
Higher Prevalence of Nonsense Pathogenic DMD Variants in a Single-Center Cohort from Brazil: A Genetic Profile Study That May Guide the Choice of Disease-Modifying Treatments

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
Clinical Characteristics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and Mutations: One Gene, Several Proteins, Multiple Phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete Cloning of the Duchenne Muscular Dystrophy (DMD) CDNA and Preliminary Genomic Organization of the DMD Gene in Normal and Affected Individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7,000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395. [Google Scholar] [CrossRef]
- Schneider, N.B.; Roos, E.C.; Staub, A.L.P.; Bevilacqua, I.P.; de Almeida, A.C.; de Camargo Martins, T.; Ramos, N.B.; Loze, P.; Saute, J.A.M.; Etges, A.P.B.d.S.; et al. Estimated Costs for Duchenne Muscular Dystrophy Care in Brazil. Orphanet. J. Rare Dis. 2023, 18, 159. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, P.A.D.; Machado-Costa, M.C.; Manzoli, G.N.; Ferreira, L.S.; Rodrigues, M.C.S.; Bueno, L.S.M.; Saute, J.A.M.; Pinto Vairo, F.; Matte, U.S.; Siebert, M.; et al. Genetic Profile of Brazilian Patients with Dystrophinopathies. Clin. Genet. 2017, 92, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Brandsema, J.F.; Darras, B.T. Dystrophinopathies. Semin. Neurol. 2015, 35, 369–384. [Google Scholar] [CrossRef]
- Segarra-Casas, A.; Domínguez-González, C.; Hernández-Laín, A.; Sanchez-Calvin, M.T.; Camacho, A.; Rivas, E.; Campo-Barasoain, A.; Madruga, M.; Ortez, C.; Natera-De Benito, D.; et al. Genetic Diagnosis of Duchenne and Becker Muscular Dystrophy through MRNA Analysis: New Splicing Events. J. Med. Genet. 2023, 60, 615–619. [Google Scholar] [CrossRef]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular Dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Cho, A.; Seong, M.W.; Lim, B.C.; Lee, H.J.; Byeon, J.H.; Kim, S.S.; Kim, S.Y.; Choi, S.A.; Wong, A.L.; Lee, J.; et al. Consecutive Analysis of Mutation Spectrum in the Dystrophin Gene of 507 Korean Boys with Duchenne/Becker Muscular Dystrophy in a Single Center. Muscle Nerve 2017, 55, 727–734. [Google Scholar] [CrossRef]
- Sitnik, R.; Campiotto, S.; Vainzof, M.; Pavanello, R.C.; Takata, R.I.; Zatz, M.; Passos-Bueno, M.R. Novel Point Mutations in the Dystrophin Gene. Hum. Mutat. 1997, 10, 217–222. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next Generation in Gene Variant Databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Censo Demográfico. 2022. Available online: https://censo2022.ibge.gov.br/ (accessed on 10 October 2023).
- Santos, S.; Kok, F.; Weller, M.; de Paiva, F.R.L.; Otto, P.A. Inbreeding Levels in Northeast Brazil: Strategies for the Prospecting of New Genetic Disorders. Genet. Mol. Biol. 2010, 33, 220–223. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Den Dunnen, J.T. Copy Number Variation in the Genome; the Human DMD Gene as an Example. Cytogenet. Genome Res. 2006, 115, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Z.; Yan, M.; Huang, S.; Chen, T.J.; Zhong, N. Similarity of DMD Gene Deletion and Duplication in the Chinese Patients Compared to Global Populations. Behav. Brain Funct. 2008, 4, 20. [Google Scholar] [CrossRef]
