The Neurotoxicity of Nitrous Oxide: The Facts and “Putative” Mechanisms



Abstract
:1. Introduction
2. Neurotoxicity
2.1. Perinatal Brain

{kind=link}
| Reference | Species | Age | Treatment | Duration | Outcome |
|---|---|---|---|---|---|
| [16] | Rat | PND 1–14 | 75% N2O + 0.75% iso + 9 mg/kg midazolam | 2, 4, 6 h | Most vulnerable to toxicity at PND7, least vulnerable at PND14 via both extrinsic and intrinsic apoptotic pathways |
| [17] | Rat | PND 7 | 75%N2O + 0.75% iso ± nociceptive stimulus | 6 h | Anaesthesia alone cause neurotoxicity and neurobehavioural deficits, which was exacerbated by nociceptive stimulation during anaesthesia |
| [18] | Rat | PND 7 | 75% N2O + 0.75% iso + 9 mg/kg midazolam ± melatonin | 6 h | Anaesthesia caused neurotoxicity but melatonin decreased neurotoxic damage |
| [11] | Rat | PND 7 | 75% N2O + 0.75% iso + 9 mg/kg midazolam | 2, 4, 6 h | Activates Trk-dependent (thalamus) and Trk-independent, P75NTR dependent (cortex) apoptotic cascade, as well as increasing BDNF |
| [19] | Rat | 6 mo | N2O ± ketamine | Not stated | N2O toxicity same as ketamine + N2O |
| 18 mo | N2O ± ketamine | N2O toxicity not as severe as ketamine+N2O | |||
| [20] | Rat | 6 mo | 70% N2O + 1.2% iso | 2 h | Impaired learning and memory in RAM |
| 20 mo | + 30% O2 | 2 h | Impaired learning and memory in RAM | ||
| [21] | Rat | 18 mo | 70%N2O + 30% O2 | 4 h | Impaired learning and memory in RAM |
| [22] | Rat | 6 mo or 18 mo | 70% N2O + 1.2% iso | 2 h | Aged rats had sustained learning impairment, young rats did not |
| [23] | Rhesus | PND 5–6 | 70% N2O ± 1% iso | 8 h | Alone, no neuronal damage but together caused ↑ caspase-3, Fluoro-Jade-C staining |
2.2. Aged Brain
| Reference | Species | Age | Treatment | Duration | Outcome |
|---|---|---|---|---|---|
| [30] | Rat | Adult | 150% N2O | Varied | N2O acts similar to NMDA antagonists, suggesting it is also an NMDA antagonist |
| [31] | Rat | Adult | 150% N2O | 1–16 h | Vacuoles present in PC-RSC. Maximal at 3 h+ exposure, persistent after 8 h+ exposure |
| [32] | Rat | Adult | 50% N2O | 5–80 min | Half-life of hepatic MS inactivation = 5.4 min |
| Human | 70% N2O during surgery | 30–290 min | Half-life of hepatic MS inactivation = 46 min | ||
| [33] | Human | Adult | Occupational N2O exposure | Varied | Increased N2O exposure correlates with increased oxidative DNA damage |
| [34] | Human | Adult | 70% N2O during surgery | Varied | Increased levels of DNA damage and post-operative wound infection |
| [35] | Human | 3mo | 60% N2O during surgery (case study) | 45 + 270 min | Severe cerebral atrophy, seizures, and apnoea resulting in death |
| [36] | Human | Adult | N2O during dental surgery (case study) | Not stated | Progressive numbness and ataxia, treated successfully with vitamin B12 injections |
3. Molecular Mechanisms of Action
4. Mechanisms of Neurotoxicity
4.1. NMDA Antagonism
4.2. Homocysteine Imbalance
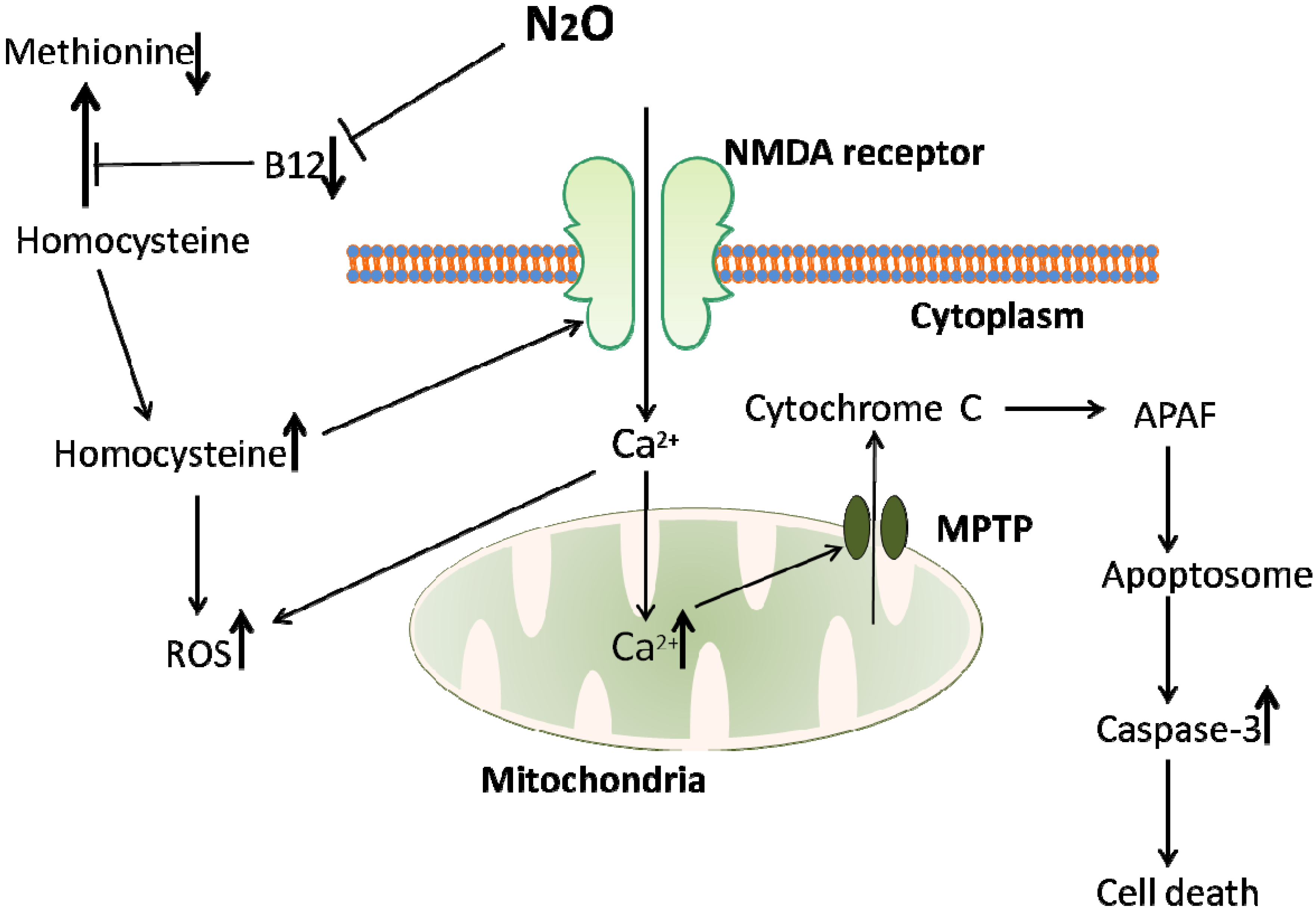
4.3. Reactive Oxygen Species and Mitochondrial Dysfunction
4.4. In Combination with Other Anaesthetics
5. Strategies to Minimize Toxicity of N2O
