The Robustness of Pathway Analysis in Identifying Potential Drug Targets in Non-Small Cell Lung Carcinoma

Abstract
:1. Introduction
2. Experimental Section

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Title | Number of Arrays | Date |
|---|---|---|---|
| E-GEOD-6044 [20] | Transcription profiling of human lung cancers | 47 | 14 June 2008 |
| E-GEOD-18842 [21] | Transcription profiling of NSCLC | 91 | 1 October 2010 |
| E-GEOD-19188 [21] | Transcription profiling of human lung cancer | 156 | 28 May 2010 |
| E-GEOD-40275 [22] | Gene expression of normal lung tissue and patients with SCLC or NSCLC | 84 | 25 August 2012 |
| E-GEOD-43458 [23] | Gene expression profiling of lung adenocarcinomas and normal lung | 110 | 6 August 2013 |
| E-GEOD-50081 [24] | Validation of histology independent prognostic signature for early stage NSCLC | 181 | 22 September 2013 |
3. Results and Discussion
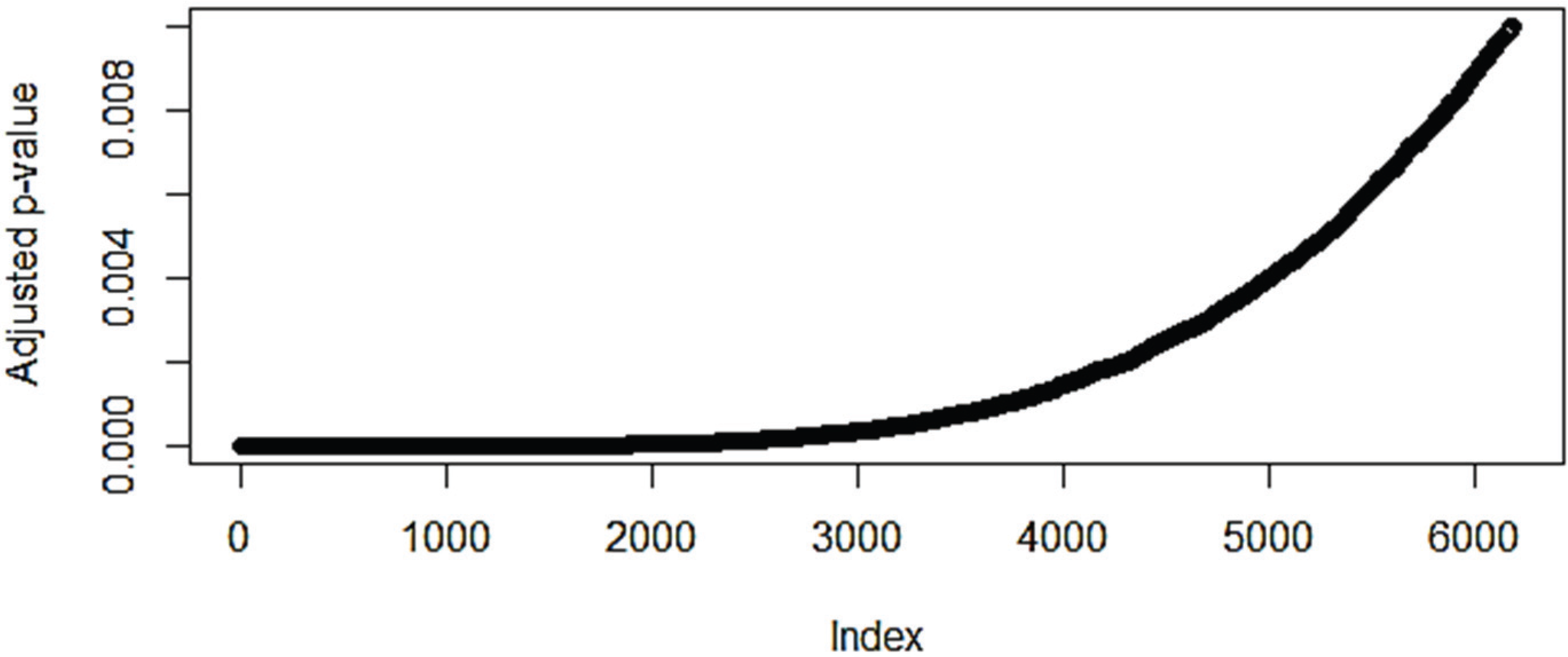
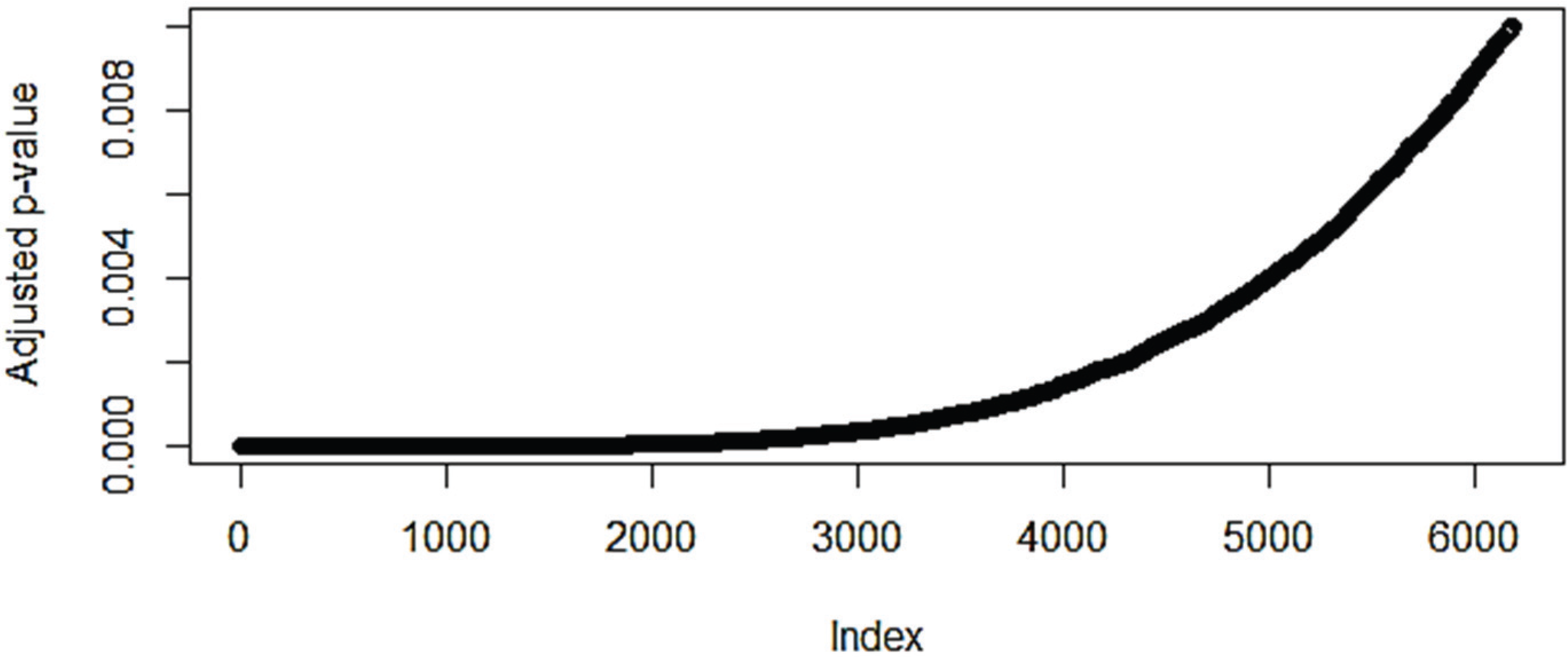
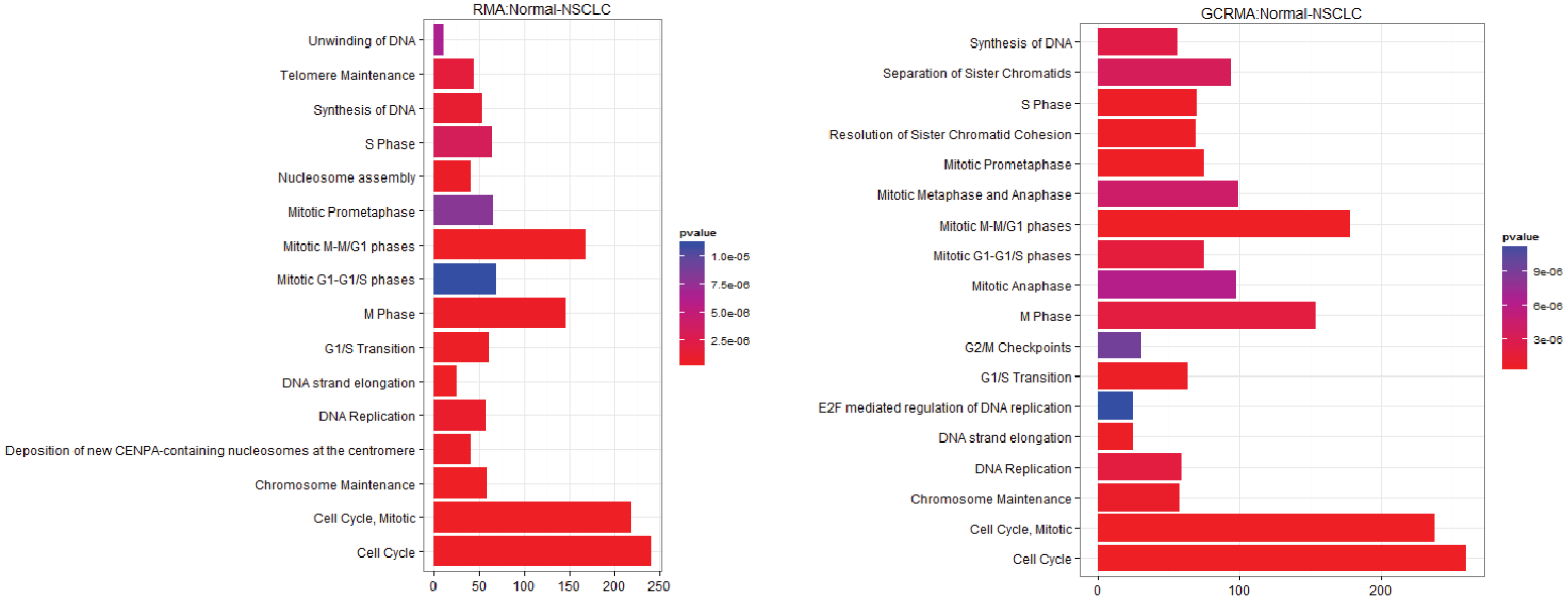
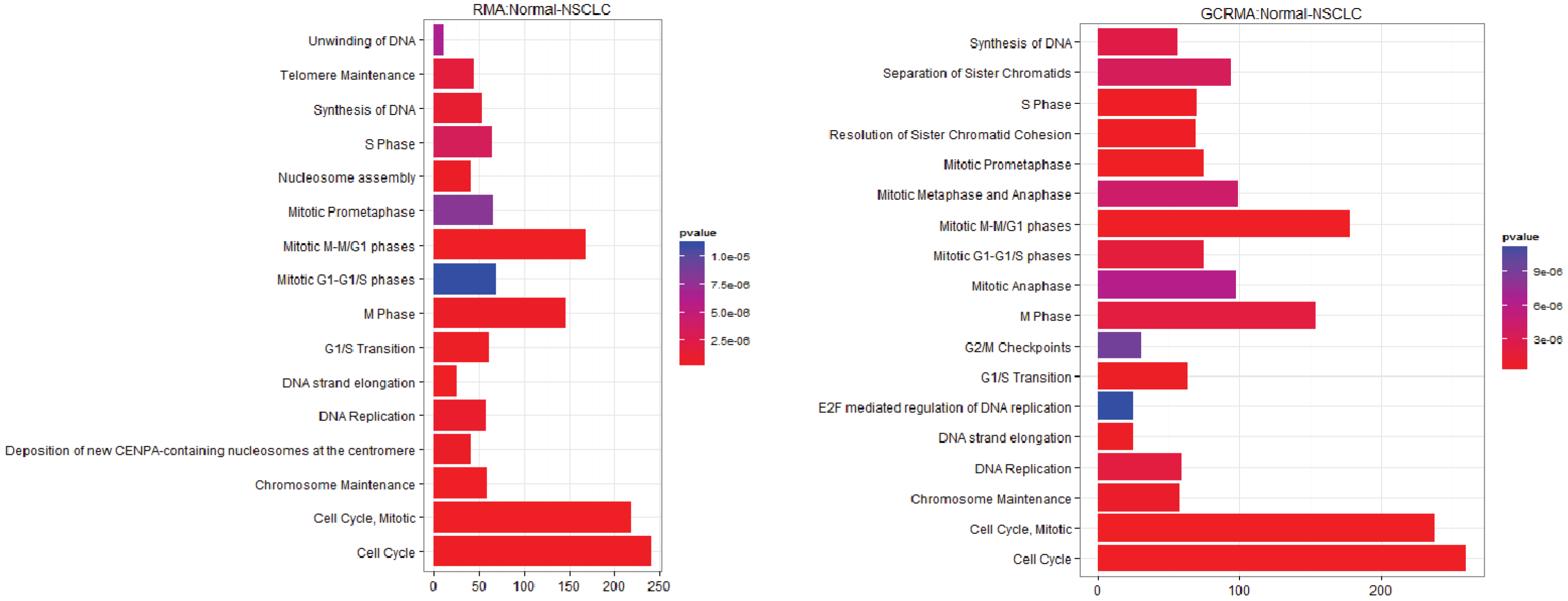
3.1. The Effect of Normalization on the Number of Differentially Expressed Genes