- Lai, P.S.; Takeshima, Y.; Adachi, K.; Van Tran, K.; Nguyen, H.T.; Low, P.S.; Matsuo, M. Comparative Study on Deletions of the Dystrophin Gene in Three Asian Populations. J. Hum. Genet. 2002, 47, 552–555. [Google Scholar] [CrossRef]
- Luce, L.; Carcione, M.; Mazzanti, C.; Buonfiglio, P.I.; Dalamón, V.; Mesa, L.; Dubrovsky, A.; Corderí, J.; Giliberto, F. Theragnosis for Duchenne Muscular Dystrophy. Front. Pharmacol. 2021, 12, 648390. [Google Scholar] [CrossRef]
- López-Hernández, L.B.; Gómez-Díaz, B.; Luna-Angulo, A.B.; Anaya-Segura, M.; Bunyan, D.J.; Zúñiga-Guzman, C.; Escobar-Cedillo, R.E.; Roque-Ramírez, B.; Ruano-Calderón, L.A.; Rangel-Villalobos, H.; et al. Comparison of Mutation Profiles in the Duchenne Muscular Dystrophy Gene among Populations: Implications for Potential Molecular Therapies. Int. J. Mol. Sci. 2015, 16, 5334–5346. [Google Scholar] [CrossRef]
- García-Acero, M.; Pineda, T.; Guerra-Torres, M.; García-Robles, R.; Ayala-Ramírez, P.; Buitrago, T.; Poveda, A.; Suárez-Obando, F. Análisis Del Espectro Mutacional de La Distrofia Muscular de Duchenne En Un Grupo de Pacientes Colombianos. Neurol. Argent. 2018, 10, 137–146. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; den Dunnen, J.T. Phenotype Predictions for Exon Deletions/Duplications: A User Guide for Professionals and Clinicians Using Becker and Duchenne Muscular Dystrophy as Examples. Hum. Mutat. 2019, 40, 1630–1633. [Google Scholar] [CrossRef]
- Mah, J.; Selby, K.; Campbell, C.; Nadeau, A.; Tarnopolsky, M.; McCormick, A.; Dooley, J.; Kolski, H.; Skalsky, A.; Smith, R.; et al. A Population-Based Study of Dystrophin Mutations in Canada. Can. J. Neurol. Sci. 2011, 38, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Triana-Fonseca, P.; Parada-Márquez, J.F.; Silva-Aldana, C.T.; Zambrano-Arenas, D.; Arias-Gomez, L.L.; Morales-Fonseca, N.; Medina-Méndez, E.; Restrepo, C.M.; Silgado-Guzmán, D.F.; Fonseca-Mendoza, D.J. Genetic Profile of the Dystrophin Gene Reveals New Mutations in Colombian Patients Affected with Muscular Dystrophinopathy. Appl. Clin. Genet. 2021, 14, 399–408. [Google Scholar] [CrossRef]
- Alcántara-Ortigoza, M.A.; Reyna-Fabián, M.E.; González-Del Angel, A.; Estandia-Ortega, B.; Bermúdez-López, C.; Cruz-Miranda, G.M.; Ruíz-García, M. Predominance of Dystrophinopathy Genotypes in Mexican Male Patients Presenting as Muscular Dystrophy with A Normal Multiplex Polymerase Chain Reaction DMD Gene Result: A Study Including Targeted Next-Generation Sequencing. Genes 2019, 10, 856. [Google Scholar] [CrossRef]
- Baskin, B.; Banwell, B.; Al Khater, R.; Hawkins, C.; Ray, P.N. Becker Muscular Dystrophy Caused by an Intronic Mutation Reducing the Efficiency of the Splice Donor Site of Intron 26 of the Dystrophin Gene. Neuromuscul. Disord. 2009, 19, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne Muscular Dystrophy Mutation Database: An Overview of Mutation Types and Paradoxical Cases That Confirm the Reading-Frame Rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Zhang, S.; Zhang, H.; Fang, S.; Dong, Y.; Zhang, Y.; Hao, W.; Wu, S.; Zhao, Y. Comprehensive Genetic Characteristics of Dystrophinopathies in China. Orphanet J. Rare Dis. 2018, 13, 109. [Google Scholar] [CrossRef]
