
SNP Analysis and Whole Exome Sequencing: Their Application in the Analysis of a Consanguineous Pedigree Segregating Ataxia

Abstract
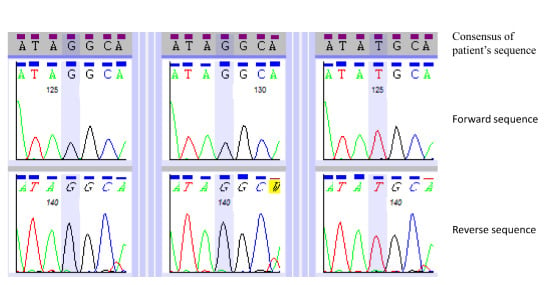
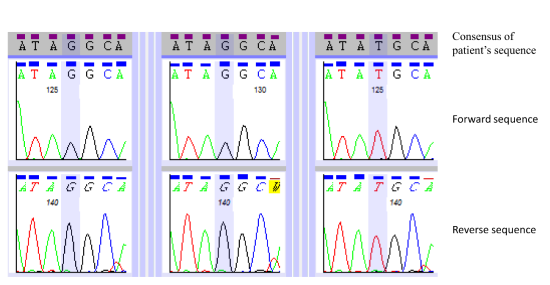
:
1. Introduction
2. Case Presentation
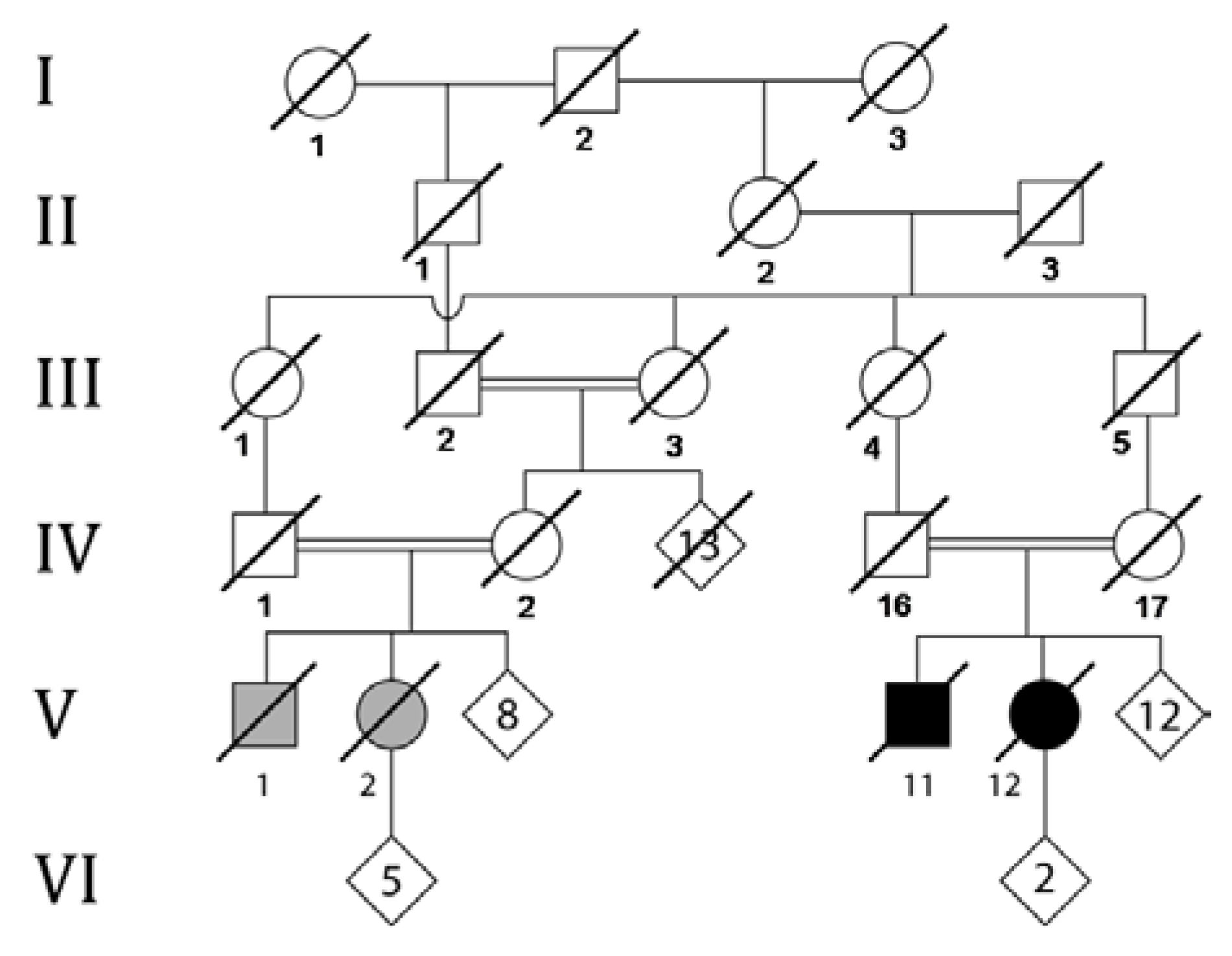
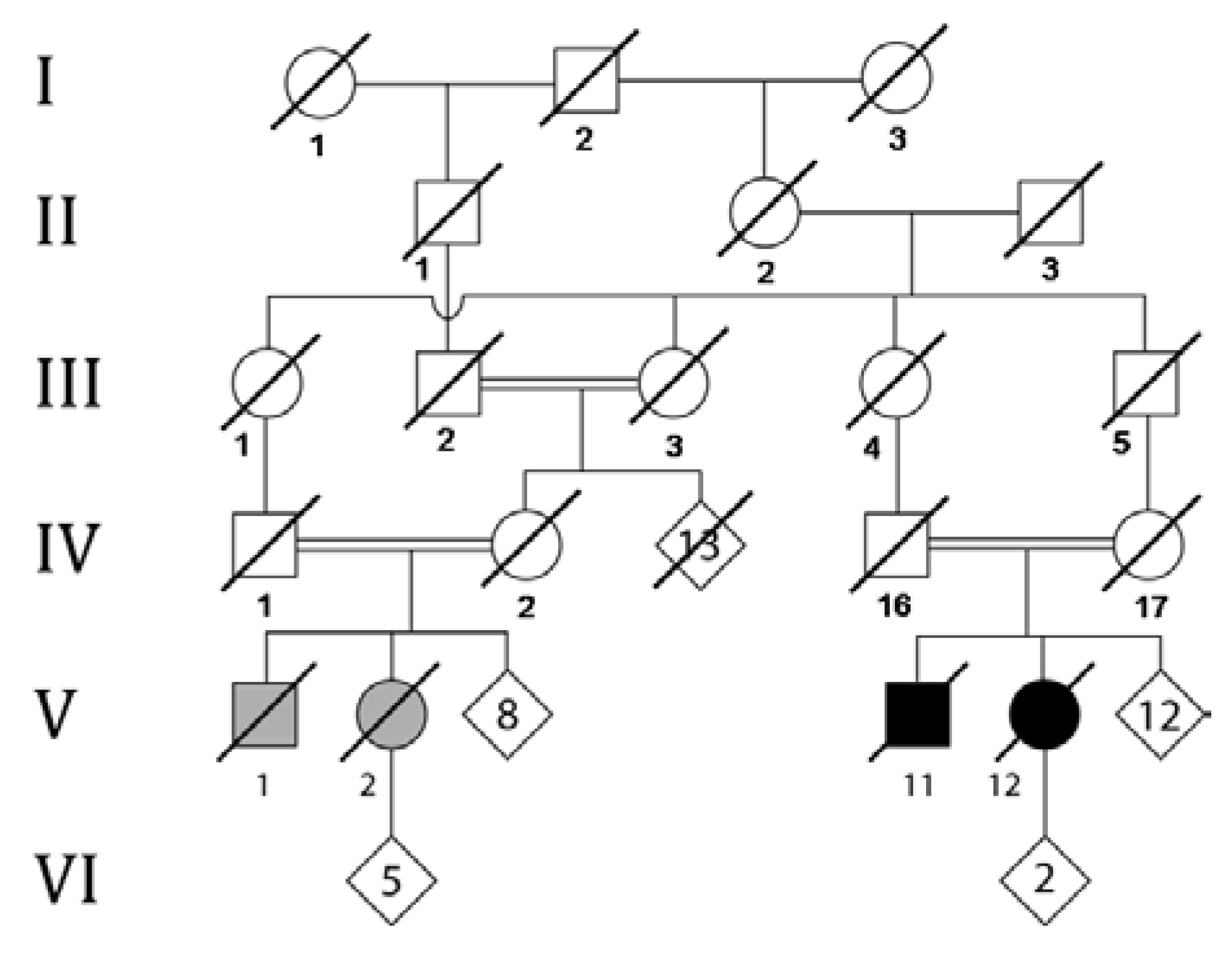
3. Molecular Studies
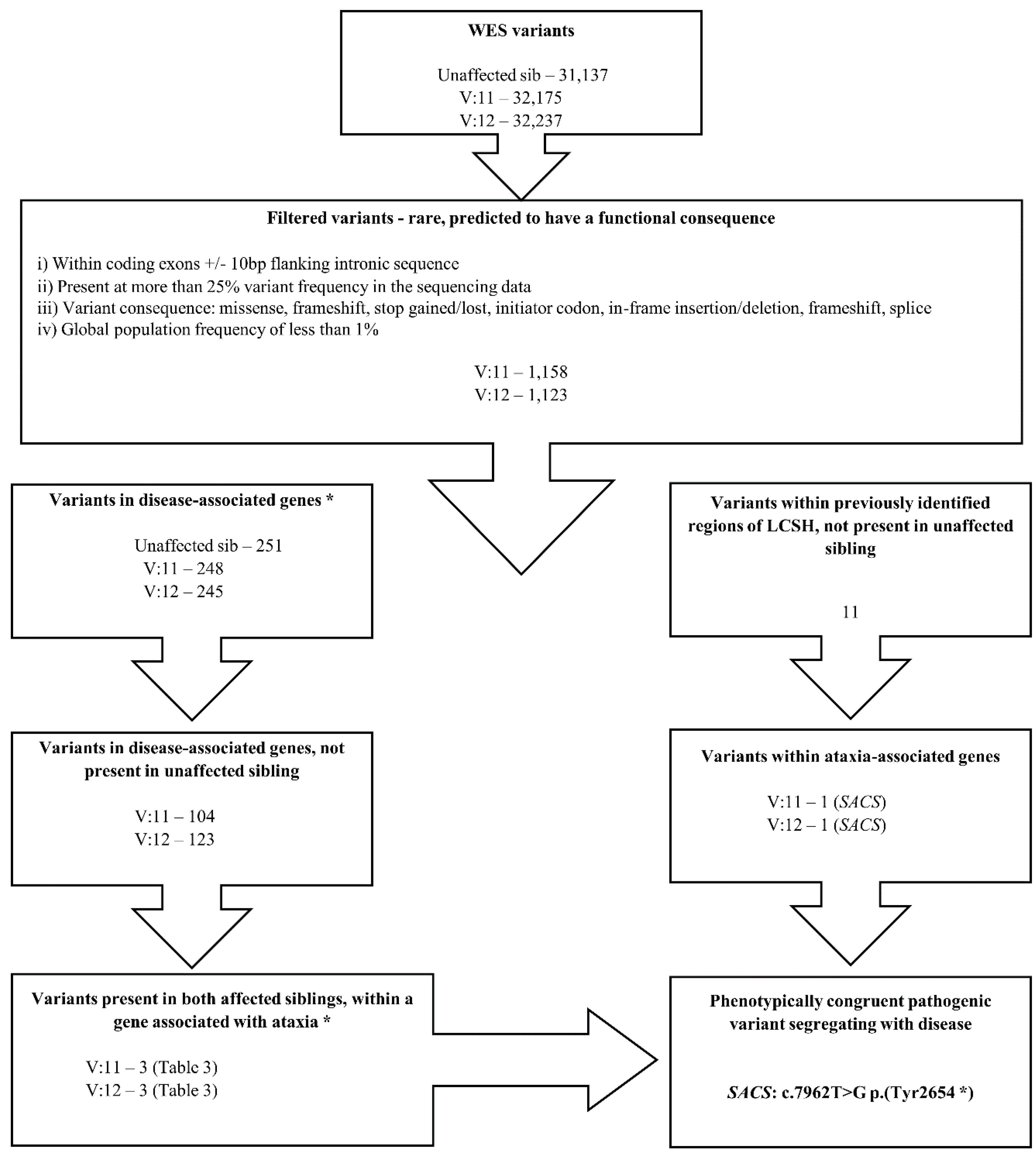
3.1. SNP Analysis
3.2. WES
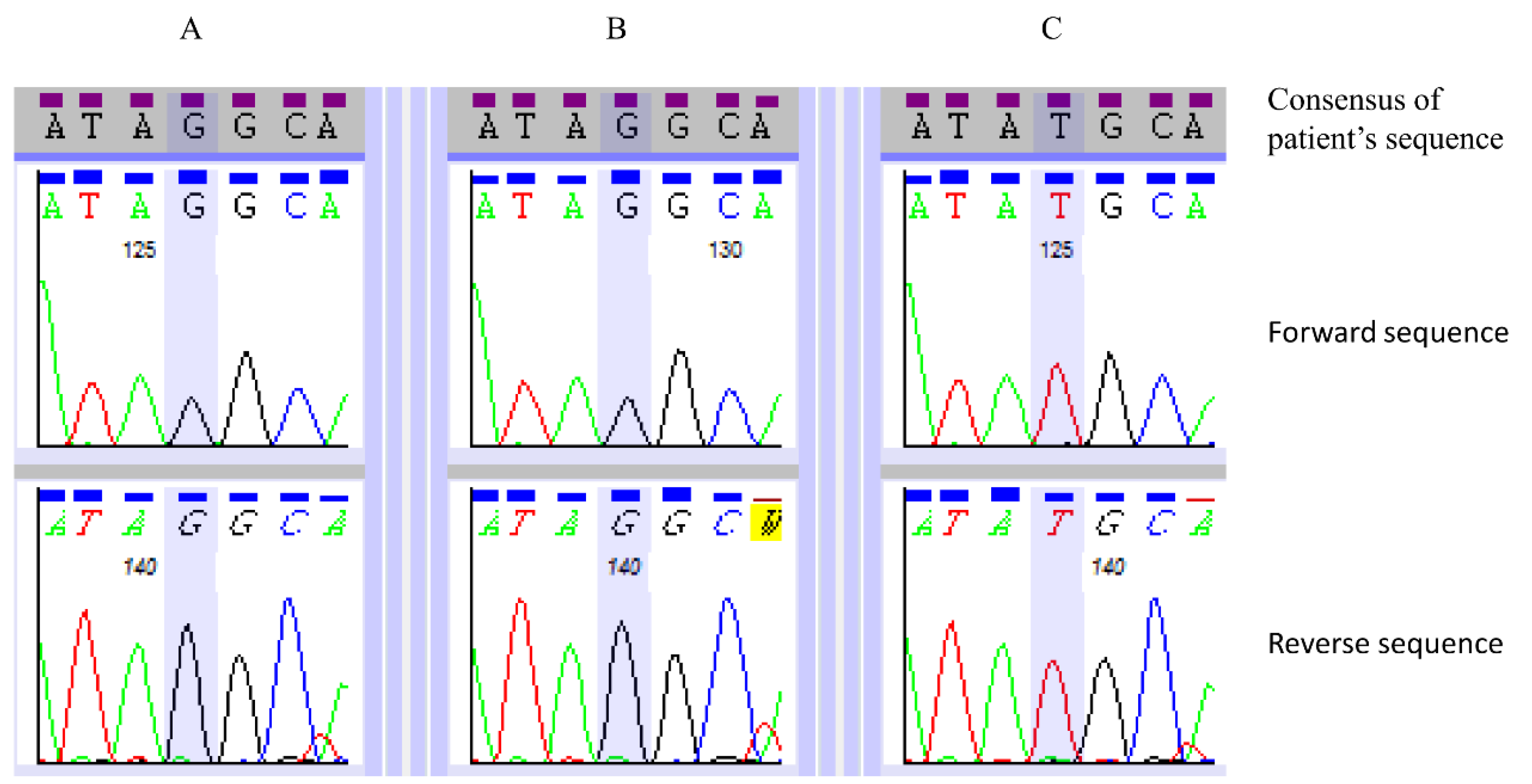
3.3. Primer Design and Sanger-based Sequencing
4. Results


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
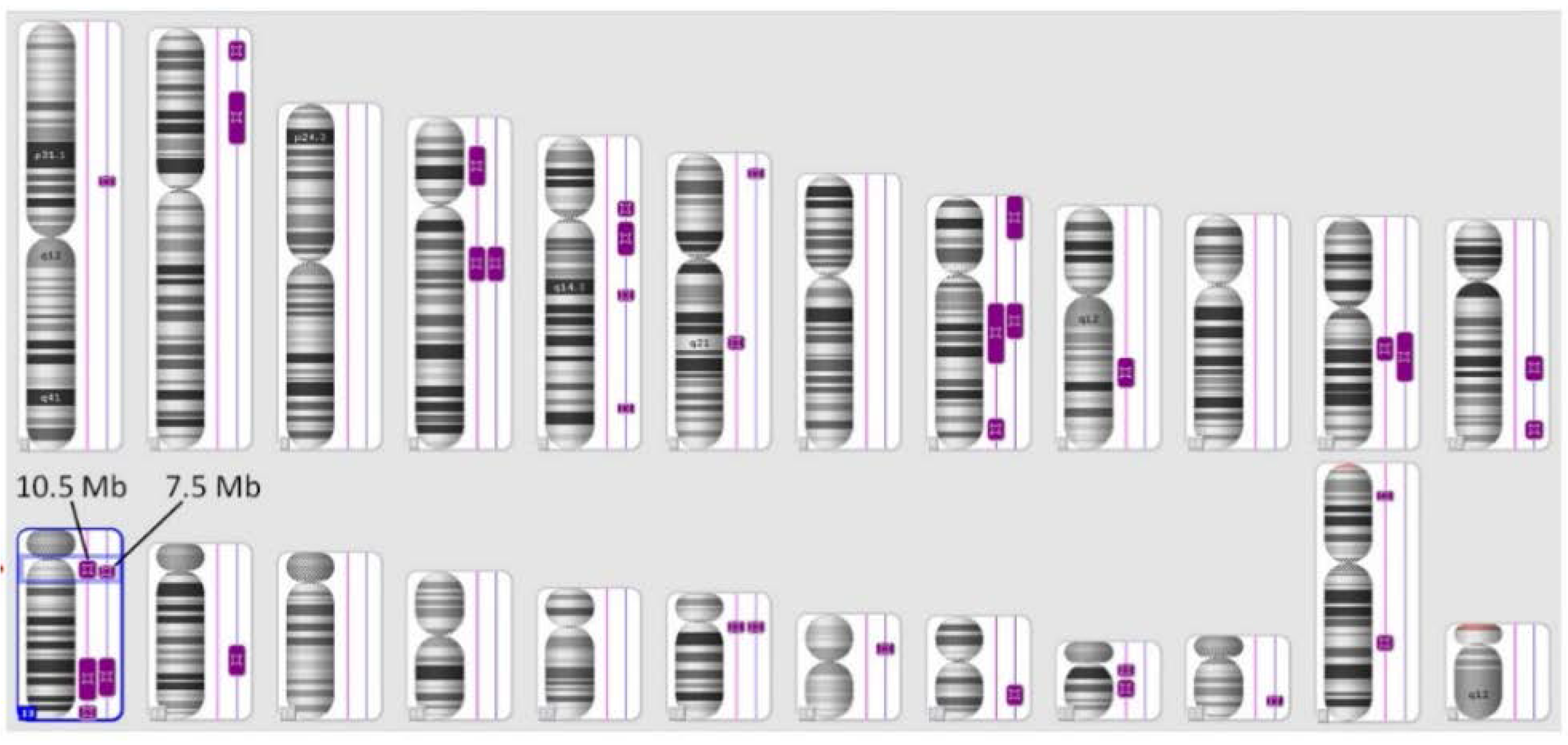
| Chromosome | Cytoband | Chromosome Coordinates (hg18) | Size (kb) | ||
|---|---|---|---|---|---|
| Start | End | Start | End | ||