| Dataset | Conditions | Number of Probes | Number of EntrezIDs | ||||
|---|---|---|---|---|---|---|---|
| rma | gcrma | farms | rma | gcrma | farms | ||
| E-GEOD-6044 | Normal-Adenocarcinma | 235 | 260 | 196 | 251 | 293 | 209 |
| Normal-Small | 556 | 554 | 482 | 591 | 603 | 516 | |
| Normal-Squamous | 341 | 347 | 248 | 368 | 388 | 264 | |
| Adenocarcinoma-Squamous | 763 | 556 | 579 | 821 | 593 | 622 | |
| E-GEOD-18842 | Normal-NSCLC | 6497 | 5951 | 5520 | 6481 | 6028 | 5599 |
| E-GEOD-19188 | Healthy-Tumor | 29,727 | 20,904 | 17,242 | 31,998 | 22,636 | 18,741 |
| Healthy-Tumor | 2000 | 2000 | 2000 | 2135 | 2132 | 2110 | |
| E-GEOD-40275 | Normal-Adenocarcinoma | 13,255 | 16,387 | ||||
| 2000 | 2418 | ||||||
| Normal-Small Cell | 14,942 | 18,559 | |||||
| 1500 | 1947 | ||||||
| Normal-Metastatic | 7132 | 8897 | |||||
| 2000 | 2492 | ||||||
| Normal-Squamous | 11,543 | 14,339 | |||||
| 2000 | 2455 | ||||||
| Adenocarcinoma-Squamous | 274 | 362 | |||||
| Adenocarcinoma-Metastatic | 6619 | 8278 | |||||
| 2000 | 2455 | ||||||
| E-GEOD-43458 | Normal-Adenocarcinoma | 12,800 | 7099 | 14,186 | 7734 | ||
| E-GEOD-50081 | Adenocarcinoma-Squamous | 7769 | 6227 | 6181 | 8393 | 6728 | 6643 |