- Elangkovan, N.; Dickson, G. Gene Therapy for Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, S303–S316. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Neuromuscular, Rehabilitation, Endocrine, and Gastrointestinal and Nutritional Management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes 2020, 11, 837. [Google Scholar] [CrossRef]
- Takeda, S.; Clemens, P.R.; Hoffman, E.P. Exon-Skipping in Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, S343–S358. [Google Scholar] [CrossRef]
- Bello, L.; Morgenroth, L.P.; Gordish-Dressman, H.; Hoffman, E.P.; McDonald, C.M.; Cirak, S. DMD Genotypes and Loss of Ambulation in the CINRG Duchenne Natural History Study. Neurology 2016, 87, 401. [Google Scholar] [CrossRef] [PubMed]
- Zambon, A.A.; Ayyar Gupta, V.; Ridout, D.; Manzur, A.Y.; Baranello, G.; Trucco, F.; Muntoni, F.; Tirupath, S.; Douglas, M.; McFetridge, J.; et al. Peak Functional Ability and Age at Loss of Ambulation in Duchenne Muscular Dystrophy. Dev. Med. Child Neurol. 2022, 64, 979–988. [Google Scholar] [CrossRef]
- Darras, B.T.; Urion, D.K.; Ghosh, P.S. Dystrophinopathies. In GeneReviews®; University of Washington: Seattle, WA, USA, 2022; p. 35. [Google Scholar]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J.I. The Incidence and Evolution of Cardiomyopathy in Duchenne Muscular Dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne Muscular Dystrophy: Improvements in Life Expectancy since 1967 and the Impact of Home Nocturnal Ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Awano, H.; Zhang, Z.; Sakuma, M.; Kitaaki, S.; Matsumoto, M.; Nagai, M.; Sato, I.; Imanishi, T.; Hayashi, N.; et al. Cardiac Dysfunction in Duchenne Muscular Dystrophy Is Less Frequent in Patients with Mutations in the Dystrophin Dp116 Coding Region Than in Other Regions. Circ. Genom. Precis. Med. 2018, 11, E001782. [Google Scholar] [CrossRef]
- Gloss, D.; Moxley, R.T.; Ashwal, S.; Oskoui, M. Practice Guideline Update Summary: Corticosteroid Treatment of Duchenne Muscular Dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016, 86, 465. [Google Scholar] [CrossRef]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the Treatment of Duchenne Muscular Dystrophy. Cochrane Database Syst. Rev. 2016, 2016, CD003725. [Google Scholar] [CrossRef] [PubMed]
- McAdam, L.C.; Mayo, A.L.; Alman, B.A.; Biggar, W.D. The Canadian Experience with Long Term Deflazacort Treatment in Duchenne Muscular Dystrophy. Acta Myol. 2012, 31, 16. [Google Scholar]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-Term Effects of Glucocorticoids on Function, Quality of Life, and Survival in Patients with Duchenne Muscular Dystrophy: A Prospective Cohort Study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Ricotti, V.; Ridout, D.A.; Scott, E.; Quinlivan, R.; Robb, S.A.; Manzur, A.Y.; Muntoni, F. On Behalf of the NorthStar Clinical Network Long-Term Benefits and Adverse Effects of Intermittent versus Daily Glucocorticoids in Boys with Duchenne Muscular Dystrophy. J. Neurol. Neurosurg Psychiatry 2013, 84, 698–705. [Google Scholar] [CrossRef]
- Traynor, K. Deflazacort Approved for Duchenne Muscular Dystrophy. Am. J. Health-Syst. Pharm. 2017, 74, 368. [Google Scholar] [CrossRef]