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Brotman, M.; Cullen, S.C. Supplementation with demerol during nitrous oxide anesthesia. Anesthesiology 1949, 10, 696–705. [Google Scholar] [CrossRef]
- Becker, D.E.; Rosenberg, M. Nitrous oxide and the inhalation anesthetics. Anesth. Prog. 2008, 55, 124–130. [Google Scholar] [CrossRef]
- Santos, M.; Kuncar, V.; Martinez-Taboada, F.; Tendillo, F.J. Large concentrations of nitrous oxide decrease the isoflurane minimum alveolar concentration sparing effect of morphine in the rat. Anesth. Analg. 2005, 100, 404–408. [Google Scholar] [CrossRef]
- Jakobsson, I.; Heidvall, M.; Davidson, S. The sevoflurane-sparing effect of nitrous oxide: A clinical study. Acta Anaesthesiol. Scand. 1999, 43, 411–414. [Google Scholar]
- Carstoniu, J.; Levytam, S.; Norman, P.; Daley, D.; Katz, J.; Sandler, A.N. Nitrous oxide in early labor. Safety and analgesic efficacy assessed by a double-blind, placebo-controlled study. Anesthesiology 1994, 80, 30–35. [Google Scholar] [CrossRef]
- Leslie, K.; Myles, P.S.; Chan, M.T.; Forbes, A.; Paech, M.J.; Peyton, P.; Silbert, B.S.; Williamson, E. Nitrous oxide and long-term morbidity and mortality in the ENIGMA trial. Anesth. Analg. 2011, 112, 387–393. [Google Scholar] [CrossRef]
- Sanders, R.D.; Weimann, J.; Maze, M. Biologic effects of nitrous oxide: A mechanistic and toxicologic review. Anesthesiology 2008, 109, 707–722. [Google Scholar] [CrossRef]
- Myles, P.S.; Leslie, K.; Chan, M.T.; Forbes, A.; Paech, M.J.; Peyton, P.; Silbert, B.S.; Pascoe, E.; Group, E.T. Avoidance of nitrous oxide for patients undergoing major surgery: A randomized controlled trial. Anesthesiology 2007, 107, 221–231. [Google Scholar] [CrossRef]
- Truxell, E.M.; Molina, J.C.; Spear, N.E. Ethanol intake in the juvenile, adolescent, and adult rat: Effects of age and prior exposure to ethanol. Alcohol. Clin. Exp. Res. 2007, 31, 755–765. [Google Scholar] [CrossRef]
- Clancy, B.; Finlay, B.L.; Darlington, R.B.; Anand, K.J. Extrapolating brain development from experimental species to humans. Neurotoxicology 2007, 28, 931–937. [Google Scholar] [CrossRef]
- Lu, L.X.; Yon, J.H.; Carter, L.B.; Jevtovic-Todorovic, V. General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis 2006, 11, 1603–1615. [Google Scholar] [CrossRef]
- Jevtovic-Todorovic, V.; Hartman, R.E.; Izumi, Y.; Benshoff, N.D.; Dikranian, K.; Zorumski, C.F.; Olney, J.W.; Wozniak, D.F. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J. Neurosci. 2003, 23, 876–882. [Google Scholar]
- Shu, Y.; Patel, S.M.; Pac-Soo, C.; Fidalgo, A.R.; Wan, Y.; Maze, M.; Ma, D. Xenon pretreatment attenuates anesthetic-induced apoptosis in the developing brain in comparison with nitrous oxide and hypoxia. Anesthesiology 2010, 113, 360–368. [Google Scholar] [CrossRef]
- Ikonomidou, C. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Yon, J.H.; Daniel-Johnson, J.; Carter, L.B.; Jevtovic-Todorovic, V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience 2005, 135, 815–827. [Google Scholar] [CrossRef]
- Shu, Y.; Zhou, Z.; Wan, Y.; Sanders, R.D.; Li, M.; Pac-Soo, C.K.; Maze, M.; Ma, D. Nociceptive stimuli enhance anesthetic-induced neuroapoptosis in the rat developing brain. Neurobiol. Dis. 2012, 45, 743–750. [Google Scholar] [CrossRef]
