
Differential DNA Methylation from Autistic Children Enriches Evidence for Genes Associated with ASD and New Candidate Genes

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
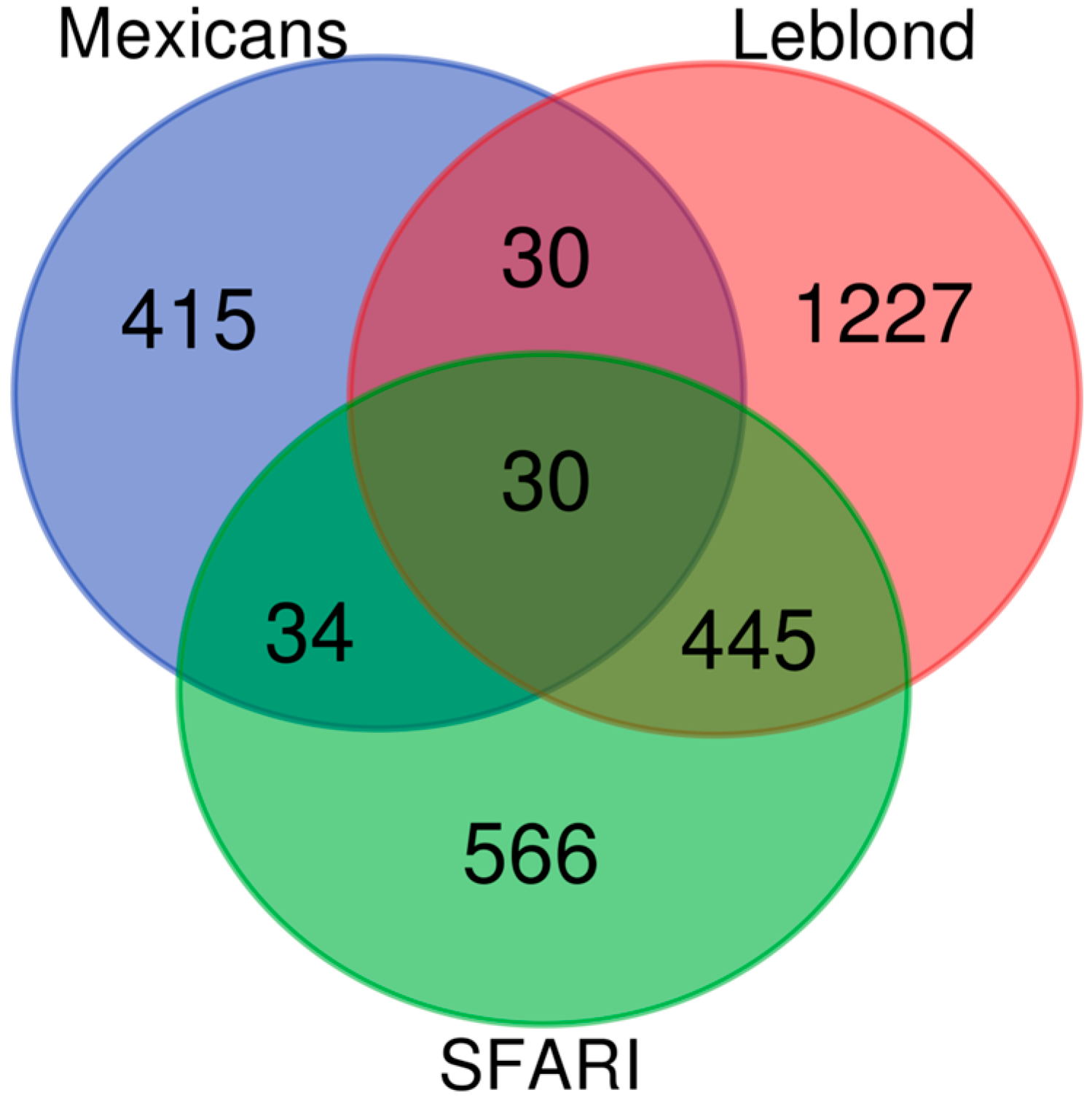
3. Results