| 2000 | 2000 | 2000 | 2121 | 2132 | 2148 | ||
| Adenocarcinoma-Mixed | 231 | 437 | 168 | 249 | 463 | 186 | |
| Squamous-Mixed | 1 | 42 | 0 | 1 | 44 | 0 | |
3.2. Pathway Analysis
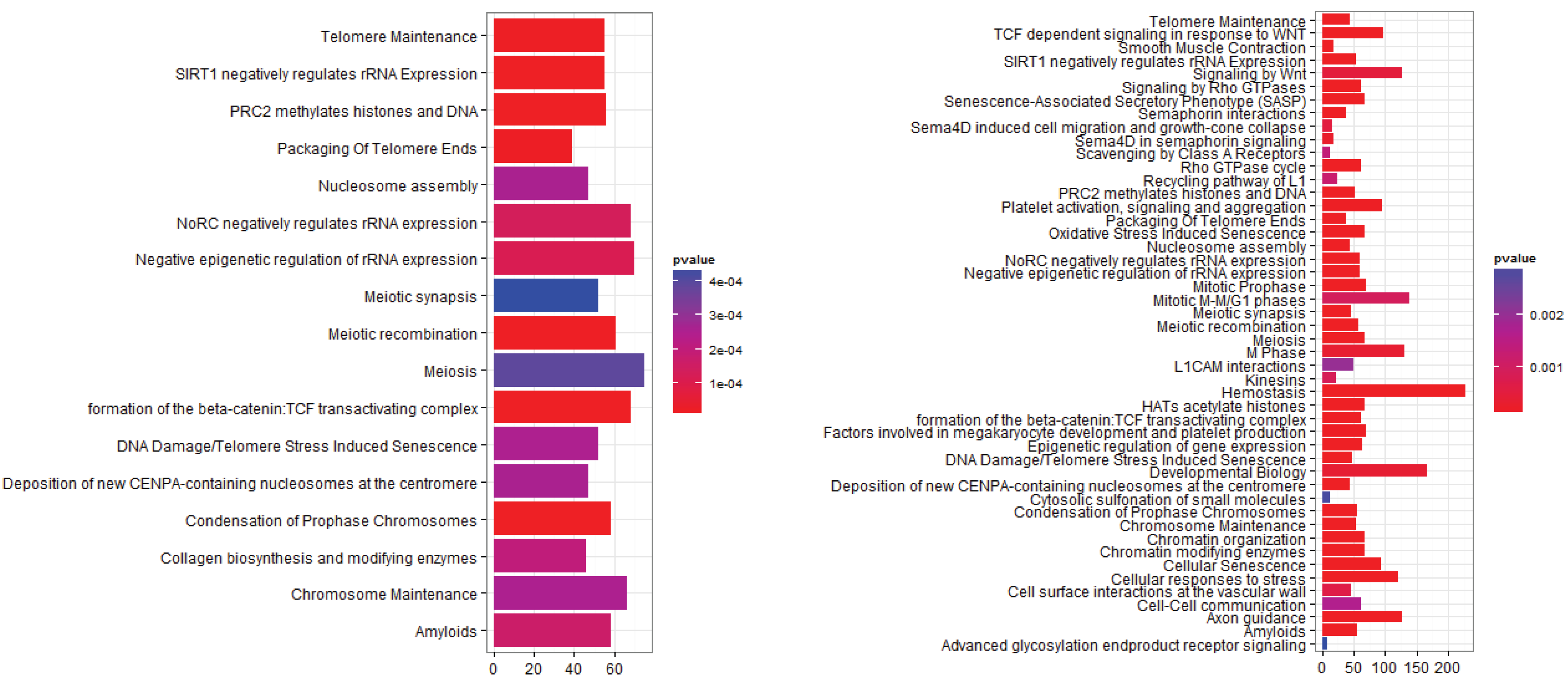
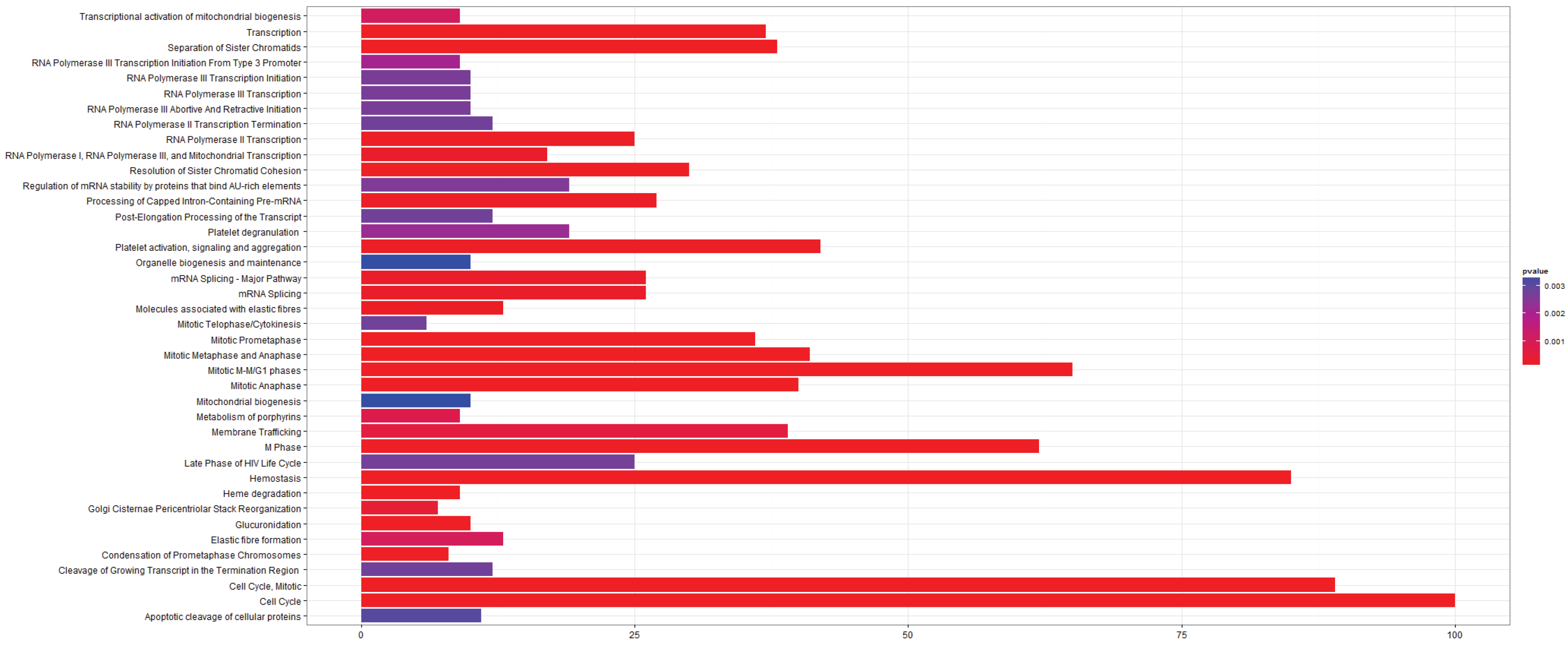
3.2.1. Pathways that Are Differentially Expressed between Normal Lung Tissue and Tumors

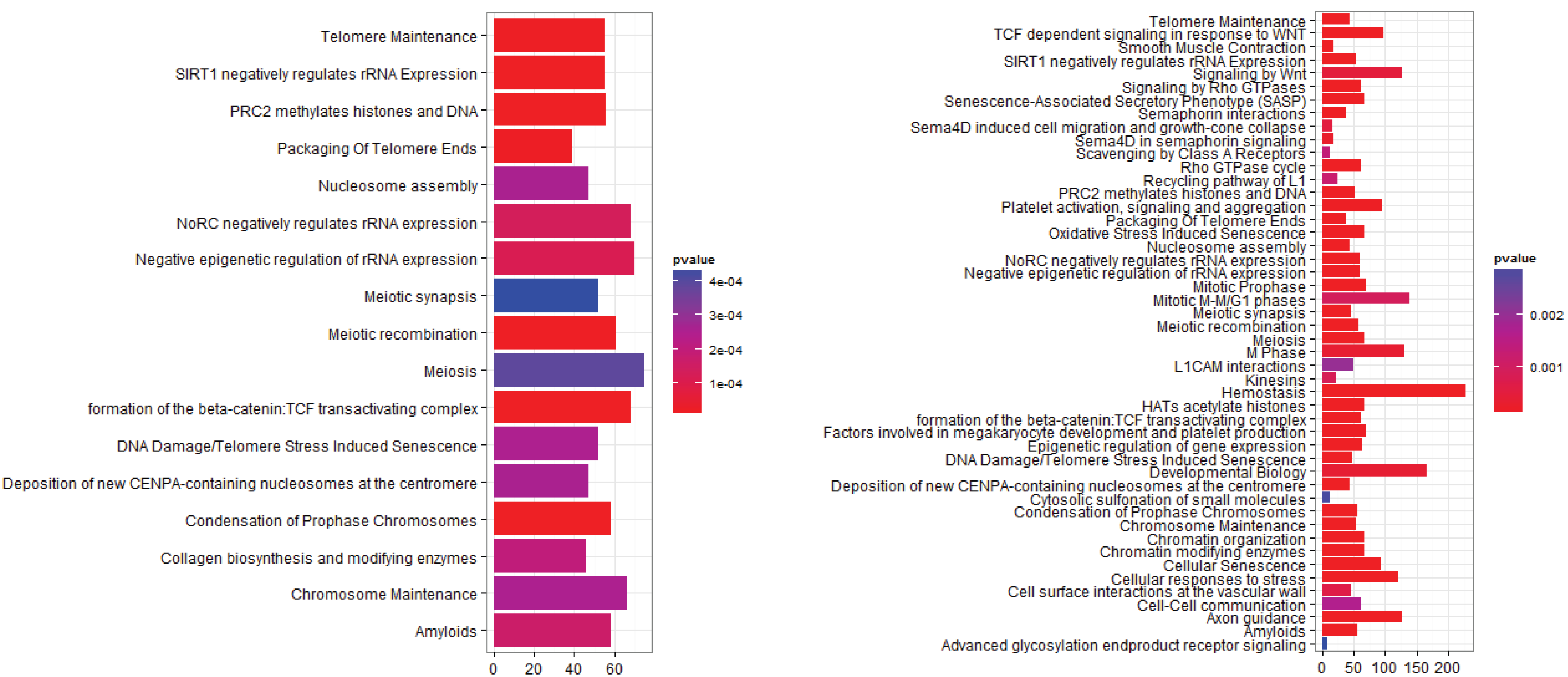
3.2.2. Pathways that Are Differentially Expressed between Normal Lung Tissue and Adenocarcinoma
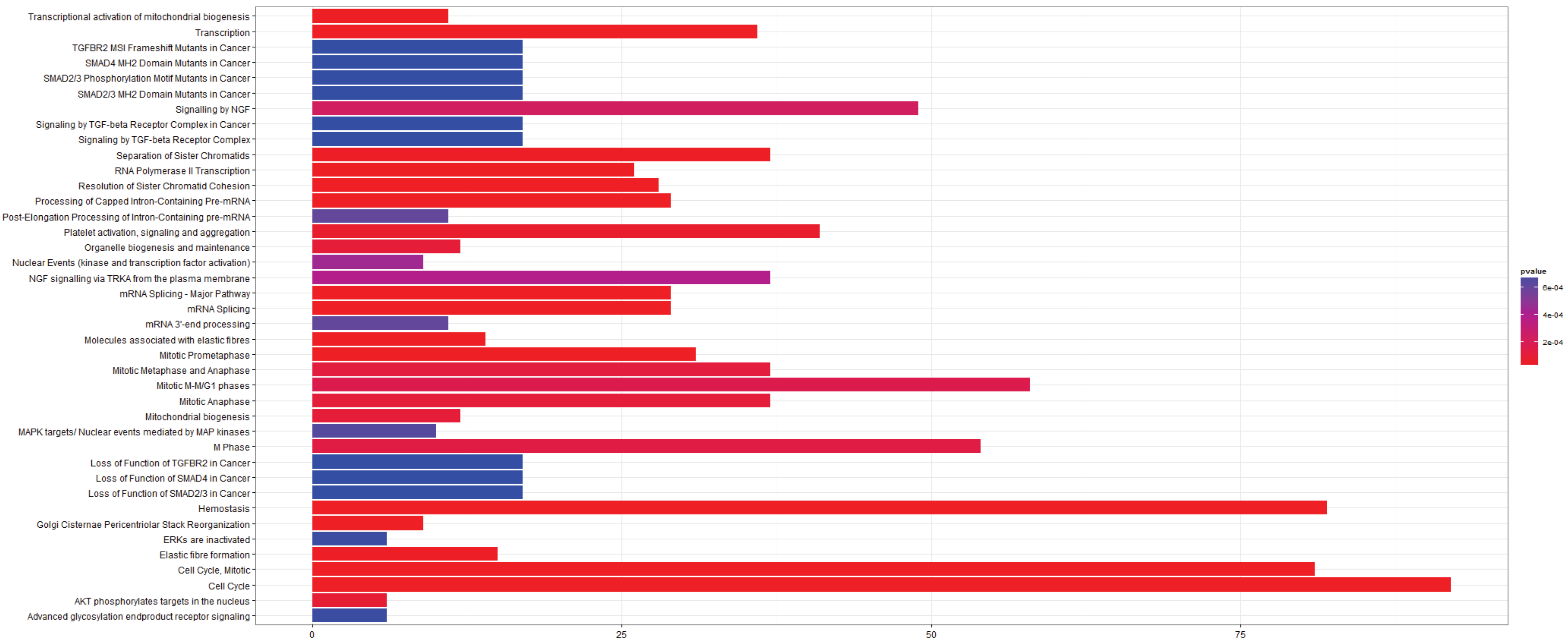