| 4 | q13.3 | q22.2 | 75,140,321 | 94,467,450 | 19,327 |
| 8 | q12.2 | q21.1 | 62,360,831 | 82,468,093 | 20,107 |
| 11 | q13.3 | q14.1 | 70,171,744 | 83,268,529 | 13,097 |
| 13 | q12.11 | q12.3 | 21,382,994 | 28,897,752 | 7,515 |
| 13 | q22.3 | q32.3 | 77,509,929 | 100,244,550 | 22,735 |
| 18 | q11.1 | q11.2 | 16,803,434 | 23,248,050 | 6,445 |
| Total | - | - | - | - | 89,226 |
| Gene | Phenotype | Inheritance |
|---|---|---|
| COQ2 | Multiple-system atrophy | AR, AD |
| GRID2 | Autosomal recessive spinocerebellar ataxia-18 | AR |
| TTPA | Ataxia with isolated vitamin E deficiency | AR |
| CYP7B1 | Autosomal recessive spastic paraplegia-5A | AR |
| PEX2 | Peroxisome biogenesis disorder-5B | AR |
| SACS | Autosomal recessive spastic ataxia of Charlevoix-Saguenay | AR |
| ATP8A2 | Cerebellar ataxia, mental retardation, and disequilibrium syndrome-4 | AR |
| Gene | Nucleotide Change | Amino Acid Change | Variant State | Located in a Region of LCSH Common to Both Affected Siblings | MAF * | Comments |
|---|---|---|---|---|---|---|
| CACNA1A | NM_023035.2: c.793C>G | NP_075461.2: p.(Gln265Glu) | Het | No | 0.0 | Point mutations in CACNA1A are consistent with phenotype. Doesn’t fit apparent inheritance pattern in this pedigree. |
| SACS | NM_014363.4: c.7962T>G | NP_003045.2: p.(Tyr2654*) | Hom | Yes | 0.0 | Truncating mutation. Homozygous in both affected siblings. Consistent with phenotype. |
| ZNF592 | NM_014630.2: c.3023G>A | NP_055445.2: p.(Arg1008Gln) | Het | No | 4.523 × 10−5 | Incomplete concordance with phenotype; AR disorder, single het variant detected only. |

5. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anheim, M.; Tranchant, C.; Koenig, M. The autosomal recessive cerebellar ataxias. N. Engl. J. Med. 2012, 366, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Hamza, W.; Pacha, L.A.; Hamadouche, T.; Muller, J.; Drouot, N.; Ferrat, F.; Makri, S.; Chaouch, M.; Tazir, M.; Koenig, M.; et al. Molecular and clinical study of a cohort of 110 Algerian patients with autosomal recessive ataxia. BMC Med. Genet. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Embiruçu, E.K.; Martyn, M.L.; Schlesinger, D.; Kok, F. Autosomal recessive ataxias: 20 Types, and counting. Arq. Neuropsiquiatr. 2009, 67, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Gros-Louis, F.; Dupré, N.; Dion, P.; Fox, M.A.; Laurent, S.; Verreault, S.; Sanes, J.R.; Bouchard, J.P.; Rouleau, G.A. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat. Genet. 2007, 39, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Engert, J.C.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000, 24, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Savitsky, K.; Platzer, M.; Ziv, Y.; Helbitz, T.; Nehls, M.; Boehm, T.; Rosenthal, A.; Shiloh, Y.; Rotman, G. Genomic organization of the ATM gene. Genomics. 1996, 33, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Cytogenetics Assay Protocol. Available online: http://media.affymetrix.com/support/downloads/manuals/cyto_assay_usermanual.pdf (accessed on 28 January 2015).
- UCSC Genome Browser. Available online: http://genome.ucsc.edu (accessed on 25 April 2015).
- SNP Check 3. Available online: https://secure.ngrl.org.uk/SNPCheck/snpcheck.htm;jsessionid=5DDF10FED54806045E1D1F12B34F3017 (accessed on 21 October 2015).
- Love, J.M.; Prosser, D.; Love, D.R.; Chintakindi, K.P.; Dalal, A.B.; Aggarwal, S. A novel glycine decarboxylase gene mutation in an Indian family with nonketotic hyperglycinemia. J. Child. Neurol. 2014, 29, 122–127. [Google Scholar] [CrossRef] [PubMed]
- ExAC Browser. Available online: http://exac.broadinstitute.org (accessed on 21 October 2015).
- HGMD Pro. Available online: https://portal.biobase-international.com/cgi-bin/portal/login.cgi?redirect_url=/hgmd/pro/start.php (accessed on 21 March 2015).