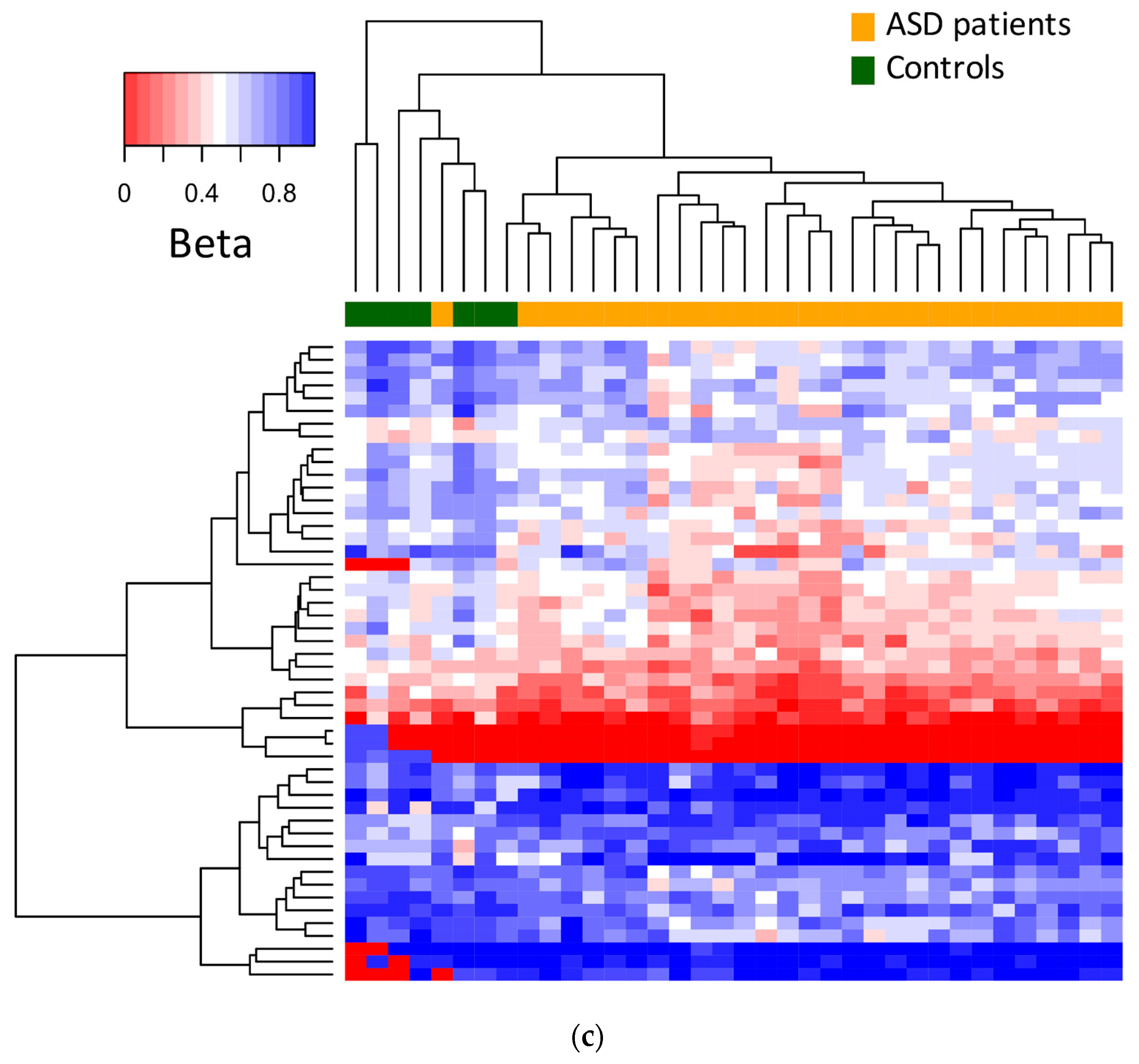
3.1. Differential DNA Methylation
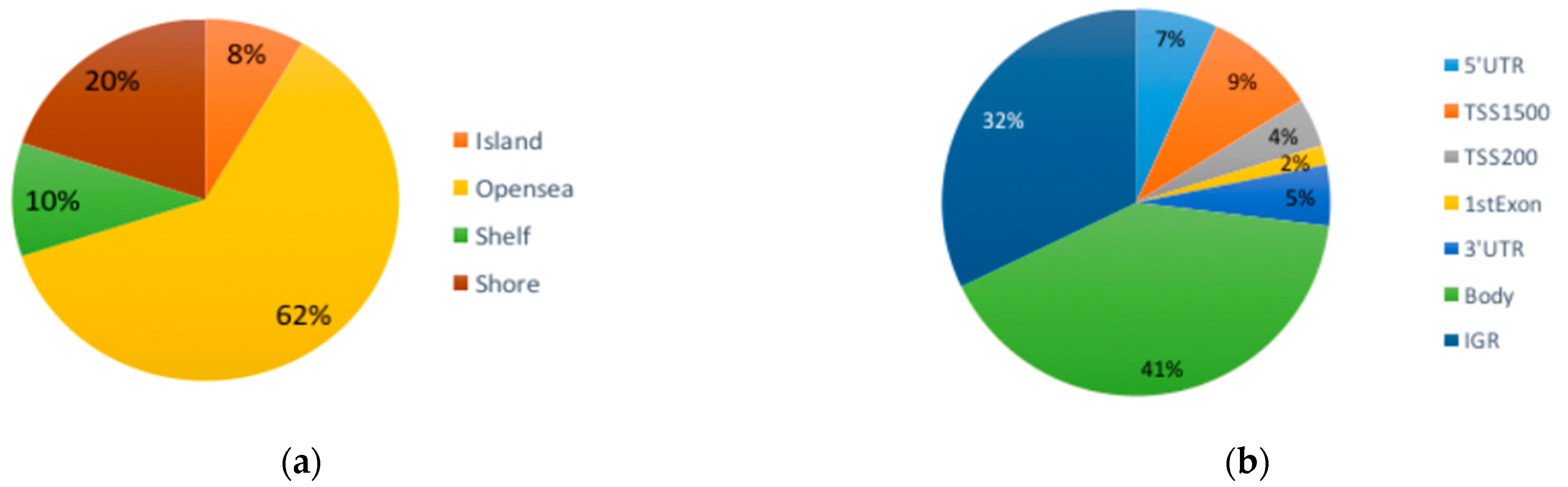
3.2. Functional Analysis of DMCs
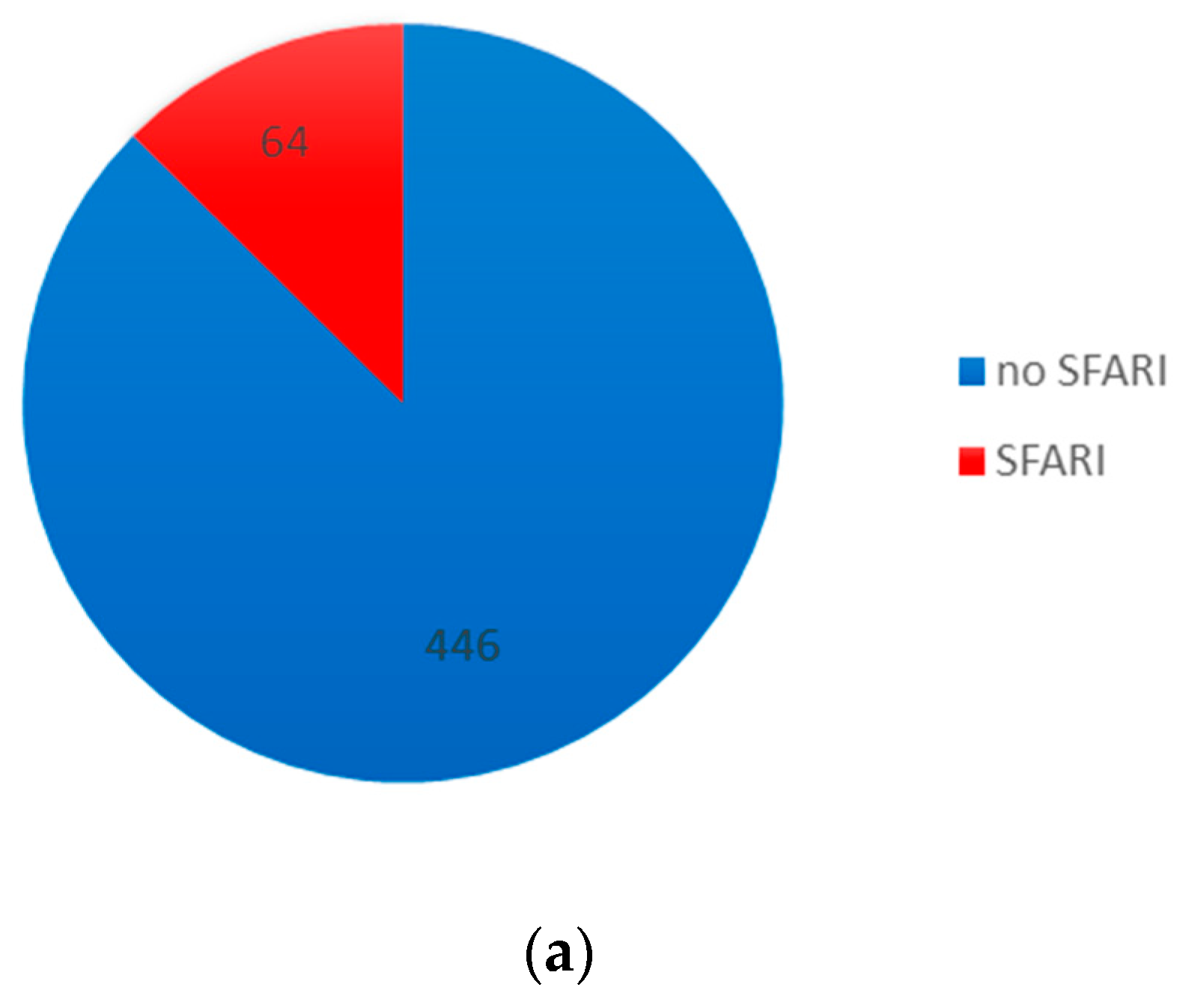
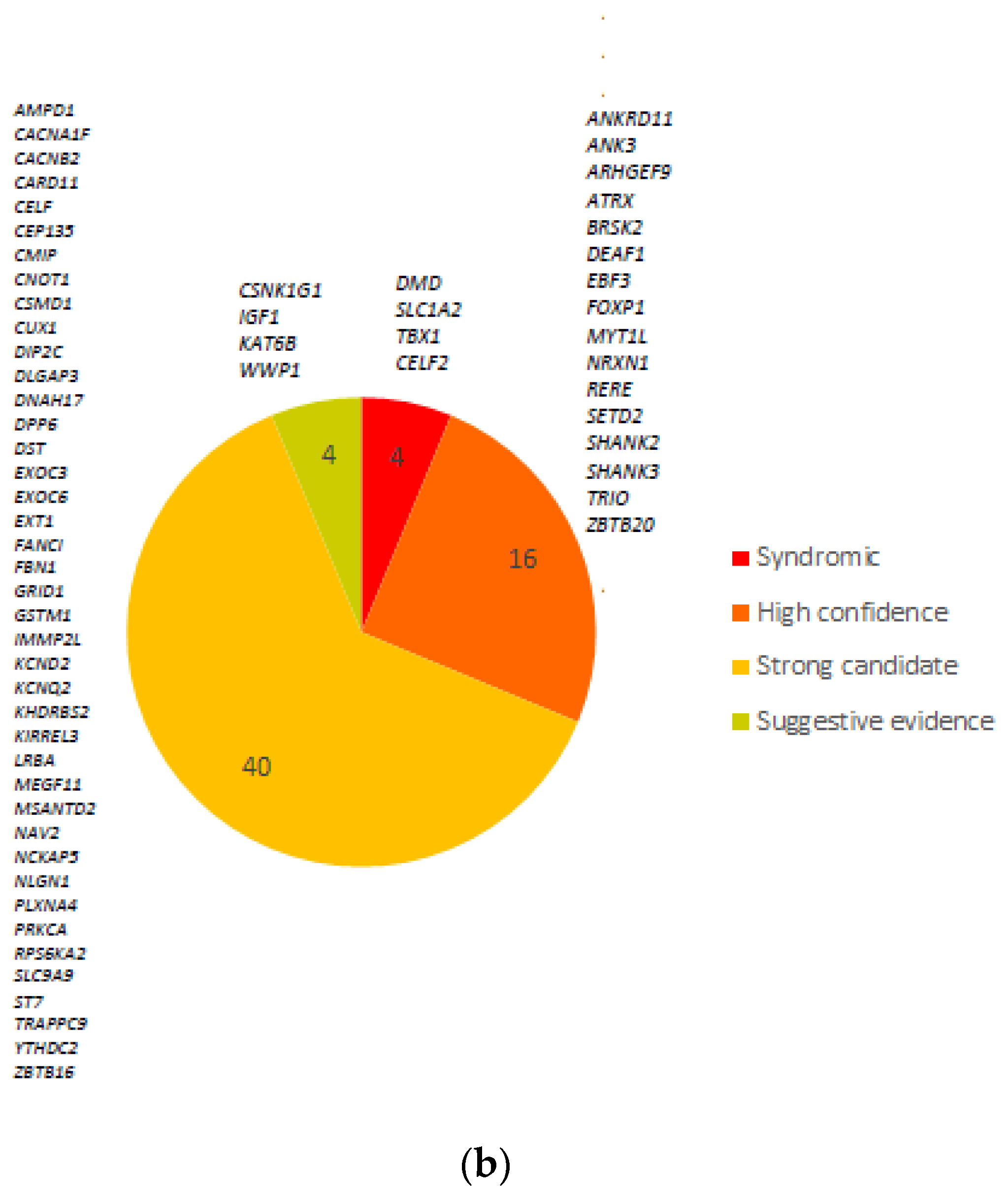
3.3. Comparison with ASD and Neurodevelopmental Gene Databases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/autism-spectrum-disorders (accessed on 25 March 2023).
- Fombonne, E.; Marcin, C.; Manero, A.C.; Bruno, R.; Diaz, C.; Villalobos, M.; Ramsay, K.; Nealy, B. Prevalence of Autism Spectrum Disorders in Guanajuato, Mexico: The Leon survey. J. Autism Dev. Disord. 2016, 46, 1669–1685. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Mughal, S.; Faizy, R.M.; Saadabadi, A. Autism Spectrum Disorder; en StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023; [En línea]. Available online: http://www.ncbi.nlm.nih.gov/books/NBK525976/ (accessed on 9 September 2023).