- Griggs, R.C.; Miller, J.P.; Greenberg, C.R.; Fehlings, D.L.; Pestronk, A.; Mendell, J.R.; Moxley, R.T.; King, W.; Kissel, J.T.; Cwik, V.; et al. Efficacy and Safety of Deflazacort vs Prednisone and Placebo for Duchenne Muscular Dystrophy. Neurology 2016, 87, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Herr, B.E.; Reha, A.; Elfring, G.; Atkinson, L.; Cwik, V.; Mccoll, E.; Tawil, R.; Pandya, S.; Mcdermott, M.P.; et al. Corticosteroids in Duchenne Muscular Dystrophy: Major Variations in Practice. Muscle Nerve 2013, 48, 27–31. [Google Scholar] [CrossRef] [PubMed]
- de Queiroz Campos Araujo, A.P.; Saute, J.A.M.; Drumond Fortes, C.P.D.; França, M.C.; Pereira, J.A.; de Albuquerque, M.A.V.; de Siqueira Carvalho, A.A.; Cavalcanti, E.B.U.; Miranda Covaleski, A.P.P.; Fagondes, S.C.; et al. Update of the Brazilian Consensus Recommendations on Duchenne Muscular Dystrophy. Arq. Neuro-Psiquiatr. 2023, 81, 81–94. [Google Scholar] [CrossRef]
- McDonald, C.M.; Sajeev, G.; Yao, Z.; McDonnell, E.; Elfring, G.; Souza, M.; Peltz, S.W.; Darras, B.T.; Shieh, P.B.; Cox, D.A.; et al. Deflazacort vs Prednisone Treatment for Duchenne Muscular Dystrophy: A Meta-Analysis of Disease Progression Rates in Recent Multicenter Clinical Trials. Muscle Nerve 2020, 61, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Osorio, A.N.; Muntoni, F.; Buccella, F.; Desguerre, I.; Kirschner, J.; Tulinius, M.; de Resende, M.B.D.; Morgenroth, L.P.; Gordish-Dressman, H.; et al. Safety and Effectiveness of Ataluren in Patients with Nonsense Mutation DMD in the STRIDE Registry Compared with the CINRG Duchenne Natural History Study (2015–2022): 2022 Interim Analysis. J. Neurol. 2023, 270, 3896–3913. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; Barohn, R.J.; Bertini, E.; Chabrol, B.; Comi, G.P.; Darras, B.T.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Iannaccone, S.T.; et al. Meta-Analyses of Ataluren Randomized Controlled Trials in Nonsense Mutation Duchenne Muscular Dystrophy. J. Comp. Eff. Res. 2020, 9, 973–984. [Google Scholar] [CrossRef] [PubMed]
- EUCTR2009-013169-24-GB; A Phase 2a Study of Ataluren (PTC124) in Nonambulatory Patients with Nonsense–Mutation-Mediated Duchenne/Becker Muscular Dystrophy—Study of Ataluren in Nonambulatory Patients with DMD/BMD. Available online: https://trialsearch.who.int/Trial2.aspx?TrialID=EUCTR2009-013169-24-GB (accessed on 25 September 2023).
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in Patients with Nonsense Mutation Duchenne Muscular Dystrophy (ACT DMD): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- McDonald, C.M.; Wu, S.; Gulati, S.; Komaki, H.; Escobar, R.E.; Kostera-Pruszczyk, A.; Vlodavets, D.; Chae, J.-H.; Jong, Y.-J.; Karachunski, P.; et al. Safety and Efficacy of Ataluren in NmDMD Patients from Study 041, a Phase 3, Randomized, Double-Blind, Placebo-Controlled Trial (PL5.001). 2023, p. 2374. Available online: https://www.mdaconference.org/abstract-library/safety-and-efficacy-of-ataluren-in-nmdmd-patients-from-study-041-a-phase-3-randomized-double-blind-placebo-controlled-trial/ (accessed on 25 September 2023).
- EMA Recommends Non-Renewal of Authorisation of Duchenne Muscular Dystrophy Medicine Translarna|European Medicines Agency. Available online: https://www.ema.europa.eu/en/news/ema-recommends-non-renewal-authorisation-duchenne-muscular-dystrophy-medicine-translarna (accessed on 10 October 2023).