- Mutalyzer 2.0.13 Name Checker. Available online: https://mutalyzer.nl/name-checker (accessed on 21 October 2015).
- Bouchard, J.P.; Barbeau, A.; Bouchard, R.; Bouchard, R.W. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci. 1978, 5, 61–69. [Google Scholar] [PubMed]
- Richter, A.; Rioux, J.D.; Bouchard, J.P.; Mercier, J.; Mathieu, J.; Ge, B.; Poirier, J.; Julien, D.; Gyapay, G.; Weissenbach, J.; et al. Location score and haplotype analyses of the locus for autosomal recessive spastic ataxia of Charlevoix-Saguenay, in chromosome region 13q11. Am. J. Hum. Genet. 1999, 64, 768–775. [Google Scholar] [CrossRef] [PubMed]
- El Euch-Fayache, G.; Lalani, I.; Amouri, R.; Turki, I.; Ouahchi, K.; Hung, W.Y.; Belal, S.; Siddique, T.; Hentati, F. Phenotypic features and genetic findings in sacsin-related autosomal recessive ataxia in Tunisia. Arch. Neurol. 2003, 60, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, C.; Banfi, S.; Orio, M.; Gasparini, P.; Monticelli, A.; Scarano, V.; Santorelli, F.M.; Perretti, A.; Santoro, L.; De Michele, G.; et al. A novel mutation in SACS gene in a family from southern Italy. Neurology 2004, 62, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, S.; Meijer, R.P.; Pijl, B.J.; Timmermans, J.; Cruysberg, J.R.; Bos, M.M.; Schelhaas, H.J.; van de Warrenburg, B.P.; Knoers, N.V.; Scheffer, H.; et al. ARSACS in the Dutch population: A frequent cause of early-onset cerebellar ataxia. Neurogenetics 2008, 9, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Sergers, K.; Bouquiaux, O.; Wang, F.C.; Janin, N.; Andris, C.; Shimazaki, H.; Sakoe, K.; Nakano, I.; Takiyama, Y. Novel SACS mutation in a Belgian family with sacsin-related ataxia. J. Neurol. Sci. 2008, 264, 73–76. [Google Scholar] [PubMed]
- Crisculo, C.; Saccà, F.; De Michele, G.; Mancini, P.; Combarros, O.; Intante, J.; Garcia, A.; Banfi, S.; Filla, A.; Berciano, J. Novel mutation of SACS gene in a Spanish family with autosomal recessive spastic ataxia. Mov. Disord. 2005, 20, 1358–1361. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Ozgul, R.K.; Poisson, V.C.; Topaloglu, H. Private SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) families from Turkey. Neurogenetics. 2004, 5, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Novel SACS mutations identified by whole exome sequencing in a Norwegian family with autosomal recessive spastic ataxia of Charlevoix-Saguenay. PLoS ONE 2013, 8, e66145.
- Takado, Y.; Hara, K.; Shimohata, T.; Tokiguchi, S.; Onodera, O.; Nishizawa, M. New mutation in the non-gigantic exon of SACS in Japanese siblings. Mov. Disord. 2007, 22, 748–749. [Google Scholar] [CrossRef] [PubMed]
- Anheim, M.; Fleury, M.; Monga, B.; Laugel, V.; Chaigne, D.; Rodier, G.; Ginglinger, E.; Boulay, C.; Courtois, S.; Drouot, N.; et al. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: Implications for clinical management. Neurogenetics 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, H.; Takiyama, Y.; Sakoe, K.; Ando, Y.; Nakano, I. A phenotype without spasticity in sacsin-related ataxia. Neurology 2005, 64, 2129–2131. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, H.; Sakoe, K.; Niijima, K.; Nakano, I.; Takiyama, Y. An unusual case of a spasticity-lacking phenotype with a novel SACS mutation. J. Neurol. Sci. 2007, 255, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Baets, J.; Deconinck, T.; Smets, K.; Goossens, D.; Van den Bergh, P.; Dahan, K.; Schmedding, E.; Santens, P.; Rasic, V.M.; Van Damme, P.; et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 2010, 28, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, W.M.; Egger, J.I.; Ahmed, A.I.; Kremer, B.P.; Vermeer, S.; van de Warrenburg, B.P. Cerebellar cognitive