ahmed, S. Evolving generalized Voronoi diagrams for accurate cellular image segmentation. Cytom. A 2010, 77, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Kozak, K.; Rinn, B.; Leven, O.; Emmenlauer, M. Strategies and Solutions to Maintain and Retain Data from High Content Imaging, Analysis, and Screening Assays. Methods Mol. Biol. 2018, 1683, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Berliocchi, L.; Chiappini, C.; Adornetto, A.; Gentile, D.; Cerri, S.; Russo, R.; Bagetta, G.; Corasaniti, M.T. Early LC3 lipidation induced by d-limonene does not rely on mTOR inhibition, ERK activation and ROS production and it is associated with reduced clonogenic capacity of SH-SY5Y neuroblastoma cells. Phytomedicine 2018, 40, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, L.; Gomez-Lechon, M.J.; Perez-Cataldo, G.; Castell, J.V.; Donato, M.T. HepG2 cells simultaneously expressing five P450 enzymes for the screening of hepatotoxicity: Identification of bioactivable drugs and the potential mechanism of toxicity involved. Arch. Toxicol. 2013, 87, 1115–1127. [Google Scholar] [CrossRef]
- Cosgrove, B.D.; King, B.M.; Hasan, M.A.; Alexopoulos, L.G.; Farazi, P.A.; Hendriks, B.S.; Griffith, L.G.; Sorger, P.K.; Tidor, B.; Xu, J.J.; et al. Synergistic drug-cytokine induction of hepatocellular death as an in vitro approach for the study of inflammation-associated idiosyncratic drug hepatotoxicity. Toxicol. Appl. Pharmacol. 2009, 237, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Jamieson, J.D.; Marroquin, L.D.; Nadanaciva, S.; Xu, J.J.; Dunn, M.C.; Smith, A.R.; Will, Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol. Sci. 2008, 103, 335–345. [Google Scholar] [CrossRef]
- Xu, J.J.; Henstock, P.V.; Dunn, M.C.; Smith, A.R.; Chabot, J.R.; de Graaf, D. Cellular imaging predictions of clinical drug-induced liver injury. Toxicol. Sci. 2008, 105, 97–105. [Google Scholar] [CrossRef]
- Mennecozzi, M.; Landesmann, B.; Palosaari, T.; Harris, G.; Whelan, M. Sex differences in liver toxicity-do female and male human primary hepatocytes react differently to toxicants in vitro? PLoS ONE 2015, 10, e0122786. [Google Scholar] [CrossRef]
- Tolosa, L.; Carmona, A.; Castell, J.V.; Gomez-Lechon, M.J.; Donato, M.T. High-content screening of drug-induced mitochondrial impairment in hepatic cells: Effects of statins. Arch. Toxicol. 2015, 89, 1847–1860. [Google Scholar] [CrossRef]
- Tolosa, L.; Jimenez, N.; Pelecha, M.; Castell, J.V.; Gomez-Lechon, M.J.; Donato, M.T. Long-term and mechanistic evaluation of drug-induced liver injury in Upcyte human hepatocytes. Arch. Toxicol. 2019, 93, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Wink, S.; Hiemstra, S.W.; Huppelschoten, S.; Klip, J.E.; van de Water, B. Dynamic imaging of adaptive stress response pathway activation for prediction of drug induced liver injury. Arch. Toxicol. 2018, 92, 1797–1814. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Negro, A.; Herrera, G.; Castell, J.V.; O’Connor, J.E.; Gomez-Lechon, M.J. Cytometric analysis for drug-induced steatosis in HepG2 cells. Chem. Biol. Interact. 2009, 181, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Hedley, D.W.; Chow, S. Evaluation of methods for measuring cellular glutathione content using flow cytometry. Cytometry 1994, 15, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Naguib, Y.M. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Wink, S.; Hiemstra, S.; Huppelschoten, S.; Danen, E.; Niemeijer, M.; Hendriks, G.; Vrieling, H.; Herpers, B.; van de Water, B. Quantitative high content imaging of cellular adaptive stress response pathways in toxicity for chemical safety assessment. Chem. Res. Toxicol. 2014, 27, 338–355. [Google Scholar] [CrossRef]
- Wink, S.; Hiemstra, S.; Herpers, B.; van de Water, B. High-content imaging-based BAC-GFP toxicity pathway reporters to assess chemical adversity liabilities. Arch. Toxicol. 2017, 91, 1367–1383. [Google Scholar] [CrossRef]