- Yon, J.H.; Carter, L.B.; Reiter, R.J.; Jevtovic-Todorovic, V. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol. Dis. 2006, 21, 522–530. [Google Scholar] [CrossRef]
- Beals, J.K.; Carter, L.B.; Jevtovic-Todorovic, V. Neurotoxicity of nitrous oxide and ketamine is more severe in aged than in young rat brain. Ann. N. Y. Acad. Sci. 2003, 993, 123–124. [Google Scholar] [CrossRef]
- Culley, D.J.; Baxter, M.G.; Yukhananov, R.; Crosby, G. Long-term impairment of acquisition of a spatial memory task following isoflurane-nitrous oxide anesthesia in rats. Anesthesiology 2004, 100, 309–314. [Google Scholar] [CrossRef]
- Culley, D.J.; Raghavan, S.V.; Waly, M.; Baxter, M.G.; Yukhananov, R.; Deth, R.C.; Crosby, G. Nitrous oxide decreases cortical methionine synthase transiently but produces lasting memory impairment in aged rats. Anesth. Analg. 2007, 105, 83–88. [Google Scholar] [CrossRef]
- Culley, D.J.; Baxter, M.; Yukhananov, R.; Crosby, G. The memory effects of general anesthesia persist for weeks in young and aged rats. Anesth. Analg. 2003, 96, 1004–1009. [Google Scholar]
- Zou, X.; Liu, F.; Zhang, X.; Patterson, T.A.; Callicott, R.; Liu, S.; Hanig, J.P.; Paule, M.G.; Slikker, W., Jr.; Wang, C. Inhalation anesthetic-induced neuronal damage in the developing rhesus monkey. Neurotoxicol. Teratol. 2011, 33, 592–597. [Google Scholar] [CrossRef]
- Wang, C.; Sadovova, N.; Hotchkiss, C.; Fu, X.; Scallet, A.C.; Patterson, T.A.; Hanig, J.; Paule, M.G.; Slikker, W., Jr. Blockade of N-methyl-d-aspartate receptors by ketamine produces loss of postnatal day 3 monkey frontal cortical neurons in culture. Toxicol. Sci. 2006, 91, 192–201. [Google Scholar] [CrossRef]
- Slikker, W., Jr.; Zou, X.; Hotchkiss, C.E.; Divine, R.L.; Sadovova, N.; Twaddle, N.C.; Doerge, D.R.; Scallet, A.C.; Patterson, T.A.; Hanig, J.P.; et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol. Sci. 2007, 98, 145–158. [Google Scholar] [CrossRef]
- Paule, M.G.; Li, M.; Allen, R.R.; Liu, F.; Zou, X.; Hotchkiss, C.; Hanig, J.P.; Patterson, T.A.; Slikker, W., Jr.; Wang, C. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol. Teratol. 2011, 33, 220–230. [Google Scholar] [CrossRef]
- Loepke, A.W.; Soriano, S.G. An assessment of the effects of general anesthetics on developing brain structure and neurocognitive function. Anesth. Analg. 2008, 106, 1681–1707. [Google Scholar] [CrossRef]
- Hollmen, A.I.; Jouppila, R.; Koivisto, M.; Maatta, L.; Pihlajaniemi, R.; Puukka, M.; Rantakyla, P. Neurologic activity of infants following anesthesia for cesarean section. Anesthesiology 1978, 48, 350–356. [Google Scholar] [CrossRef]
- Noguchi, K.K.; Nemmers, B.; Farber, N.B. Age has a similar influence on the susceptibility to NMDA antagonist-induced neurodegeneration in most brain regions. Brain Res. Dev. Brain Res. 2005, 158, 82–91. [Google Scholar] [CrossRef]
- Jevtovic-Todorovic, V.; Todorovic, S.M.; Mennerick, S.; Powell, S.; Dikranian, K.; Benshoff, N.; Zorumski, C.F.; Olney, J.W. Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant and neurotoxin. Nat. Med. 1998, 4, 460–463. [Google Scholar] [CrossRef]