- Gomes, G.R.F.; Mariano, T.C.; Braga, V.L.L.; Ribeiro, E.M.; Guimarães, I.P.; Pereira, K.S.A.F.; Nóbrega, P.R.; Pessoa, A.L.S. Bailey-Bloch Congenital Myopathy in Brazilian Patients: A Very Rare Myopathy with Malignant Hyperthermia Susceptibility. Brain Sci. 2023, 13, 1184. [Google Scholar] [CrossRef]
- De Paiva, A.R.B.; Pessoa, A.L.S.; Nóbrega, P.R.; Moreno, C.A.M.; Lynch, D.S.; Taniguti, L.M.; Kitajima, J.P.; Freua, F.; Della-Ripa, B.; Cunha, P.; et al. Ceroid Lipofuscinosis Type 5: Novel Pathogenic Variants and Unexpected Phenotypic Findings. J. Neurol. Neurosurg Psychiatry 2023, 94, 405–408. [Google Scholar] [CrossRef]
- Nóbrega, P.R.; Morais, J.L.A.; Ferreira, A.M.; de Medeiros, A.D.; Duarte, B.A.; Rangel, D.M.; Lima, F.O.; de Paiva, A.R.B.; Paim-Marques, L.; Kok, F.; et al. Aseptic Meningitis in Fabry Disease Due to a Novel GLA Variant: An Expanded Phenotype? Neurol. Sci. 2023, 44, 319–327. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
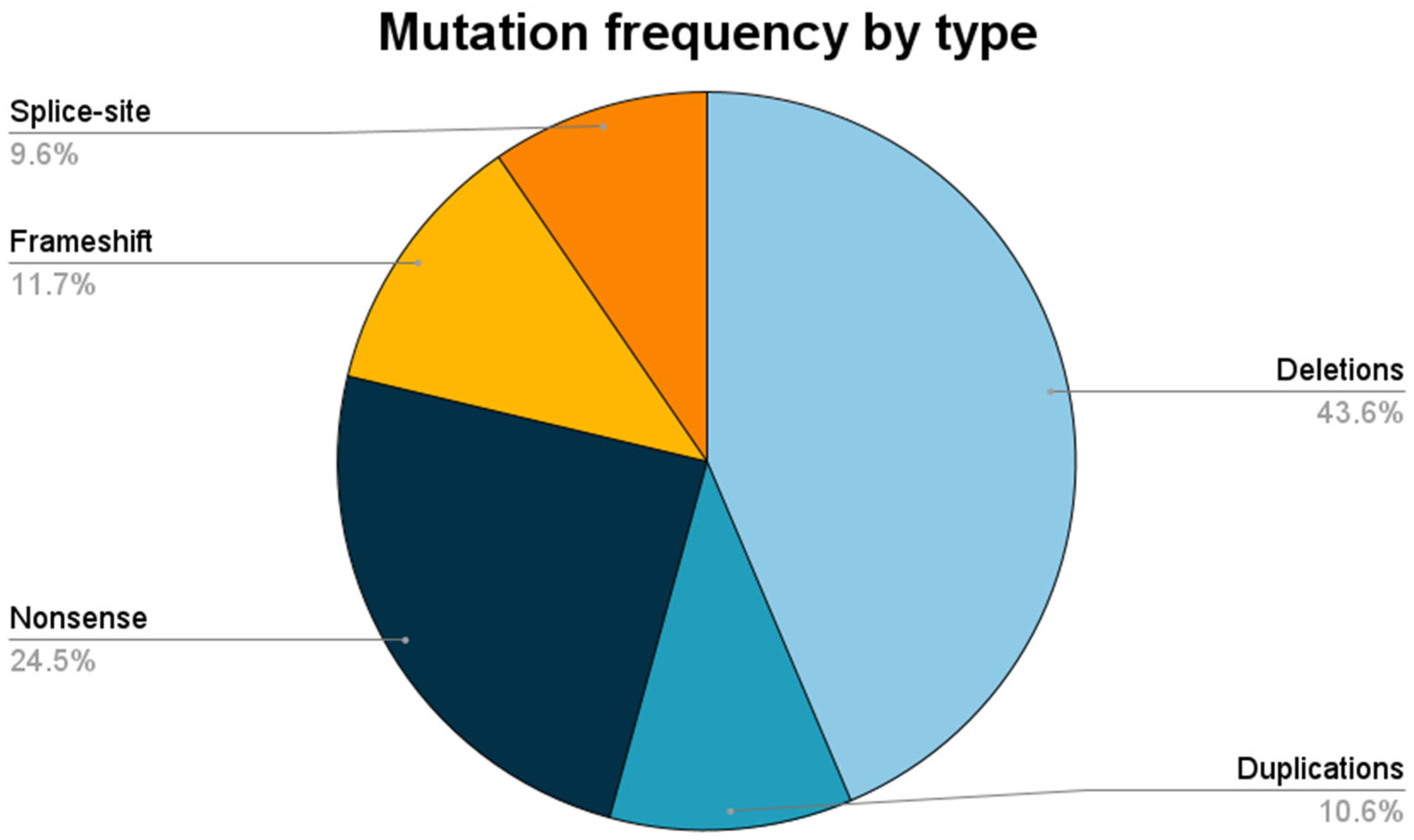
| Type of Pathogenic Variant | DMD (n = 86; 91.5%) | DMB (n = 8; 8.5%) | All Patients (n = 94; 100%) |
|---|---|---|---|
| LARGE | |||
| Deletions | 36 (41.9%) | 5 (62.5%) | 41 (43.6%) |
| Duplications | 9 (10.5%) | 1 (12.5%) | 10 (10.6%) |
| SMALL | |||
| Nonsense | 23 (26.7%) | 0 | 23 (24.5%) |
| Frameshift | 11 (12.8%) | 0 | 11 (11.7%) |
| Splice-site | 7 (8.1%) | 2 (25.0%) | 9 (9.6%) |
| Mutation | Families | Nucleotide Change | Protein Change | ClinVar | dbSNP |
|---|---|---|---|---|---|
| Nonsense | 2 | c.8038 C > T | p.(Arg2680*) | 217213 | rs863225011 |
| Nonsense | 2 | c.433 C > T | p.(Arg145*) | 11225 | rs128626235 |
| Nonsense | 2 | c.6283 C > T | p.(Arg2095*) | 94697 | rs398124008 |
| Nonsense | 1 | c.453 T > G | p.(Try151*) | 803948 | rs1603437254 |
| Splice-site | 1 | c.3603 + 3A > T | 409882 | rs1060502615 | |
| Nonsense | 1 | c.3151C > T | p.(Arg1051*) | 94576 | rs398123929 |
| Frameshift | 1 | c.4314_4315delAA | p.(Arg1439Serfs*6) | 94619 | rs398123950 |
| Nonsense | 1 | c.6292 C > T | p.(Arg2098*) | 11260 | rs128626250 |
| Nonsense | 1 | c.8608C > T | p.(Arg2870*) | 94810 | rs398124074 |
| Nonsense | 1 | c.8944G > A | p.(Arg2982*) | 11211 | rs128625229 |
