

Narcolepsy—A Neuropathological Obscure Sleep Disorder: A Narrative Review of Current Literature

 ,
,  ,
,  ,
, 


Abstract
1. Introduction
2. Epidemiology
3. Genetic and Non-Genetic Associations with Narcolepsy
3.1. HLA Genes: HLADQB1 as Risk Factor of Narcolepsy Type 1
3.2. Non-HLA Genetic Associations
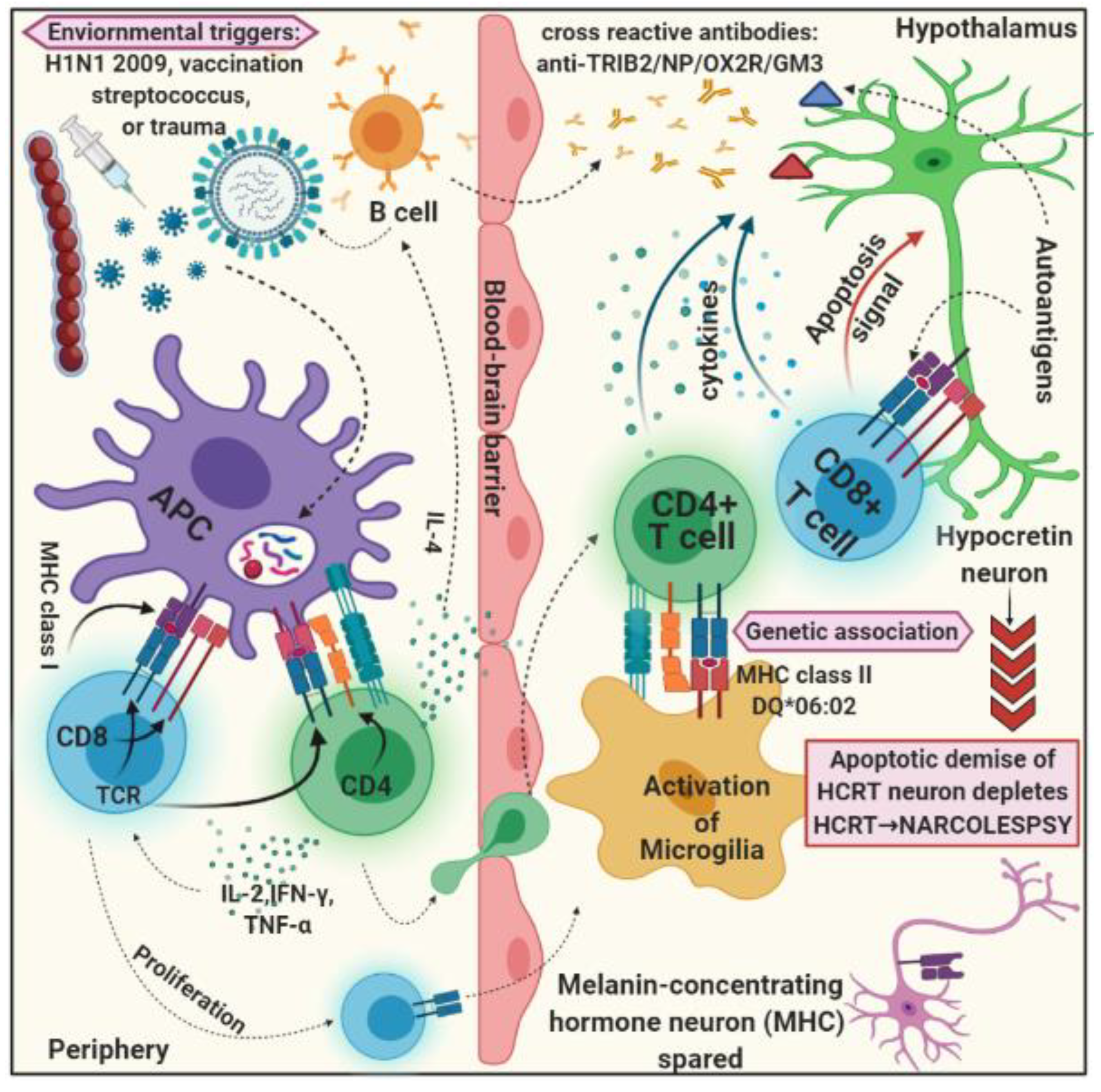
3.3. Environmental Factors
4. Inflammatory Response
4.1. Autoimmune Hypothesis
4.2. Autoantibody and T Cell Laboratory Findings
5. Narcolepsy and Neuromediator Systems
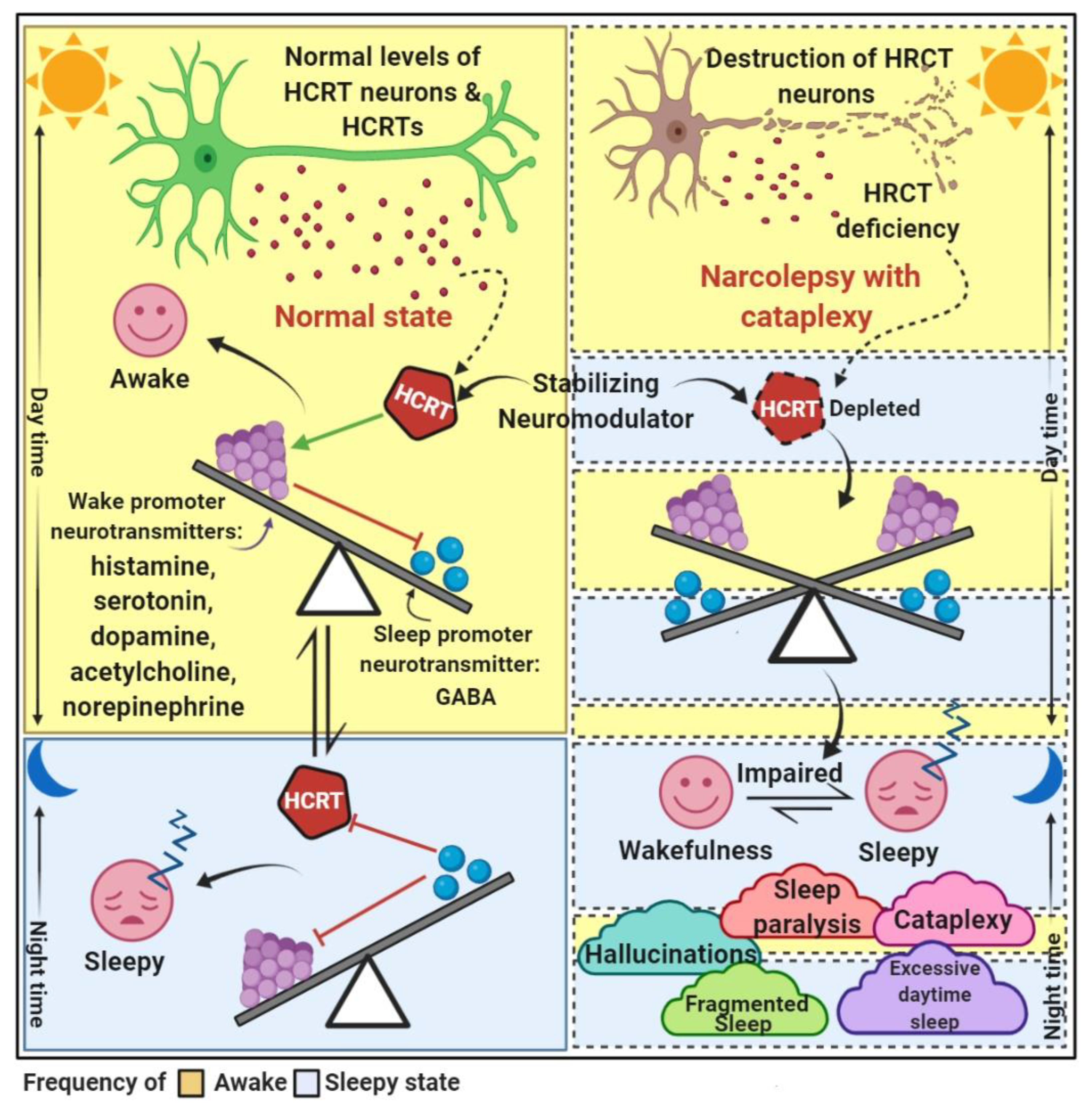
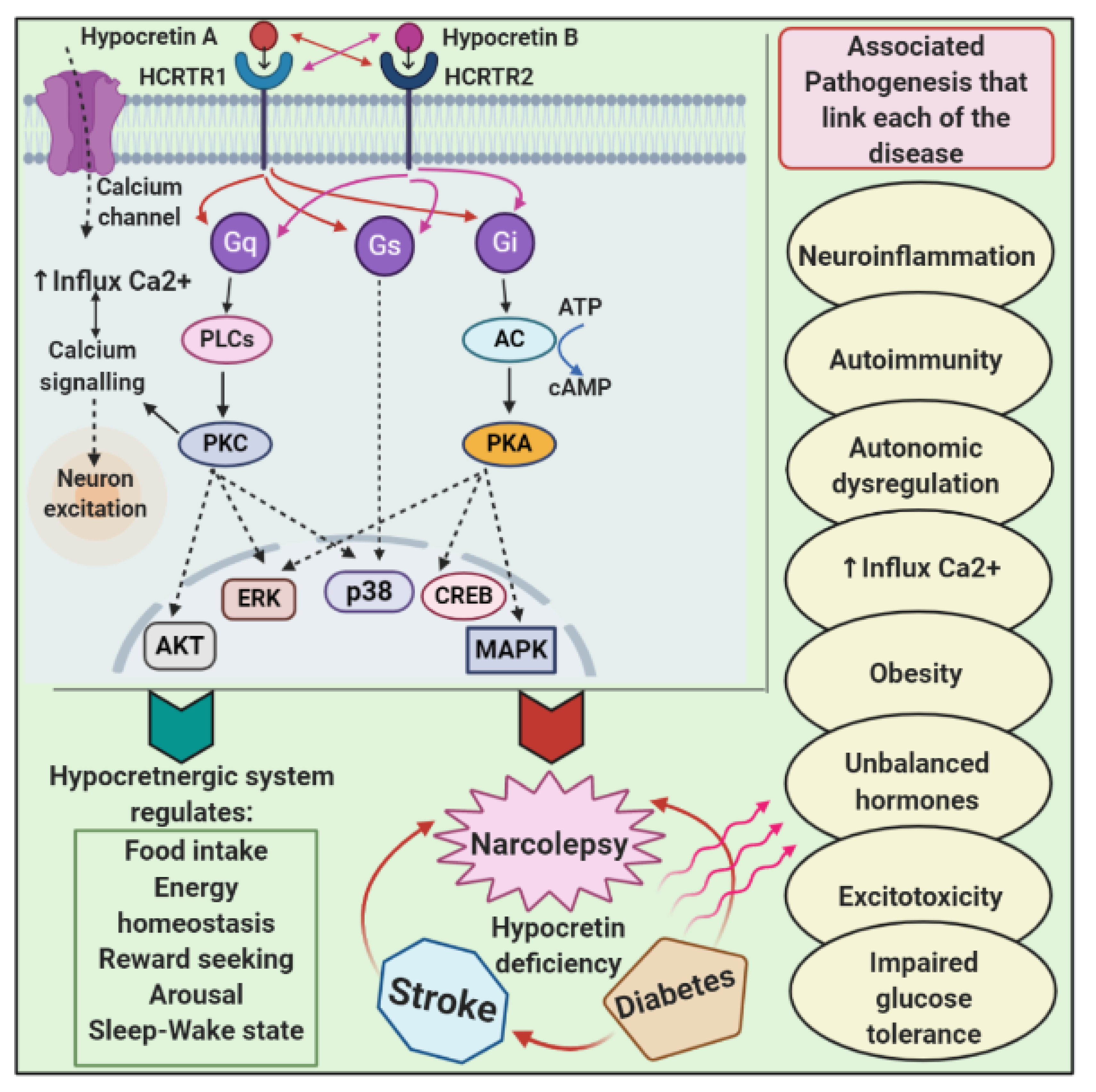
5.1. Hypocretinergic System
5.2. Dopaminergic System
5.3. Histaminergic System
6. Clinical Features
7. Pathophysiology and Links with Other Diseases
7.1. Narcolepsy and Diabetes
7.2. Narcolepsy and Ischemic Stroke
7.3. Narcolepsy and Alzheimer’s Disease (AD)
7.4. Narcolepsy and Parkinson’s Disease
8. Diagnosis and Treatment
8.1. Future Therapeutics
8.2. Prospects for Future Research

{kind=link}
{kind=link}
{kind=link}
| Sr No. | Class | Drug Candidate | Mode of Action | Improve Narcolepsy Symptoms |
|---|---|---|---|---|
| 1 | Stimulants [189,190,191,192,193] | Modafinil | Blocks several monoaminergic transporters and inhibits dopamine reuptake transporter. | Potential to cause fatal hepatotoxicity; no longer recommended. |
| - | - | Armodafinil | (R)-enantiomer of modafinil more-potent and long-lasting | Treats excessive daytime sleepiness. |
| - | - | Methylphenidate | Non-competitive dopamine reuptake blocker and, to a lesser degree, a serotonin–noradrenaline reuptake blocker. | Second-line treatment for excessive daytime sleepiness. |
| - | - | Dextro-amphetamine sulfate | Competitive dopamine transporter blocker that also blocks the vesicular mono amine transporter. | Third-line treatment for excessive daytime sleepiness. |
| - | - | Pemoline | Selectively blocks dopamine reuptake. | |
| - | - | Solriamfetol (JZP-11) | Inhibits norepinephrine-dopamine reuptake. | Treats impaired wakefulness and excessive sleepiness. |