3.2.3. Pathways that Are Differentially Expressed between Normal Lung Tissue and Squamous Cell Carcinoma
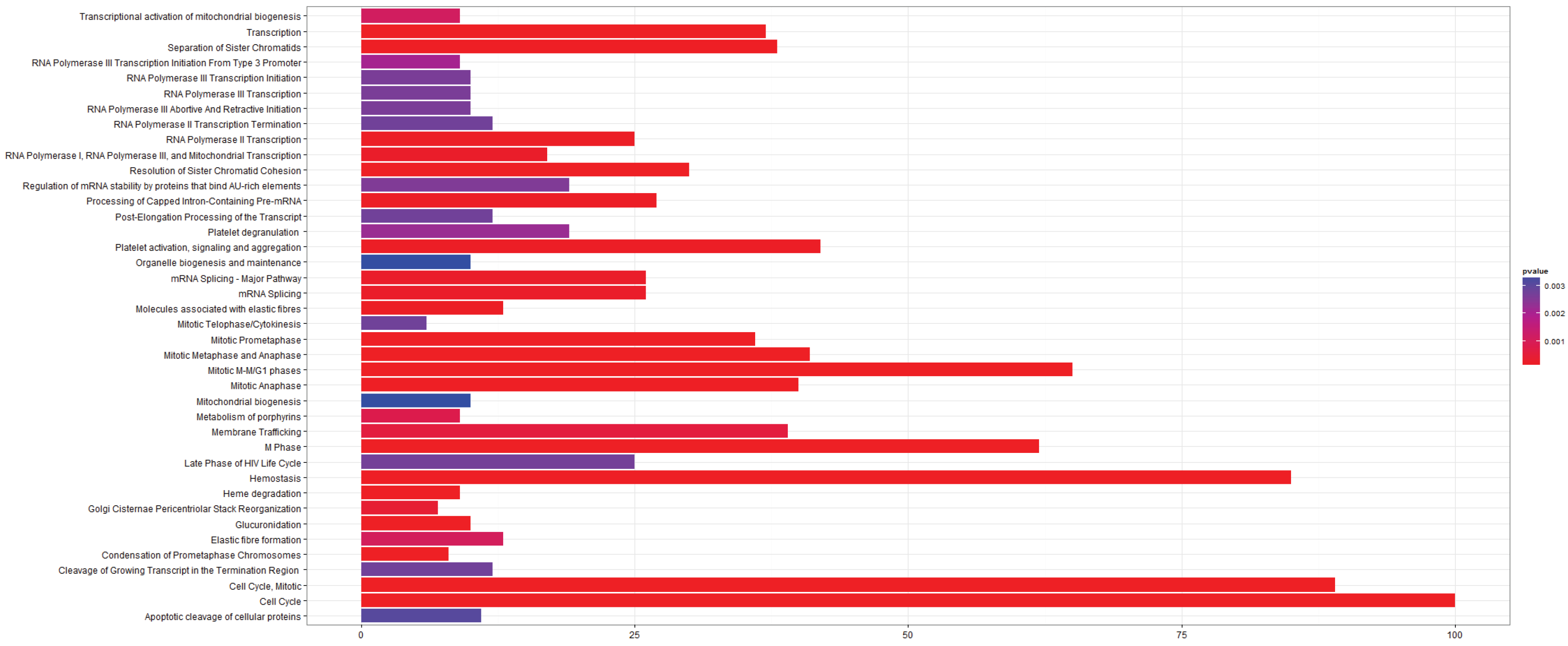
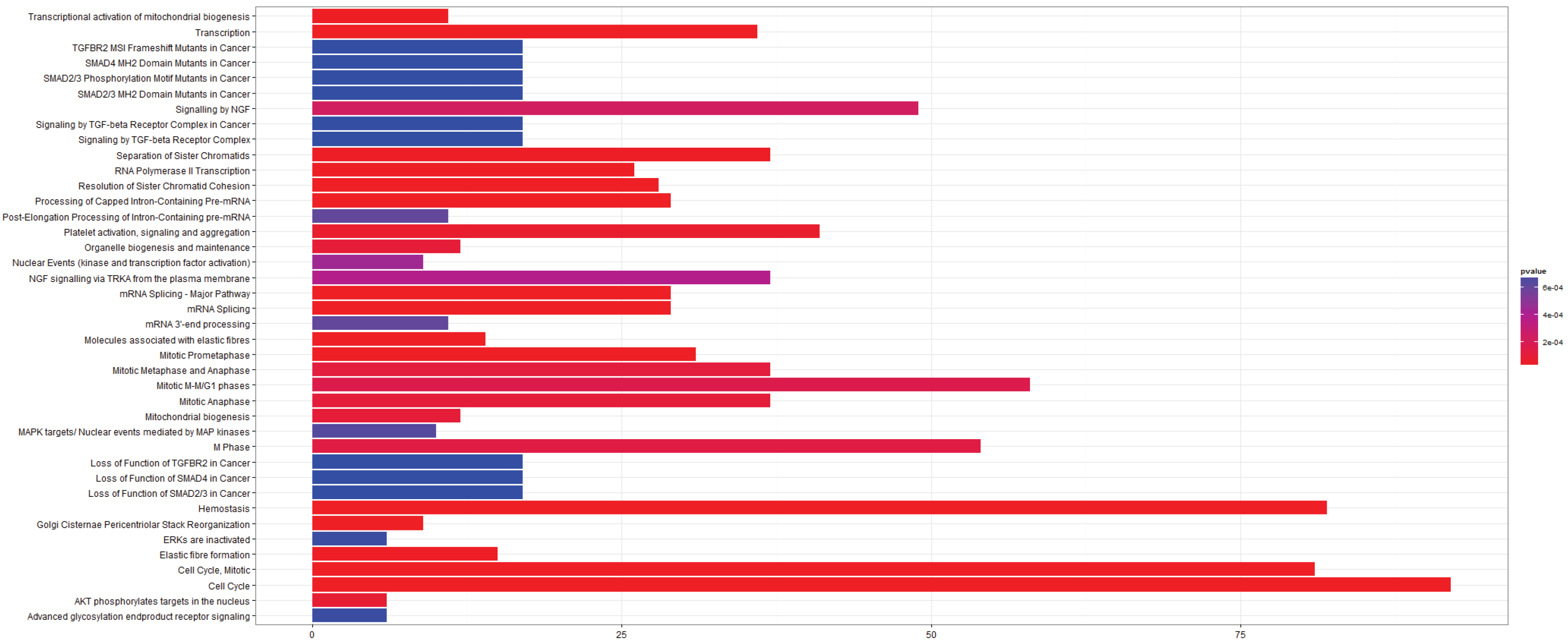
3.2.4. Pathways that Are Differentially Expressed between Adenocarcinoma and Squamous Cell Carcinoma
4. Conclusions
Supplementary Files
Supplementary File 1Author Contributions
Conflicts of Interest
References
- Support, M.C. Living after Diagnosis: Median Cancer Survival Times. Available online: http://www.macmillan.org.uk/Documents/AboutUs/Newsroom/LivingAfterCancerMedianCancerSurvivalTimes.pdf (accessed on 24 November 2011).