- Jevtovic-Todorovic, V.; Beals, J.; Benshoff, N.; Olney, J.W. Prolonged exposure to inhalational anesthetic nitrous oxide kills neurons in adult rat brain. Neuroscience 2003, 122, 609–616. [Google Scholar] [CrossRef]
- Royston, B.D.; Nunn, J.F.; Weinbren, H.K.; Royston, D.; Cormack, R.S. Rate of inactivation of human and rodent hepatic methionine synthase by nitrous oxide. Anesthesiology 1988, 68, 213–216. [Google Scholar] [CrossRef]
- Wronska-Nofer, T.; Nofer, J.R.; Jajte, J.; Dziubaltowska, E.; Szymczak, W.; Krajewski, W.; Wasowicz, W.; Rydzynski, K. Oxidative DNA damage and oxidative stress in subjects occupationally exposed to nitrous oxide (N2O). Mutat. Res. 2012, 731, 58–63. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Cheng, C.H.; Gin, T.; Leslie, K.; Myles, P.; Chan, M.T. Leukocyte DNA damage and wound infection after nitrous oxide administration: A randomized controlled trial. Anesthesiology 2013, 118, 1322–1331. [Google Scholar] [CrossRef]
- Selzer, R.R.; Rosenblatt, D.S.; Laxova, R.; Hogan, K. Adverse effect of nitrous oxide in a child with 5,10-methylenetetrahydrofolate reductase deficiency. N. Engl. J. Med. 2003, 349, 45–50. [Google Scholar] [CrossRef]
- Singer, M.A.; Lazaridis, C.; Nations, S.P.; Wolfe, G.I. Reversible nitrous oxide-induced myeloneuropathy with pernicious anemia: Case report and literature review. Muscle Nerve 2008, 37, 125–129. [Google Scholar] [CrossRef]
- Berkowitz, B.A.; Finck, A.D.; Ngai, S.H. Nitrous oxide analgesia: Reversal by naloxone and development of tolerance. J. Pharmacol. Exp. Ther. 1977, 203, 539–547. [Google Scholar]
- Branda, E.M.; Ramza, J.T.; Cahill, F.J.; Tseng, L.F.; Quock, R.M. Role of brain dynorphin in nitrous oxide antinociception in mice. Pharmacol. Biochem. Behav. 2000, 65, 217–221. [Google Scholar] [CrossRef]
- Quock, R.M.; Best, J.A.; Chen, D.C.; Vaughn, L.K.; Portoghese, P.S.; Takemori, A.E. Mediation of nitrous oxide analgesia in mice by spinal and supraspinal kappa-opioid receptors. Eur. J. Pharmacol. 1990, 175, 97–100. [Google Scholar] [CrossRef]
- Hodges, B.L.; Gagnon, M.J.; Gillespie, T.R.; Breneisen, J.R.; O’Leary, D.F.; Hara, S.; Quock, R.M. Antagonism of nitrous oxide antinociception in the rat hot plate test by site-specific mu and epsilon opioid receptor blockade. J. Pharmacol. Exp. Ther. 1994, 269, 596–600. [Google Scholar]
- Ori, C.; Fordrice, F.; London, E.D. Effects of nitrous-oxide and halothane on mu-opioid and kappa-opioid receptors in guinea-pig brain. Anesthesiology 1989, 70, 541–544. [Google Scholar] [CrossRef]
- Orestes, P.; Bojadzic, D.; Lee, J.; Leach, E.; Salajegheh, R.; Digruccio, M.R.; Nelson, M.T.; Todorovic, S.M. Free radical signalling underlies inhibition of CaV3.2 T-type calcium channels by nitrous oxide in the pain pathway. J. Physiol. 2011, 589, 135–148. [Google Scholar]
- Nagele, P.; Metz, L.B.; Crowder, C.M. Nitrous oxide (N2O) requires the N-methyl-d-aspartate receptor for its action in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2004, 101, 8791–8796. [Google Scholar] [CrossRef]
- Gruss, M.; Bushell, T.J.; Bright, D.P.; Lieb, W.R.; Mathie, A.; Franks, N.P. Two-pore-domain K+ channels are a novel target for the anesthetic gases xenon, nitrous oxide, and cyclopropane. Mol. Pharmacol. 2004, 65, 443–452. [Google Scholar] [CrossRef]