| 2 | Sodium salt of γ-hydroxybutyrate (GHB; a neurotransmitter that related to γ-aminobutyric acid (GABA) [194] | Sodium oxybate | GABAB receptor agonist that activates the GABA type B receptor and possibly its own specific GHB receptor. | First-line treatment for cataplexy. Improves “qualitative wakefulness”—fewer nightly awakenings, reduces NREM stage 1 sleep and increases slow-wave sleep, decreases arousals, and has a variable effect on latency and amount of REM sleep. Alleviates sleep paralysis, hypnagogic hallucinations. |
| 3 | Antidepressant [16,162,195,196,197,198,199] | Venlafaxine, duloxetine, reboxetine, and viloxazine (SNRI), atomoxetine (SNRI) | Blocks serotonin–noradrenaline reuptake pumps. | First-line off-label to treat cataplexy. Improves excessive daytime and night-time sleepiness. Second line to treat cataplexy. |
| Clomipramine, imipramine (TCA) | Mono-aminergic reuptake inhibition, inhibit the reuptake of catecholamines. | It increases muscle tone and suppresses REM sleep. Second line to treat cataplexy. | ||
| Selegiline (MOAI) | Monoamine oxidase type B inhibitor. | Preferred initial choice for treatment of excessive daytime sleepiness. Can be used to treat cataplexy. | ||
| Fluoxetine, Femoxetine, Citalopram (SSRI) | Blocks serotonin reuptake pumps. | Second line or third line to treat cataplexy. | ||
| 4 | Psychoactive drugs [200,201] | Benzodiazepines and hypnotics, Triazolam, Ambien, clonazepam | Enhances the effect of the neurotransmitter gamma-aminobutyric acid (GABA) at the GABAA receptor. | Second-line disrupted nocturnal sleep. Decreases arousal at night and sleep fragmentation. It improves cataplexy and sleep paralysis. |
| 5 | H3 blockers [202] | Pitolisant | histamine H3 receptor antagonists (inverse agonist). | Improves excessive daytime sleepiness. Not recommended in the newest guidelines. |
| 6 | Hypocretin replacement therapy or hypocretin receptor agonists [203] | - | HCRTR2 agonist-YNT-185, trace amine-associated receptor 1 (TAAR1) agonist. | Improves cataplexy, excessive daytime sleepiness, sleep paralysis and hypnagogic/hypnopompic hallucinations and suppress REM sleep. |