| Nonsense | 1 | c.9337C > T | p.(Arg3113*) | 94839 | rs398124092 |
| Frameshift | 1 | c.141dupG | p.(Arg48Glufs*41) | 565437 | rs1569533965 |
| Frameshift | 1 | c.2552_2553insA G > GT | p.(Asn851Lysfs*17) | ◊ | ◊ |
| Nonsense | 1 | c.10011C > A | p.(Cys3337*) | * | * |
| Frameshift | 1 | c.3295_3296delCA | p.(Gln1099Asp fs*11) | * | * |
| Frameshift | 1 | c.5131del | p.(Gln1711Serfs*10) | 1685728 | |
| Nonsense | 1 | c.Gln3037C > T | p.(Gln3037*) | 1322249 | |
| Nonsense | 1 | c.133C > T | p.(Gln45*) | 196372 | rs794727499 |
| Frameshift | 1 | c.3533_3536delAAGA | p.(Glu1178Glyfs*22) | 803892 | rs1603633864 |
| Frameshift | 1 | c.9269_9270delAG | p.(Glu3090Alafs*) | 803806 | rs1603253563 |
| Frameshift | 1 | c.3185_3192delinsTTTGTAT | p.(Lys1062llefs*10) | * | * |
| Frameshift | 1 | c.3396delA | p.(Lys1132Asnfs*20) | * | * |
| Frameshift | 1 | c.6986del | p.(Lys2329Serfs*9) | 455927 | rs398124040 |
| Nonsense | 1 | c.8744 G > A | p.(Trp2915*) | 803815 | rs1603222922 |
| Nonsense | 1 | c.9248G > A | p.(Trp3083*) | ◊ | ◊ |
| Nonsense | 1 | c.5646 C > A | p.(Tyr1882*) | ◊ | ◊ |
| Nonsense | 1 | c.6276C > A | p.(Tyr2092*) | 618598 | rs1569555987 |
| Splice-site | 1 | c.5740-1G > T | 1365986 | ||
| Splice-site | 1 | c.9362-1G > C | 1685709 | ||
| Splice-site | 1 | c.2804-1del | 496616 | rs1557374667 | |
| Splice-site | 1 | c.2169-1G > A | 803918 | rs1603635331 | |
| Splice-site | 1 | c.9286 + 2delT | * | * |
| Genotypic Spectrum | Phenotype | p Value 1 | ||
|---|---|---|---|---|
| DMD (n = 45; 88%) | DMB (n = 6; 12%) | |||
| Duplication | In-frame | 3 (7%) | 1 (17%) | <0.05 |
| Out-of-frame | 6 (13%) | 0 | ||
| Deletion | In-frame | 4 (9%) | 5 (83%) | |
| Out-of-frame | 32 (71%) | 0 | ||
| Characteristic | n (n%) | Mean (SD) |
|---|---|---|
| Age at last follow-up 1 | 12 (5.1) | |
| Loss of ambulation 2 | 21 (38.2) | |
| Age at loss of ambulation 1 | 9.45 (2.21) | |
| Low LVEF in echocardiogram study 2 | 12 (30.8) | |
| ACEI or ARB | 18 (19.1) |
| Variant | n (n%) |
|---|---|
| p.(Arg2095*) | 2 (10.5) |
| p.(Arg2680*) | 2 (10.5) |
| p.(Arg145*) | 2 (10.5) |
| p.(Try151*) | 1 (5.26) |
| p.(Arg1051*) | 1 (5.26) |
| p.(Arg3113*) | 1 (5.26) |
| p.(Trp2915*) | 1 (5.26) |