- Rylaarsdam, L.E.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Geschwind, D.H. Genetics of autism spectrum disorders. Trends Cogn. Sci. 2011, 15, 409–416. [Google Scholar] [CrossRef]
- Dias, C.M.; Walsh, C.A. Recent Advances in Understanding the Genetic Architecture of Autism. Annu. Rev. Genom. Hum. Genet. 2020, 21, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Choi, J.; Lee, W.J.; Do, J.T. Genetic and Epigenetic Etiology Underlying Autism Spectrum Disorder. J. Clin. Med. 2020, 9, 966. [Google Scholar] [CrossRef]
- Hoischen, A.; Krumm, N.; Eichler, E.E. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat. Neurosci. 2014, 17, 764–772. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Kushak, R.I.; Sengupta, A.; Winter, H.S. Interactions between the intestinal microbiota and epigenome in individuals with autism spectrum disorder. Dev. Med. Child Neurol. 2021, 64, 296–304. [Google Scholar] [CrossRef]
- Tremblay, M.W.; Jiang, Y.-H. DNA Methylation and Susceptibility to Autism Spectrum Disorder. Annu. Rev. Med. 2019, 70, 151–166. [Google Scholar] [CrossRef]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef] [PubMed]
- Grova, N.; Schroeder, H.; Olivier, J.-L.; Turner, J.D. Epigenetic and Neurological Impairments Associated with Early Life Exposure to Persistent Organic Pollutants. Int. J. Genom. 2019, 2019, 2085496. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.Y.; Meaburn, E.L.; Ronald, A.; Price, T.S.; Jeffries, A.R.; Schalkwyk, L.C.; Plomin, R.; Mill, J. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry 2014, 19, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Loke, Y.J.; Hannan, A.J.; Craig, J.M. The Role of Epigenetic Change in Autism Spectrum Disorders. Front. Neurol. 2015, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Hu, X.; Li, Y.; Ni, Y.; Xue, L. Identification of methylation markers for diagnosis of autism spectrum disorder. Metab. Brain Dis. 2022, 37, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Gadad, B.S.; Hewitson, L.; Young, K.A.; German, D.C. Neuropathology and Animal Models of Autism: Genetic and Environmental Factors. Autism Res. Treat. 2013, 2013, 731935. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.V.; Ellis, S.E.; Bakulski, K.M.; Sheppard, B.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Feinberg, A.P.; Arking, D.E.; Ladd-Acosta, C.; et al. Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder. Nat. Commun. 2017, 8, 1011. [Google Scholar] [CrossRef]
- Ciernia, A.V.; LaSalle, J. The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat. Rev. Neurosci. 2016, 17, 411–423. [Google Scholar] [CrossRef]
- Saeliw, T.; Tangsuwansri, C.; Thongkorn, S.; Chonchaiya, W.; Suphapeetiporn, K.; Mutirangura, A.; Tencomnao, T.; Hu, V.W.; Sarachana, T. Integrated genome-wide Alu methylation and transcriptome profiling analyses reveal novel epigenetic regulatory networks associated with autism spectrum disorder. Mol. Autism 2018, 9, 27. [Google Scholar] [CrossRef]
- Van Dongen, J.; Ehli, E.A.; Jansen, R.; Van Beijsterveldt, C.E.M.; Willemsen, G.; Hottenga, J.J.; Kallsen, N.A.; Peyton, S.A.; Breeze, C.E.; Kluft, C.; et al. Genome-wide analysis of DNA methylation in buccal cells: A study of monozygotic twins and mQTLs. Epigenetics Chromatin 2018, 11, 54. [Google Scholar] [CrossRef]
- McEwen, L.M.; O’donnell, K.J.; McGill, M.G.; Edgar, R.D.; Jones, M.J.; MacIsaac, J.L.; Lin, D.T.S.; Ramadori, K.; Morin, A.; Gladish, N.; et al. The PedBE clock accurately estimates DNA methylation age in pediatric buccal cells. Proc. Natl. Acad. Sci. USA 2020, 117, 23329–23335. [Google Scholar] [CrossRef] [PubMed]
- Bakulski, K.M.; Dou, J.F.; Feinberg, J.I.; Aung, M.T.; Ladd-Acosta, C.; Volk, H.E.; Newschaffer, C.J.; Croen, L.A.; Hertz-Picciotto, I.; Levy, S.E.; et al. Autism-Associated DNA Methylation at Birth From Multiple Tissues Is Enriched for Autism Genes in the Early Autism Risk Longitudinal Investigation. Front. Mol. Neurosci. 2021, 14, 775390. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism Diagnostic Interview-Revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.N. Social Responsiveness Scale. In Encyclopedia of Autism Spectrum Disorders; Volkmar, F.R., Ed.; Springer: New York, NY, USA, 2013; pp. 2919–2929. [Google Scholar]
- WebGestalt. 2023. Available online: http://www.webgestalt.org/ (accessed on 30 July 2023).
- SFARI gene. 2023. Available online: https://gene.sfari.org/ (accessed on 16 April 2023).