- Aithal, G.P. Hepatotoxicity related to antirheumatic drugs. Nat. Rev. Rheumatol. 2011, 7, 139–150. [Google Scholar] [CrossRef]
- Matsuo, K.; Sasaki, E.; Higuchi, S.; Takai, S.; Tsuneyama, K.; Fukami, T.; Nakajima, M.; Yokoi, T. Involvement of oxidative stress and immune- and inflammation-related factors in azathioprine-induced liver injury. Toxicol. Lett. 2014, 224, 215–224. [Google Scholar] [CrossRef]
- Garside, H.; Marcoe, K.F.; Chesnut-Speelman, J.; Foster, A.J.; Muthas, D.; Kenna, J.G.; Warrior, U.; Bowes, J.; Baumgartner, J. Evaluation of the use of imaging parameters for the detection of compound-induced hepatotoxicity in 384-well cultures of HepG2 cells and cryopreserved primary human hepatocytes. Toxicol. In Vitro 2014, 28, 171–181. [Google Scholar] [CrossRef]
- Pradip, A.; Steel, D.; Jacobsson, S.; Holmgren, G.; Ingelman-Sundberg, M.; Sartipy, P.; Bjorquist, P.; Johansson, I.; Edsbagge, J. High Content Analysis of Human Pluripotent Stem Cell Derived Hepatocytes Reveals Drug Induced Steatosis and Phospholipidosis. Stem Cells Int. 2016, 2016, 2475631. [Google Scholar] [CrossRef]
- Saito, J.; Okamura, A.; Takeuchi, K.; Hanioka, K.; Okada, A.; Ohata, T. High content analysis assay for prediction of human hepatotoxicity in HepaRG and HepG2 cells. Toxicol. In Vitro 2016, 33, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.; Berntsen, H.F.; Zimmer, K.E.; Frizzell, C.; Verhaegen, S.; Ropstad, E.; Connolly, L. Effects of defined mixtures of persistent organic pollutants (POPs) on multiple cellular responses in the human hepatocarcinoma cell line, HepG2, using high content analysis screening. Toxicol. Appl. Pharmacol. 2016, 294, 21–31. [Google Scholar] [CrossRef]
- Hiemstra, S.; Ramaiahgari, S.C.; Wink, S.; Callegaro, G.; Coonen, M.; Meerman, J.; Jennen, D.; van den Nieuwendijk, K.; Dankers, A.; Snoeys, J.; et al. High-throughput confocal imaging of differentiated 3D liver-like spheroid cellular stress response reporters for identification of drug-induced liver injury liability. Arch. Toxicol. 2019, 93, 2895–2911. [Google Scholar] [CrossRef]
- Rodeiro, I.; Hernandez, I.; Herrera, J.A.; Riera, M.; Donato, M.T.; Tolosa, L.; Gonzalez, K.; Ansoar, Y.; Gomez-Lechon, M.J.; Vanden Berghe, W.; et al. Assessment of the cytotoxic potential of an aqueous-ethanolic extract from Thalassia testudinum angiosperm marine grown in the Caribbean Sea. J. Pharm. Pharmacol. 2018, 70, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, L.; Rodeiro, I.; Donato, M.T.; Herrera, J.A.; Delgado, R.; Castell, J.V.; Gomez-Lechon, M.J. Multiparametric evaluation of the cytoprotective effect of the Mangifera indica L. stem bark extract and mangiferin in HepG2 cells. J. Pharm. Pharmacol. 2013, 65, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J. High-content analysis in toxicology: Screening substances for human toxicity potential, elucidating subcellular mechanisms and in vivo use as translational safety biomarkers. Basic Clin. Pharmacol. Toxicol. 2014, 115, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, P.; Lopez, O.; Amberg, A.; Pastor, M.; Sanz, F. Hepatotoxicity prediction by systems biology modeling of disturbed metabolic pathways using gene expression data. ALTEX 2017, 34, 219–234. [Google Scholar] [CrossRef]
- Garcia-Canaveras, J.C.; Castell, J.V.; Donato, M.T.; Lahoz, A. A metabolomics cell-based approach for anticipating and investigating drug-induced liver injury. Sci. Rep. 2016, 6, 27239. [Google Scholar] [CrossRef]
- Li, S.; Xia, M. Review of high-content screening applications in toxicology. Arch. Toxicol. 2019, 93, 3387–3396. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Bottger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mechanism | Compounds | References |
|---|---|---|