affective syndrome and autosomal recessive spastic of Charlevoix-Saguenay: A report of two male sibs. Psychopathology 2012, 45, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Mignarri, A.; Tessa, A.; Carluccio, M.A.; Rufa, A.; Stort, E.; Bonelli, G.; Marcotulli, C.; Santorelli, F.M.; Leonardi, L.; Casali, C. Cerebellum and neuropsychiatric disorders: Insights from ARSACS. Neurol. Sci. 2014, 35, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, J.; Richter, A.; Melancon, S.; Mathieu, J.; Michaud, J. Autosomal recessive spastic ataxia (Charlevoic-Saguenay). In Handbook of Ataxia Disorders; Klockgether, K., Ed.; Marcel Dekker: New York, NY, USA, 2000; pp. 311–324. [Google Scholar]
- Martin, M.H.; Bouchard, J.P.; Sylvain, M.; St-Onge, O.; Truchon, S. Autosomal recessive spastic ataxia of Charlevoic-Saguenay: A report of MR imaging in 5 patients. Am. J. Neuroradiol. 2007, 28, 1606–1608. [Google Scholar] [CrossRef] [PubMed]
- Engert, J.C.; Doré, C.; Mercier, J.; Ge, B.; Bétard, C.; Rioux, J.; Owen, C.; Bérubé, P.; Devon, K.; Birren, B.; et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): High-resolution physical and transcript map of the candidate region in chromosome region 13q11. Genomics 1999, 62, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Mrissa, N.; Belal, S.; Hamida, C.B.; Amouri, R.; Turki, I.; Mrissa, R.; Hamida, M.B.; Hentati, F. Linkage to chromosome 13q11–12 of an autosomal recessive cerebellar ataxia in a Tunisian family. Neurology 2000, 54, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Larivière, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; McKinney, R.A.; Shoubridge, E.A.; et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoic-Saguenay. Hum. Mol. Genet. 2015, 24, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Ben Hamida, M.; Belal, S.; Sirugo, G.; Ben Hamida, C.; Panayides, K.; Ionannou, P.; Beckman, J.; Mandel, J.L.; Hentati, F.; Koenig, M.; et al. Friedreich’s ataxia phenotype not linked to chromosome 9 and associated with selective autosomal recessive vitamin E deficiency in two inbred Tunisian families. Neurology 1993, 43, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- H’mida-Ben Brahim, D.; M’zahem, A.; Assoum, M.; Bouhlal, F.; Fattori, F.; Anheim, M.; Ali-Pacha, L.; Ferrat, F.; Chaouch, M.; Lagier-Tourenne, C.; et al. Molecular diagnosis of known recessive ataxias by homozygosity mapping with SNP arrays. J. Neurol. 2011, 258, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B. Exome sequencing: a transformative technology. Lancet Neurol. 2011, 10, 942–946. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nickerson, S.L.; Marquis-Nicholson, R.; Claxton, K.; Ashton, F.; Leong, I.U.S.; Prosser, D.O.; Love, J.M.; George, A.M.; Taylor, G.; Wilson, C.; et al. SNP Analysis and Whole Exome Sequencing: Their Application in the Analysis of a Consanguineous Pedigree Segregating Ataxia. Microarrays 2015, 4, 490-502. https://doi.org/10.3390/microarrays4040490
Nickerson SL, Marquis-Nicholson R, Claxton K, Ashton F, Leong IUS, Prosser DO, Love JM, George AM, Taylor G, Wilson C, et al. SNP Analysis and Whole Exome Sequencing: Their Application in the Analysis of a Consanguineous Pedigree Segregating Ataxia. Microarrays. 2015; 4(4):490-502. https://doi.org/10.3390/microarrays4040490
Chicago/Turabian StyleNickerson, Sarah L., Renate Marquis-Nicholson, Karen Claxton, Fern Ashton, Ivone U. S. Leong, Debra O. Prosser, Jennifer M. Love, Alice M. George, Graham Taylor, Callum Wilson, and et al. 2015. "SNP Analysis and Whole Exome Sequencing: Their Application in the Analysis of a Consanguineous Pedigree Segregating Ataxia" Microarrays 4, no. 4: 490-502. https://doi.org/10.3390/microarrays4040490