- O’Connell, A.D.; Morton, M.J.; Hunter, M. Two-pore domain K+ channels-molecular sensors. Biochim. Biophys. Acta 152–161.
- Heurteaux, C.; Guy, N.; Laigle, C.; Blondeau, N.; Duprat, F.; Mazzuca, M.; Lang-Lazdunski, L.; Widmann, C.; Zanzouri, M.; Romey, G.; et al. REK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 2004, 23, 2684–2695. [Google Scholar] [CrossRef]
- Alloui, A.; Zimmermann, K.; Mamet, J.; Duprat, F.; Noel, J.; Chemin, J.; Guy, N.; Blondeau, N.; Voilley, N.; Rubat-Coudert, C.; et al. REK-1, a K+ channel involved in polymodal pain perception. EMBO J. 2006, 25, 2368–2376. [Google Scholar] [CrossRef]
- Safari, A.; Emadi, F.; Jamali, E.; Borhani-Haghighi, A. Clinical and MRI manifestations of nitrous oxide induced vitamin B12 deficiency: A case report. Iran J. Neurol. 2013, 12, 111–113. [Google Scholar]
- Hsu, C.K.; Chen, Y.Q.; Lung, V.Z.; His, S.C.; Lo, H.C.; Shyu, H.Y. Myelopathy and polyneuropathy caused by nitrous oxide toxicity: A case report. Am. J. Emerg. Med. 2012, 30, 1016.e3–1016.e6. [Google Scholar]
- Lin, R.J.; Chen, H.F.; Chang, Y.C.; Su, J.J. Subacute combined degeneration caused by nitrous oxide intoxication: Case reports. Acta Neurol. Taiwanica 2011, 20, 129–137. [Google Scholar]
- Cheng, H.M.; Park, J.H.; Hernstadt, D. Subacute combined degeneration of the spinal cord following recreational nitrous oxide use. BMJ Case Rep. 2013. [Google Scholar] [CrossRef]
- Sotirchos, E.S.; Saidha, S.; Becker, D. Neurological picture. Nitrous oxide-induced myelopathy with inverted V-sign on spinal MRI. J. Neurol. Neurosurg. Psychiatry 2012, 83, 915–916. [Google Scholar] [CrossRef]
- Abraini, J.H.; David, H.N.; Nicole, O.; MacKenzie, E.T.; Buisson, A.; Lemaire, M. Neuroprotection by nitrous oxide and xenon and its relation to minimum alveolar concentration. Anesthesiology 2004, 101, 260–261. [Google Scholar]
- David, H.N.; Leveille, F.; Chazalviel, L.; MacKenzie, E.T.; Buisson, A.; Lemaire, M.; Abraini, J.H. Reduction of ischemic brain damage by nitrous oxide and xenon. J. Cereb. Blood Flow Metab. 2003, 23, 1168–1173. [Google Scholar]
- Haelewyn, B.; David, H.N.; Rouillon, C.; Chazalviel, L.; Lecocq, M.; Risso, J.J.; Lemaire, M.; Abraini, J.H. Neuroprotection by nitrous oxide: Facts and evidence. Crit. Care Med. 2008, 36, 2651–2659. [Google Scholar] [CrossRef]
- Haelewyn, B.; David, H.N.; Colloc’h, N.; Colomb, D.G., Jr.; Risso, J.J.; Abraini, J.H. Interactions between nitrous oxide and tissue plasminogen activator in a rat model of thromboembolic stroke. Anesthesiology 2011, 115, 1044–1053. [Google Scholar] [CrossRef]
- Wong, E.H.; Kemp, J.A.; Priestley, T.; Knight, A.R.; Woodruff, G.N.; Iversen, L.L. The anticonvulsant MK-801 is a potent N-methyl-d-aspartate antagonist. Proc. Natl. Acad. Sci. USA 1986, 83, 7104–7108. [Google Scholar] [CrossRef]
- McNamara, J.O.; Russell, R.D.; Rigsbee, L.; Bonhaus, D.W. Anticonvulsant and antiepileptogenic actions of MK-801 in the kindling and electroshock models. Neuropharmacology 1988, 27, 563–568. [Google Scholar]