| Direct or indirect impairment of the mitochondrial respiratory chain (MRC) | Acetylsalicylic, amiodarone, azathioprine, buprenorphine, chloroquine, lovastatin, tamoxifen, nefazodone, troglitazone | [12,16,20,21] |
| Depletion of GSH pool and/or antioxidant enzymes | Azathioprine, doxorubicin, flutamide, isoniazid, valproic acid, paracetamol (acetaminophen) | [16,19,22,23,24,25] |
| Generation of electrophilic metabolites | Amitriptyline, benoxaprofen, diclofenac flutamide, paracetamol, ticlopidine, trogitazone | [12,13,16,25,26] |
| Redox cycling-induction | Diquat, paraquat, menadione, doxorubicin, flutamide | [22,27,28] |
| Probe/Reporter | Indicator | Excitation | Emission | Reference |
|---|---|---|---|---|
| BODIPY 665/676 | Peroxyl radicals (lipid peroxidation) | 665 | 676 | [13] |
| CellROX Green | ROS production (nuclei) | 485 | 520 | [44] |
| CellROX Deep Red | ROS production (cytoplasm) | 644 | 665 | [45] |
| CM-H2DCFDA | 495 | 527 | [46,47,48] | |
| Dihydroethidium | Superoxide anion | 518 | 605 | [49] |
| mBCl | Glutathione | 390 | 478 | [46,47,48] |
| MitoSOX Red | Mitochondrial superoxide | 510 | 580 | [50,51] |
| Srnx1-GFP reporter | Nrf2 oxidative stress response | 488 | 510 | [52] |
| CHOP-GFP reporter | Endoplasmic reticulum-stress/unfolded protein response | 488 | 510 | [52] |
| p21-GFP reporter | P53 dependent DNA damage-related signalling | 488 | 510 | [52] |
| ICAM1-GFP reporter | NF-κB-mediated pro-inflammatory cytokine signalling | 488 | 510 | [52] |
| Test Model | Probes/Reporters | Drugs | Reference |
|---|---|---|---|
| PHH | DRAQ5; TMRM; CM-H2DCFDA; mBCl | 300 DILI and non-DILI compounds | [48] |
| HepG2 | Hoechst 33342; PI; TMRM; Fluo-4 AM; BODIPY 665/676 | 78 DILI and non-DILI compounds | [13] |
| HepG2 transfected with CYP adenovirus | Hoechst 33342; PI; TMRM; Fluo-4 AM; CellROX | 15 DILI and non-DILI compounds | [45] |
| HepG2, HepG2 + S9, PHH | CM-H2DFFDA, TMRE Activated caspase-3, phosphorylated histone- H3 and HSP 70/72 assays LipidTox (Phospholipids + neutral lipids) | 144 DILI and non-DILI compounds | [60] |
| Male and female PHHs | DHE, TMRE, TOTO3, Fluo4 | 6 chemicals | [49] |
| iPSC-HLCs | MitoTracker orange, carboxy-H2DCFDA, TOTO-3, Hoechst33342 | 8 toxicants | [61] |
| HepG2 and HepaRG cells | Hoechst 33342, CM-H2DCFDA, TMRM, TOTO-3 DRAQ5, mBCl, YOYO-1 | 28 DILI and non-DILI compounds | [62] |
| HepG2 | Cellomics HCS reagent; CellROX Deep Red; Hoechst 33342 | Complex mixtures of perfluorinated, brominated, and chlorinated compounds (persistent organic pollutants) | [63] |
| HepG2 reporter lines | 11 BAC-GFP reporters containing target genes, representing the oxidative stress response pathway (KEAP1/NFR2/SRXN1), the unfolded protein response and DNA damage | 30 hepatotoxicants | [57] |
| HepG2 reporter lines | SRXN1-GFP, CHOP-GFP, p21-GFP and ICAM-GFP | 118 FDA-labelled drugs | [52] |
| HepG2 reporter lines in 3D spheroids | Srxn1-GFP, NQO1-GFP, BiP-GFP, Chop-GFP, p21-GFP and Btg2-GFP | 33 compounds | [64] |
| UHH | BODIPY 493/503; LipidTOX Red Phspholipidosis, CellROX, Fluo-4 AM, Hoechst 33342; PI; MitoSOX Red, TMRM and mBCl | 15 DILI and non-DILI compounds | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donato, M.T.; Tolosa, L. High-Content Screening for the Detection of Drug-Induced Oxidative Stress in Liver Cells. Antioxidants 2021, 10, 106. https://doi.org/10.3390/antiox10010106
Donato MT, Tolosa L. High-Content Screening for the Detection of Drug-Induced Oxidative Stress in Liver Cells. Antioxidants. 2021; 10(1):106. https://doi.org/10.3390/antiox10010106
Chicago/Turabian StyleDonato, María Teresa, and Laia Tolosa. 2021. "High-Content Screening for the Detection of Drug-Induced Oxidative Stress in Liver Cells" Antioxidants 10, no. 1: 106. https://doi.org/10.3390/antiox10010106
APA StyleDonato, M. T., & Tolosa, L. (2021). High-Content Screening for the Detection of Drug-Induced Oxidative Stress in Liver Cells. Antioxidants, 10(1), 106. https://doi.org/10.3390/antiox10010106