| 7 | Non-pharmacological therapy [203] | Sleep–wake schedules | - | Single scheduled daytime nap (of about 2 h) reduces the total amount of involuntary daytime sleep. Combined with stimulant treatment, two 15 min naps per day, and regular night-time sleep schedules, it has been shown to reduce subjective drowsiness and involuntary daytime sleep. Intensity of night-time NREM sleep is improved by daytime sleep restriction. |
| 8 | - | Psychosocial guidance | - | Helps in awareness and improves social problems associated with narcolepsy |
| 9 | - | Slow-wave sleep-enhancing treatments | - | Improves sleep |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Taheri, S. The Immune Basis of Narcolepsy: What Is the Evidence? Sleep Med. Clin. 2017, 12, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Poli, F.; Overeem, S.; Lammers, G.J.; Plazzi, G.; Lecendreux, M.; Bassetti, C.L.; Dauvilliers, Y.; Keene, D.; Khatami, R.; Li, Y.; et al. Narcolepsy as an adverse event following immunization: Case definition and guidelines for data collection, analysis and presentation. Vaccine 2013, 31, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Sarkanen, T.; Alakuijala, A.; Partinen, M. Ullanlinna Narcolepsy Scale in diagnosis of narcolepsy. Sleep 2019, 42, 238. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, C.L.A.; Kallweit, U.; Vignatelli, L.; Plazzi, G.; Lecendreux, M.; Baldin, E.; Dolenc-Groselj, L.; Jennum, P.; Khatami, R.; Manconi, M.; et al. European guideline and expert statements on the management of narcolepsy in adults and children. Eur. J. Neurol. 2021, 28, 2815–2830. [Google Scholar] [CrossRef]
- Monaca, C.; Franco, P.; Philip, P.; Dauvilliers, Y. French consensus. Type 1 and type 2 Narcolepsy: Investigations and follow-up. Rev. Neurol. 2017, 173, 25–31. [Google Scholar] [CrossRef]
- Nevsimalova, S.; Vaňková, J.; Štěpánová, I.; Seemanova, E.; Mignot, E.; Nishino, S. Hypocretin deficiency in Prader-Willi syndrome. Eur. J. Neurol. 2005, 12, 70–72. [Google Scholar] [CrossRef]
- Kornum, B.R.; Knudsen, S.; Ollila, H.M.; Pizza, F.; Jennum, P.J.; Dauvilliers, Y.; Overeem, S. Narcolepsy. Nat. Rev. Dis. Primers. 2017, 3, 16100. [Google Scholar] [CrossRef]