| p.(Trp3083*) | 1 (5.26) |
| p.(Arg145*) | 1 (5.26) |
| p.(Arg2098*) | 1 (5.26) |
| p.(Arg2982*) | 1 (5.26) |
| p.(Gln3037*) | 1 (5.26) |
| p.(Gln45*) | 1 (5.26) |
| p.(Arg2870*) | 1 (5.26) |
| p.(Cys3337*) | 1 (5.26) |
| p.(Tyr1882*) | 1 (5.26) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braga, V.L.L.; Lima, D.P.; Mariano, T.C.; Lima, P.L.G.d.S.B.; Maia, A.B.d.A.; da Silva Meireles, W.W.; de Oliveira Pessoa, K.T.; de Oliveira, C.M.; Ribeiro, E.M.; Nóbrega, P.R.; et al. Higher Prevalence of Nonsense Pathogenic DMD Variants in a Single-Center Cohort from Brazil: A Genetic Profile Study That May Guide the Choice of Disease-Modifying Treatments. Brain Sci. 2023, 13, 1521. https://doi.org/10.3390/brainsci13111521
Braga VLL, Lima DP, Mariano TC, Lima PLGdSB, Maia ABdA, da Silva Meireles WW, de Oliveira Pessoa KT, de Oliveira CM, Ribeiro EM, Nóbrega PR, et al. Higher Prevalence of Nonsense Pathogenic DMD Variants in a Single-Center Cohort from Brazil: A Genetic Profile Study That May Guide the Choice of Disease-Modifying Treatments. Brain Sciences. 2023; 13(11):1521. https://doi.org/10.3390/brainsci13111521
Chicago/Turabian StyleBraga, Vitor Lucas Lopes, Danielle Pessoa Lima, Tamiris Carneiro Mariano, Pedro Lucas Grangeiro de Sá Barreto Lima, Ana Beatriz de Almeida Maia, Wallace William da Silva Meireles, Kécia Tavares de Oliveira Pessoa, Cristiane Mattos de Oliveira, Erlane Marques Ribeiro, Paulo Ribeiro Nóbrega, and et al. 2023. "Higher Prevalence of Nonsense Pathogenic DMD Variants in a Single-Center Cohort from Brazil: A Genetic Profile Study That May Guide the Choice of Disease-Modifying Treatments" Brain Sciences 13, no. 11: 1521. https://doi.org/10.3390/brainsci13111521
APA StyleBraga, V. L. L., Lima, D. P., Mariano, T. C., Lima, P. L. G. d. S. B., Maia, A. B. d. A., da Silva Meireles, W. W., de Oliveira Pessoa, K. T., de Oliveira, C. M., Ribeiro, E. M., Nóbrega, P. R., & Pessoa, A. L. S. (2023). Higher Prevalence of Nonsense Pathogenic DMD Variants in a Single-Center Cohort from Brazil: A Genetic Profile Study That May Guide the Choice of Disease-Modifying Treatments. Brain Sciences, 13(11), 1521. https://doi.org/10.3390/brainsci13111521