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Brambilla, E.; Travis, W.D.; Colby, T.; Corrin, B.; Shimosato, Y. The new world health organization classification of lung tumours. Eur. Respir. J. 2001, 18, 1059–1068. [Google Scholar] [CrossRef]
- Kuner, R. Lung cancer gene signatures and clinical perspectives. Microarrays 2013, 2, 318–339. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Perez-Moreno, P.; Brambilla, E.; Thomas, R.; Soria, J.C. Squamous cell carcinoma of the lung: Molecular subtypes and therapeutic opportunities. Clin. Cancer Res. 2012, 18, 2443–2451. [Google Scholar]
- The Clinical Lung Cancer Genome Project (CLCGP); Network Genomic Medicine (NGM). A genomics-based classification of human lung tumors. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Kendziorski, C.; Irizarry, R.; Chen, K.-S.; Haag, J.; Gould, M. On the utility of pooling biological samples in microarray experiments. Proc. Natl. Acad. Sci. USA 2005, 102, 4252–4257. [Google Scholar] [CrossRef]
- Churchill, G.A. Fundamentals of experimental design for cdna microarrays. Nat. Genet. 2002, 32, 490–495. [Google Scholar] [CrossRef]
- Allison, D.B.; Cui, X.; Page, G.P.; Sabripour, M. Microarray data analysis: From disarray to consolidation and consensus. Nat. Rev. Genet. 2006, 7, 55–65. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X. Distinct types of diffuse large b-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Aguilar-Ruiz, J.S.; Azuaje, F. Knowledge discovery in lymphoma cancer from gene–expression. In Intelligent Data Engineering and Automated Learning–Ideal 2004; Springer: Berlin, Germany, 2004; pp. 31–38. [Google Scholar]
- Knudsen, S. Guide to Analysis of DNA Microarray Data; John Wiley & Sons: Hokoben, NJ, USA, 2005. [Google Scholar]
- Stekel, D. Microarray Bioinformatics; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef]
- Dalby, A.R.; Emam, I.; Franke, R. Analysis of gene expression data from non-small cell lung carcinoma cell lines reveals distinct sub-classes from those identified at the phenotype level. PLoS One 2012, 7, e50253. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar]
- Parkinson, H.; Kapushesky, M.; Kolesnikov, N.; Rustici, G.; Shojatalab, M.; Abeygunawardena, N.; Berube, H.; Dylag, M.; Emam, I.; Farne, A. Arrayexpress update—From an archive of functional genomics experiments to the atlas of gene expression. Nucleic Acids Res. 2009, 37, D868–D872. [Google Scholar] [CrossRef]
- Rohrbeck, A.; Neukirchen, J.; Rosskopf, M.; Pardillos, G.G.; Geddert, H.; Schwalen, A.; Gabbert, H.E.; von Haeseler, A.; Pitschke, G.; Schott, M. Gene expression profiling for molecular distinction and characterization of laser captured primary lung cancers. J. Transl. Med. 2008, 6, 69. [Google Scholar] [CrossRef]
- Wang, D.; Moothart, D.R.; Lowy, D.R.; Qian, X. The expression of glyceraldehyde-3-phosphate dehydrogenase associated cell cycle (gacc) genes correlates with cancer stage and poor survival in patients with solid tumors. PLoS One 2013, 8, e61262. [Google Scholar]
- Kastner, S.; Voss, T.; Keuerleber, S.; Glöckel, C.; Freissmuth, M.; Sommergruber, W. Expression of g protein-coupled receptor 19 in human lung cancer cells is triggered by entry into s-phase and supports g2–m cell-cycle progression. Mol. Cancer Res. 2012, 10, 1343–1358. [Google Scholar] [CrossRef]
- Kabbout, M.; Garcia, M.M.; Fujimoto, J.; Liu, D.D.; Woods, D.; Chow, C.W.; Mendoza, G.; Momin, A.A.; James, B.P.; Solis, L. Ets2 mediated tumor suppressive function and met oncogene inhibition in human non–small cell lung cancer. Clin. Cancer Res. 2013, 19, 3383–3395. [Google Scholar] [CrossRef]
- Der, S.D.; Sykes, J.; Pintilie, M.; Zhu, C.Q.; Strumpf, D.; Liu, N.; Jurisica, I.; Shepherd, F.A.; Tsao, M.S. Validation of a histology-independent prognostic gene signature for early-stage, non-small-cell lung cancer including stage ia patients. J. Thorac. Oncol. 2014, 9, 59–64. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A language for data analysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Berlin, Germany, 2005; pp. 397–420. [Google Scholar]
- Yu, G. Reactome pathway analysis. Homo 2012, 1266738, 1266738. [Google Scholar]
- González-Barón, M.; Fernández-Chacón, M.; Fernández-Chacón, J.; Ordóñez, A.; Girón, C. Hemostasis and cancer. Rev. Clin. Esp. 1989, 184, 84–87. [Google Scholar]
- Garnier, D.; Magnus, N.; D’Asti, E.; Hashemi, M.; Meehan, B.; Milsom, C.; Rak, J. Genetic pathways linking hemostasis and cancer. Thromb. Res. 2012, 129, S22–S29. [Google Scholar]