- Sateia, M.J. International classification of sleep disorders-third edition: Highlights and modifications. Chest 2014, 146, 1387–1394. [Google Scholar] [CrossRef]
- Plazzi, G.; Ruoff, C.; Lecendreux, M.; Dauvilliers, Y.; Rosen, C.L.; Black, J.; Parvataneni, R.; Guinta, D.; Wang, Y.G.; Mignot, E. Treatment of paediatric narcolepsy with sodium oxybate: A double-blind, placebo-controlled, randomised-withdrawal multicentre study and open-label investigation. Lancet Child Adolesc. Health 2018, 2, 483–494. [Google Scholar] [CrossRef]
- Vuorela, A.; Freitag, T.L.; Leskinen, K.; Pessa, H.; Härkönen, T.; Stracenski, I.; Kirjavainen, T.; Olsen, P.; Saarenpää-Heikkilä, O.; Ilonen, J.; et al. Enhanced influenza A H1N1 T cell epitope recognition and cross-reactivity to protein-O-mannosyltransferase 1 in Pandemrix-associated narcolepsy type 1. Nat. Commun. 2021, 12, 2283. [Google Scholar] [CrossRef]
- Bogan, R.K.; Thorpy, M.J.; Dauvilliers, Y.; Partinen, M.; Del Rio Villegas, R.; Foldvary-Schaefer, N.; Skowronski, R.; Tang, L.; Skobieranda, F.; Šonka, K. Efficacy and safety of calcium, magnesium, potassium, and sodium oxybates (lower-sodium oxybate [LXB]; JZP-258) in a placebo-controlled, double-blind, randomized withdrawal study in adults with narcolepsy with cataplexy. Sleep 2021, 44, 206. [Google Scholar] [CrossRef] [PubMed]
- Sarkanen, T.O.; Alakuijala, A.P.E.; Dauvilliers, Y.A.; Partinen, M.M. Incidence of narcolepsy after H1N1 influenza and vaccinations: Systematic review and meta-analysis. Sleep Med. Rev. 2018, 38, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Goel, D.; Farney, R.; Walker, J. Narcolepsy: A case from India with polysomnographic findings. Neurol. India 2012, 60, 79–81. [Google Scholar] [CrossRef]
- Scammell, T.E. The neurobiology, diagnosis, and treatment of narcolepsy. Ann. Neurol. 2003, 53, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Mignot, E. Genetic and familial aspects of narcolepsy. Neurology 1998, 50, S16–S22. [Google Scholar] [CrossRef]