- Fix, A.S.; Horn, J.W.; Wightman, K.A.; Johnson, C.A.; Long, G.G.; Storts, R.W.; Farber, N.; Wozniak, D.F.; Olney, J.W. Neuronal vacuolization and necrosis induced by the noncompetitive N-methyl-d-aspartate (NMDA) antagonist MK(+)801 (dizocilpine maleate): A light and electron microscopic evaluation of the rat retrosplenial cortex. Exp. Neurol. 1993, 123, 204–215. [Google Scholar] [CrossRef]
- Wiescholleck, V.; Manahan-Vaughan, D. Long-lasting changes in hippocampal synaptic plasticity and cognition in an animal model of NMDA receptor dysfunction in psychosis. Neuropharmacology 2013, 74, 48–58. [Google Scholar] [CrossRef]
- Giovannini, M.G.; Mutolo, D.; Bianchi, L.; Michelassi, A.; Pepeu, G. NMDA receptor antagonists decrease GABA outflow from the septum and increase acetylcholine outflow from the hippocampus: A microdialysis study. J. Neurosci. 1994, 14, 1358–1365. [Google Scholar]
- Kim, S.H.; Price, M.T.; Olney, J.W.; Farber, N.B. Excessive cerebrocortical release of acetylcholine induced by NMDA antagonists is reduced by GABAergic and alpha2-adrenergic agonists. Mol. Psychiatry 1999, 4, 344–352. [Google Scholar]
- Shichino, T.; Murakawa, M.; Adachi, T.; Arai, T.; Miyazaki, Y.; Mori, K. Effects of inhalation anaesthetics on the release of acetylcholine in the rat cerebral cortex in vivo. Br. J. Anaesth. 1998, 80, 365–370. [Google Scholar] [CrossRef]
- Strominger, N.L.; Hori, N.; Carpenter, D.O.; Tan, Y.; Folger, W.H. Effects of acetylcholine and GABA on neurons in the area postrema of Suncus murinus brainstem slices. Neurosci. Lett. 2001, 309, 77–80. [Google Scholar] [CrossRef]
- Olney, J.W.; Newcomer, J.W.; Farber, N.B. NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 1999, 33, 523–533. [Google Scholar] [CrossRef]
- Wozniak, D.F.; Dikranian, K.; Ishimaru, M.J.; Nardi, A.; Corso, T.D.; Tenkova, T.; Olney, J.W.; Fix, A.S. Disseminated corticolimbic neuronal degeneration induced in rat brain by MK-801: Potential relevance to Alzheimer’s disease. Neurobiol. Dis. 1998, 5, 305–322. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Jevtovic-Todorovic, V.; Selke, G.; Melson, A.K.; Hershey, T.; Craft, S.; Olney, J.W. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology 1999, 20, 106–118. [Google Scholar]
- Homayoun, H.; Moghaddam, B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 2007, 27, 11496–11500. [Google Scholar] [CrossRef]
- Javitt, D.C. Glutamatergic theories of schizophrenia. Isr. J. PsychiatryRelat. Sci. 2010, 47, 4–16. [Google Scholar]
- Nunn, J.F. Clinical aspects of the interaction between nitrous oxide and vitamin B12. Br. J. Anaesth. 1987, 59, 3–13. [Google Scholar] [CrossRef]
- Mayer, E.L.; Jacobsen, D.W.; Robinson, K. Homocysteine and coronary atherosclerosis. J. Am. Coll. Cardiol. 1996, 27, 517–527. [Google Scholar] [CrossRef]
- Blacher, J.; Demuth, K.; Guerin, A.P.; Vadez, C.; Moatti, N.; Safar, M.E.; London, G.M. Association between plasma homocysteine concentrations and cardiac hypertrophy in end-stage renal disease. J. Nephrol. 1999, 12, 248–255. [Google Scholar]
- Badner, N.H.; Drader, K.; Freeman, D.; Spence, J.D. The use of intraoperative nitrous oxide leads to postoperative increases in plasma homocysteine. Anesth. Analg. 1998, 87, 711–713. [Google Scholar]