- Jain, S.; Harris, J.; Ware, J. Platelets linking hemostasis and cancer. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2362–2367. [Google Scholar] [CrossRef]
- Weinberg, R. The Biology of Cancer; Garland Science: London, UK, 2013. [Google Scholar]
- Smits, V.A.; Klompmaker, R.; Arnaud, L.; Rijksen, G.; Nigg, E.A.; Medema, R.H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2000, 2, 672–676. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. [Google Scholar] [CrossRef]
- Wilsker, D.; Bunz, F. Loss of ataxia telangiectasia mutated– and rad3-related function potentiates the effects of chemotherapeutic drugs on cancer cell survival. Mol. Cancer Ther. 2007, 6, 1406–1413. [Google Scholar] [CrossRef]
- Herman-Antosiewicz, A.; Stan, S.D.; Hahm, E.R.; Xiao, D.; Singh, S.V. Activation of a novel ataxia-telangiectasia mutated and rad3 related/checkpoint kinase 1–dependent prometaphase checkpoint in cancer cells by diallyl trisulfide, a promising cancer chemopreventive constituent of processed garlic. Mol. Cancer Ther. 2007, 6, 1249–1261. [Google Scholar] [CrossRef]
- Calvo, M.B.; Figueroa, A.; Pulido, E.G.; Campelo, R.G.; Aparicio, L.A. Potential role of sugar transporters in cancer and their relationship with anticancer therapy. Int. J. Endocrinol. 2010, 2010, 205357. [Google Scholar] [CrossRef]
- Raveh, S.; Gavert, N.; Ben-Ze’ev, A. L1 cell adhesion molecule (l1cam) in invasive tumors. Cancer Lett. 2009, 282, 137–145. [Google Scholar] [CrossRef]
- Zhang, H.; Wong, C.; Wei, H.; Gilkes, D.; Korangath, P.; Chaturvedi, P.; Schito, L.; Chen, J.; Krishnamachary, B.; Winnard, P. Hif-1-dependent expression of angiopoietin-like 4 and l1cam mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2011, 31, 1757–1770. [Google Scholar] [CrossRef]
- Cho, W.C.; Yip, T.T.; Yip, C.; Yip, V.; Thulasiraman, V.; Ngan, R.K.; Yip, T.T.; Lau, W.H.; Au, J.S.; Law, S.C. Identification of serum amyloid a protein as a potentially useful biomarker to monitor relapse of nasopharyngeal cancer by serum proteomic profiling. Clin. Cancer Res. 2004, 10, 43–52. [Google Scholar] [CrossRef]
- Biran, H.; Friedman, N.; Neumann, L.; Pras, M.; Shainkin-Kestenbaum, R. Serum amyloid A (SAA) variations in patients with cancer: Correlation with disease activity, stage, primary site, and prognosis. J. Clin. Pathol. 1986, 39, 794–797. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. RHO–gtpases and cancer. Nat. Rev. Cancer 2002, 2, 133–142. [Google Scholar] [CrossRef]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Van Noort, M.; Clevers, H. TCF transcription factors, mediators of wnt-signaling in development and cancer. Dev. Biol. 2002, 244, 1–8. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P. The β-catenin/tcf-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef]
- Golembesky, A.K.; Gammon, M.D.; North, K.E.; Bensen, J.T.; Schroeder, J.C.; Teitelbaum, S.L.; Neugut, A.I.; Santella, R.M. Peroxisome proliferator-activated receptor-alpha (PPARA) genetic polymorphisms and breast cancer risk: A long island ancillary study. Carcinogenesis 2008, 29, 1944–1949. [Google Scholar] [CrossRef]
- Tsubouchi, Y.; Sano, H.; Kawahito, Y.; Mukai, S.; Yamada, R.; Kohno, M.; Inoue, K.I.; Hla, T.; Kondo, M. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor-γ agonists through induction of apoptosis. Biochem. Biophys. Res. Commun. 2000, 270, 400–405. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Richards, W.G.; Staunton, J.; Li, C.; Monti, S.; Vasa, P.; Ladd, C.; Beheshti, J.; Bueno, R.; Gillette, M. Classification of human lung carcinomas by mrna expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. USA 2001, 98, 13790–13795. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalby, A.; Bailey, I. The Robustness of Pathway Analysis in Identifying Potential Drug Targets in Non-Small Cell Lung Carcinoma. Microarrays 2014, 3, 212-225. https://doi.org/10.3390/microarrays3040212
Dalby A, Bailey I. The Robustness of Pathway Analysis in Identifying Potential Drug Targets in Non-Small Cell Lung Carcinoma. Microarrays. 2014; 3(4):212-225. https://doi.org/10.3390/microarrays3040212
Chicago/Turabian StyleDalby, Andrew, and Ian Bailey. 2014. "The Robustness of Pathway Analysis in Identifying Potential Drug Targets in Non-Small Cell Lung Carcinoma" Microarrays 3, no. 4: 212-225. https://doi.org/10.3390/microarrays3040212
APA StyleDalby, A., & Bailey, I. (2014). The Robustness of Pathway Analysis in Identifying Potential Drug Targets in Non-Small Cell Lung Carcinoma. Microarrays, 3(4), 212-225. https://doi.org/10.3390/microarrays3040212