- Akintomide, G.S.; Rickards, H. Narcolepsy: A review. Neuropsychiatr. Dis. Treat. 2011, 7, 507–518. [Google Scholar] [CrossRef]
- Simmonds, M.J.; Howson, J.M.M.; Heward, J.M.; Carr-Smith, J.; Franklyn, J.A.; Todd, J.A.; Gough, S.C.L. A novel and major association of HLA-C in Graves’ disease that eclipses the classical HLA-DRB1 effect. Hum. Mol. Genet. 2007, 16, 2149–2153. [Google Scholar] [CrossRef]
- Lundström, E.; Källberg, H.; Smolnikova, M.; Ding, B.; Rönnelid, J.; Alfredsson, L.; Klareskog, L.; Padyukov, L. Gough. Opposing effects of hla-drb1 * 13 alleles on the risk of developing anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 2009, 60, 924–930. [Google Scholar] [CrossRef]
- Knip, M.; Siljander, H.; Ilonen, J.; Simell, O.; Veijola, R. Role of humoral beta-cell autoimmunity in type 1 diabetes. Pediatr. Diabetes 2016, 17, 17–24. [Google Scholar] [CrossRef]
- Singal, D.P.; Blajchman, M.A. Histocompatibility (HL A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes 1973, 22, 429–432. [Google Scholar] [CrossRef]
- Sonti, S.; Grant, S.F.A. Leveraging genetic discoveries for sleep to determine causal relationships with common complex traits. Sleep 2022, 45, 180. [Google Scholar] [CrossRef] [PubMed]
- Peacock, J.; Benca, R.M. Narcolepsy: Clinical features, co-morbidities & treatment. Indian J. Med. Res. 2010, 131, 338–349. [Google Scholar] [PubMed]
- Tafti, M. Genetic aspects of normal and disturbed sleep. Sleep Med. 2009, 10, S17–S21. [Google Scholar] [CrossRef]
- Kachooei-Mohaghegh-Yaghoobi, L.; Rezaei-Rad, F.; Sadeghniiat-Haghighi, K.; Zamani, M. The impact of the HLA DQB1 gene and amino acids on the development of narcolepsy. Int. J. Neurosci. 2020, 22, 706–713. [Google Scholar] [CrossRef]
- Bernard-Valnet, R.; Yshii, L.; Quériault, C.; Nguyen, X.H.; Arthaud, S.; Rodrigues, M.; Canivet, A.; Morel, A.-L.; Matthys, A.; Bauer, J.; et al. CD8 T cell-mediated killing of orexinergic neurons induces a narcolepsy-like phenotype in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 10956–10961. [Google Scholar] [CrossRef] [PubMed]