- Nagele, P.; Tallchief, D.; Blood, J.; Sharma, A.; Kharasch, E.D. Nitrous oxide anesthesia and plasma homocysteine in adolescents. Anesth. Analg. 2011, 113, 843–848. [Google Scholar]
- Nappo, F.; de Rosa, N.; Marfella, R.; de Lucia, D.; Ingrosso, D.; Perna, A.F.; Farzati, B.; Giugliano, D. Impairment of endothelial functions by acute hyperhomocysteinemia and reversal by antioxidant vitamins. JAMA 1999, 281, 2113–2118. [Google Scholar] [CrossRef]
- Ermens, A.A.; Refsum, H.; Rupreht, J.; Spijkers, L.J.; Guttormsen, A.B.; Lindemans, J.; Ueland, P.M.; Abels, J. Monitoring cobalamin inactivation during nitrous oxide anesthesia by determination of homocysteine and folate in plasma and urine. Clin. Pharmacol. Ther. 1991, 49, 385–393. [Google Scholar] [CrossRef]
- Badner, N.H.; Freeman, D.; Spence, J.D. Preoperative oral B vitamins prevent nitrous oxide-inducedpostoperative plasma homocysteine increases. Anesth. Analg. 2001, 93, 1507–1510. [Google Scholar] [CrossRef]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef]
- Seshadri, S. Elevated plasma homocysteine levels: Risk factor or risk marker for the development of dementia and Alzheimer’s disease? J. Alzheimers Dis. 2006, 9, 393–398. [Google Scholar]
- Troen, A.M.; Shea-Budgell, M.; Shukitt-Hale, B.; Smith, D.E.; Selhub, J.; Rosenberg, I.H. B-vitamin deficiency causes hyperhomocysteinemia and vascular cognitive impairment in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12474–12479. [Google Scholar]
- Nagele, P.; Zeugswetter, B.; Wiener, C.; Burger, H.; Hupfl, M.; Mittlbock, M.; Fodinger, M. Influence of methylenetetrahydrofolate reductase gene polymorphisms on homocysteine concentrations after nitrous oxide anesthesia. Anesthesiology 2008, 109, 36–43. [Google Scholar] [CrossRef]
- Nygard, O.; Vollset, S.E.; Refsum, H.; Stensvold, I.; Tverdal, A.; Nordrehaug, J.E.; Ueland, M.; Kvale, G. Total plasma homocysteine and cardiovascular risk profile. The Hordaland Homocysteine Study. JAMA 1995, 274, 1526–1533. [Google Scholar] [CrossRef]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef]
- Akaike, A.; Katsuki, H.; Kume, T.; Maeda, T. Reactive oxygen species in NMDA receptor-mediated glutamate neurotoxicity. Parkinsonism Relat. Disord. 1999, 5, 203–207. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Hastings, T.G. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 1995, 15, 3318–3327. [Google Scholar]
- Dugan, L.L.; Sensi, S.L.; Canzoniero, L.M.; Handran, S.D.; Rothman, S.M.; Lin, T.S.; Goldberg, M.P.; Choi, D.W. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J. Neurosci. 1995, 15, 6377–6388. [Google Scholar]
- Gordeeva, A.V.; Zvyagilskaya, R.A.; Labas, Y.A. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry 2003, 68, 1077–1080. [Google Scholar]
- Hernandez-Fonseca, K.; Cardenas-Rodriguez, N.; Pedraza-Chaverri, J.; Massieu, L. Calcium-dependent production of reactive oxygen species is involved in neuronal damage induced during glycolysis inhibition in cultured hippocampal neurons. J. Neurosci. Res. 2008, 86, 1768–1780. [Google Scholar] [CrossRef]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar]
- Adam-Vizi, V.; Starkov, A.A. Calcium and mitochondrial reactive oxygen species generation: How to read the facts. J. Alzheimers Dis. 2010, 20, S413–S426. [Google Scholar]