- Leblond, C.S.; Le, T.-L.; Malesys, S.; Cliquet, F.; Tabet, A.-C.; Delorme, R.; Rolland, T.; Bourgeron, T. Operative list of genes associated with autism and neurodevelopmental disorders based on database review. Mol. Cell Neurosci. 2021, 113, 103623. [Google Scholar] [CrossRef] [PubMed]
- Lansdon, L.A.; Darbro, B.W.; Petrin, A.L.; Hulstrand, A.M.; Standley, J.M.; Brouillette, R.B.; Long, A.; Mansilla, M.A.; Cornell, R.A.; Murray, J.C.; et al. Identification of Isthmin 1 as a Novel Clefting and Craniofacial Patterning Gene in Humans. Genetics 2018, 208, 283–296. [Google Scholar] [CrossRef]
- Wu, Y.; Liang, X.; Ni, J.; Zhao, R.; Shao, S.; Lu, S.; Han, W.; Yu, L. Effect of ISM1 on the Immune Microenvironment and Epithelial-Mesenchymal Transition in Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9, 681240. [Google Scholar] [CrossRef]
- Menghuan, L.; Yang, Y.; Qianhe, M.; Na, Z.; Shicheng, C.; Bo, C.; XueJie, Y.I. Advances in research of biological functions of Isthmin-1. J. Cell Commun. Signal. 2023, 17, 507–521. [Google Scholar] [CrossRef]
- Valle-Rios, R.; Maravillas-Montero, J.L.; Burkhardt, A.M.; Martinez, C.; Buhren, B.A.; Homey, B.; Gerber, P.A.; Robinson, O.; Hevezi, P.; Zlotnik, A. Isthmin 1 Is a Secreted Protein Expressed in Skin, Mucosal Tissues, and NK, NKT, and Th17 Cells. J. Interf. Cytokine Res. 2014, 34, 795–801. [Google Scholar] [CrossRef]
- Boni, C.; Laudanna, C.; Sorio, C. A Comprehensive Review of Receptor-Type Tyrosine-Protein Phosphatase Gamma (PTPRG) Role in Health and Non-Neoplastic Disease. Biomolecules 2022, 12, 84. [Google Scholar] [CrossRef]
- Di Maio, A.; De Rosa, A.; Pelucchi, S.; Garofalo, M.; Marciano, B.; Nuzzo, T.; Gardoni, F.; Isidori, A.M.; Di Luca, M.; Errico, F.; et al. Analysis of mRNA and Protein Levels of CAP2, DLG1 and ADAM10 Genes in Post-Mortem Brain of Schizophrenia, Parkinson’s and Alzheimer’s Disease Patients. Int. J. Mol. Sci. 2022, 23, 1539. [Google Scholar] [CrossRef]
- Won, S.Y.; Lee, P.; Kim, H.M. Synaptic organizer: Slitrks and type IIa receptor protein tyrosine phosphatases. Curr. Opin. Struct. Biol. 2019, 54, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Aruga, J.; Yokota, N.; Mikoshiba, K. Human SLITRK family genes: Genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 2003, 315, 87–94. [Google Scholar] [CrossRef]
- Yim, Y.S.; Kwon, Y.; Nam, J.; Yoon, H.I.; Lee, K.; Kim, D.G.; Kim, E.; Kim, C.H.; Ko, J. Slitrks control excitatory and inhibitory synapse formation with LAR receptor protein tyrosine phosphatases. Proc. Natl. Acad. Sci. USA 2013, 110, 4057–4062. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, Z.; Ghaderian, S.M.H.; Najmabadi, H.; Omrani, M.D. Understanding the Molecular Basis of Fragile X Syndrome Using Differentiated Mesenchymal Stem Cells. Iran. J. Child Neurol. 2022, 16, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Isoherranen, N.; Zhong, G. Biochemical and physiological importance of the CYP26 retinoic acid hydroxylases. Pharmacol. Ther. 2019, 204, 107400. [Google Scholar] [CrossRef]
- The Brainstorm Consortium; Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; et al. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef]