- Ollila, H.M.; Ravel, J.M.; Han, F.; Faraco, J.; Lin, L.; Zheng, X.; Plazzi, G.; Dauvilliers, Y.; Pizza, F.; Hong, S.-C.; et al. HLA-DPB1 and HLA class I confer risk of and protection from narcolepsy. Am. J. Hum. Genet. 2015, 96, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Lin, L.; Warby, S.C.; Faraco, J.; Li, J.; Dong, S.X.; An, P.; Zhao, L.; Wang, L.H.; Li, Q.Y.; et al. Narcolepsy onset is seasonal and increased following the 2009 H1N1 pandemic in china. Ann. Neurol. 2011, 70, 410–417. [Google Scholar] [CrossRef]
- Partinen, M.; Hublin, C.; Kaprio, J.; Koskenvuo, M.; Guilleminault, C. Twin studies in narcolepsy. Sleep 1994, 17, S13–S16. [Google Scholar] [CrossRef][Green Version]
- Bourgin, P.; Zeitzer, J.M.; Mignot, E. CSF hypocretin-1 assessment in sleep and neurological disorders. Lancet Neurol. 2008, 7, 649–662. [Google Scholar] [CrossRef]
- Aran, A.; Lin, L.; Nevsimalova, S.; Plazzi, G.; Hong, S.C.; Weiner, K.; Zeitzer, J.; Mignot, E. Elevated anti-streptococcal antibodies in patients with recent narcolepsy onset. Sleep 2009, 32, 979–983. [Google Scholar] [CrossRef]
- Dale, R.C. Post-streptococcal autoimmune disorders of the central nervous system. Dev. Med. Child Neurol. 2005, 47, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, N.; Perrini, P.; Marani, W.; Chaurasia, B.; Corsalini, M.; Scarano, A.; Rapone, B. Multiple Brain Abscesses of Odontogenic Origin. May Oral Microbiota Affect Their Development? A Review of the Current Literature. Appl. Sci. 2021, 11, 3316. [Google Scholar] [CrossRef]
- Longstreth, W.T.; Ton, T.G.N.; Koepsell, T.D. Narcolepsy and streptococcal infections. Sleep 2009, 32, 1548. [Google Scholar] [CrossRef] [PubMed]
- Dauvilliers, Y.; Montplaisir, J.; Cochen, V.; Desautels, A.; Einen, M.; Lin, L.; Kawashima, M.; Bayard, S.; Monaca, C.; Tiberge, M.; et al. Post-H1N1 narcolepsy-cataplexy. Sleep 2010, 33, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Granath, F.; Gedeborg, R.; Smedje, H.; Feltelius, N. Change in risk for narcolepsy over time and impact of definition of onset date following vaccination with AS03 adjuvanted pandemic A/H1N1 influenza vaccine (Pandemrix) during the 2009 H1N1 influenza pandemic. Pharmacoepidemiol. Drug