- Herrmann, W.; Obeid, R. Homocysteine: A biomarker in neurodegenerative diseases. Clin. Chem. Lab. Med. 2011, 49, 435–441. [Google Scholar]
- Zhuo, J.M.; Wang, H.; Pratico, D. Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmaco. Sci. 2011, 32, 562–571. [Google Scholar] [CrossRef]
- Zieminska, E.; Lazarewicz, J.W. Excitotoxic neuronal injury in chronic homocysteine neurotoxicity studied in vitro: The role of NMDA and group I metabotropic glutamate receptors. Acta Neurobiol. Exp. 2006, 66, 301–309. [Google Scholar]
- Camello-Almaraz, C.; Gomez-Pinilla, P.J.; Pozo, M.J.; Camello, P.J. Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. Cell Physiol. 2006, 291, C1082–C1088. [Google Scholar] [CrossRef]
- Boscolo, A.; Starr, J.A.; Sanchez, V.; Lunardi, N.; DiGruccio, M.R.; Ori, C.; Erisir, A.; Trimmer, P.; Bennett, J.; Jevtovic-Todorovic, V. The abolishment of anesthesia-induced cognitive impairment by timely protection of mitochondria in the developing rat brain: The importance of free oxygen radicals and mitochondrial integrity. Neurobiol. Dis. 2012, 45, 1031–1041. [Google Scholar] [CrossRef]
- Zhen, Y.; Dong, Y.; Wu, X.; Xu, Z.; Lu, Y.; Zhang, Y.; Norton, D.; Tian, M.; Li, S.; Xie, Z. Nitrous oxide plus isoflurane induces apoptosis and increases beta-amyloid protein levels. Anesthesiology 2009, 111, 741–752. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Herlenius, E.; Lagercrantz, H. Development of neurotransmitter systems during critical periods. Exp. Neurol. 2004, 190, S8–S21. [Google Scholar] [CrossRef]
- Murawska-Cialowicz, E.; Januszewska, L.; Zuwala-Jagiello, J.; Milczarska, J.; Zawadzki, M.; Paprocka-Borowicz, M.; Wierzbicka-Damska, I. Melatonin decreases homocysteine level in blood of rats. J. Physiol. Pharmacol. 2008, 59, 717–729. [Google Scholar]
- Baydas, G.; Koz, S.T.; Tuzcu, M.; Nedzvetsky, V.S. Melatonin prevents gestational hyperhomocysteinemia-associated alterations in neurobehavioral developments in rats. J. Pineal Res. 2008, 44, 181–188. [Google Scholar] [CrossRef]
- Hancock, S.M.; Nathanson, M.H. Nitrous oxide or remifentanil for the “at risk” brain. Anaesthesia 2004, 59, 313–315. [Google Scholar] [CrossRef]
- Volmanen, P.; Akural, E.; Raudaskoski, T.; Ohtonen, P.; Alahuhta, S. Comparison of remifentanil and nitrous oxide in labour analgesia. Acta Anaesthesiol. Scand. 2005, 49, 453–458. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Savage, S.; Ma, D. The Neurotoxicity of Nitrous Oxide: The Facts and “Putative” Mechanisms. Brain Sci. 2014, 4, 73-90. https://doi.org/10.3390/brainsci4010073
Savage S, Ma D. The Neurotoxicity of Nitrous Oxide: The Facts and “Putative” Mechanisms. Brain Sciences. 2014; 4(1):73-90. https://doi.org/10.3390/brainsci4010073
Chicago/Turabian StyleSavage, Sinead, and Daqing Ma. 2014. "The Neurotoxicity of Nitrous Oxide: The Facts and “Putative” Mechanisms" Brain Sciences 4, no. 1: 73-90. https://doi.org/10.3390/brainsci4010073
APA StyleSavage, S., & Ma, D. (2014). The Neurotoxicity of Nitrous Oxide: The Facts and “Putative” Mechanisms. Brain Sciences, 4(1), 73-90. https://doi.org/10.3390/brainsci4010073