- Grotzinger, A.D.; Mallard, T.T.; Akingbuwa, W.A.; Ip, H.F.; Adams, M.J.; Lewis, C.M.; McIntosh, A.M.; Grove, J.; Dalsgaard, S.; Lesch, K.-P.; et al. Genetic architecture of 11 major psychiatric disorders at biobehavioral, functional genomic and molecular genetic levels of analysis. Nat. Genet. 2022, 54, 548–559. [Google Scholar] [CrossRef]
- Hammerschlag, A.R.; De Leeuw, C.A.; Middeldorp, C.M.; Polderman, T.J.C. Synaptic and brain-expressed gene sets relate to the shared genetic risk across five psychiatric disorders. Psychol. Med. 2020, 50, 1695–1705. [Google Scholar] [CrossRef]
- Zoccante, L.; Ciceri, M.L.; Gozzi, L.A.; Di Gennaro, G.; Zerman, N. The “Connectivome Theory”: A New Model to Understand Autism Spectrum Disorders. Front. Psychiatry 2022, 12, 794516. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.M.; Satterstrom, F.K.; Peng, M.; Brand, H.; Collins, R.L.; Dong, S.; Wamsley, B.; Klei, L.; Wang, L.; Hao, S.P.; et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 2022, 54, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, X.; Li, X.-L.; Towers, A.; Cao, X.; Wang, P.; Bowman, R.; Yang, H.; Goldstein, J.; Li, Y.-J.; et al. Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum. Mol. Genet. 2013, 23, 1563–1578. [Google Scholar] [CrossRef] [PubMed]
- Braun, P.R.; Han, S.; Hing, B.; Nagahama, Y.; Gaul, L.N.; Heinzman, J.T.; Grossbach, A.J.; Close, L.; Dlouhy, B.J.; Howard, M.A.; et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry 2019, 9, 47. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Mean Age (Years) | SD | Diagnosis | Therapy | Medication | Formal Schooling |
|---|---|---|---|---|---|---|
| Cases (29) | 5.1 | 1.1 | ASD | Yes:69% ND:31% | Yes:48.3% No:20.7% ND:31.0% | Yes:55.2% No:17.2% ND:27.6% |
| Controls (7) | 5.7 | 1.1 | None | None | None | Yes: 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Marín, M.E.; Castro Martínez, X.H.; Centeno Cruz, F.; Barajas-Olmos, F.; Náfate López, O.; Gómez Cotero, A.G.; Orozco, L.; Nicolini Sánchez, H. Differential DNA Methylation from Autistic Children Enriches Evidence for Genes Associated with ASD and New Candidate Genes. Brain Sci. 2023, 13, 1420. https://doi.org/10.3390/brainsci13101420
Morales-Marín ME, Castro Martínez XH, Centeno Cruz F, Barajas-Olmos F, Náfate López O, Gómez Cotero AG, Orozco L, Nicolini Sánchez H. Differential DNA Methylation from Autistic Children Enriches Evidence for Genes Associated with ASD and New Candidate Genes. Brain Sciences. 2023; 13(10):1420. https://doi.org/10.3390/brainsci13101420
Chicago/Turabian StyleMorales-Marín, Mirna Edith, Xochitl Helga Castro Martínez, Federico Centeno Cruz, Francisco Barajas-Olmos, Omar Náfate López, Amalia Guadalupe Gómez Cotero, Lorena Orozco, and Humberto Nicolini Sánchez. 2023. "Differential DNA Methylation from Autistic Children Enriches Evidence for Genes Associated with ASD and New Candidate Genes" Brain Sciences 13, no. 10: 1420. https://doi.org/10.3390/brainsci13101420
APA StyleMorales-Marín, M. E., Castro Martínez, X. H., Centeno Cruz, F., Barajas-Olmos, F., Náfate López, O., Gómez Cotero, A. G., Orozco, L., & Nicolini Sánchez, H. (2023). Differential DNA Methylation from Autistic Children Enriches Evidence for Genes Associated with ASD and New Candidate Genes. Brain Sciences, 13(10), 1420. https://doi.org/10.3390/brainsci13101420