Saf. 2019, 28, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Viste, R.; Lie, B.A.; Viken, M.K.; Rootwelt, T.; Knudsen-Heier, S.; Kornum, B.R. Narcolepsy type 1 patients have lower levels of effector memory CD4+ T cells compared to their siblings when controlling for H1N1-(Pandemrix™)-vaccination and HLA DQB1∗06:02 status. Sleep Med. 2021, 85, 271–279. [Google Scholar] [CrossRef]
- Ton, T.G.N.; Longstreth, W.T.; Koepsell, T.D. Environmental toxins and risk of narcolepsy among people with HLA DQB1*0602. Environ. Res. 2010, 110, 565–570. [Google Scholar] [CrossRef][Green Version]
- Viola-Saltzman, M.; Watson, N.F. Traumatic brain injury and sleep disorders. Neurol. Clin. 2012, 30, 1299–1312. [Google Scholar] [CrossRef]
- van den Pol, A.N.; Gao, X.B.; Obrietan, K.; Kilduff, T.S.; Belousov, A.B. Presynaptic and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. J. Neurosci. 1998, 18, 7962–7971. [Google Scholar] [CrossRef]
- Peyron, C.; Faraco, J.; Rogers, W.; Ripley, B.; Overeem, S.; Charnay, Y.; Nevsimalova, S.; Aldrich, M.; Reynolds, D.; Albin, R.; et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med. 2000, 6, 991–997. [Google Scholar] [CrossRef]
- Thannickal, T.C.; Moore, R.Y.; Nienhuis, R.; Ramanathan, L.; Gulyani, S.; Aldrich, M.; Cornford, M.; Siegel, J.M. Reduced number of hypocretin neurons in human narcolepsy. Neuron 2000, 27, 469–474. [Google Scholar] [CrossRef]
- Xi, M.C.; Morales, F.R.; Chase, M.H. Effects on sleep and wakefulness of the injection of hypocretin-1 (orexin-A) into the laterodorsal tegmental nucleus of the cat. Brain Res. 2001, 901, 259–264. [Google Scholar] [CrossRef]
- Lecendreux, M.; Churlaud, G.; Pitoiset, F.; Regnault, A.; Tran, T.A.; Liblau, R.; Klatzmann, D.; Rosenzwajg, M. Narcolepsy Type 1 Is Associated with a Systemic Increase and Activation of Regulatory T Cells and with a Systemic Activation of Global T Cells. PLoS ONE 2017, 12, e0169836. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.M.; Fang, J.; Taishi, P.; Chen, Z.; Kushikata, T.; Gardi, J. Sleep—A physiologic role for IL-1β and TNF-αa. Ann. New York Acad. Sci. 1998, 856, 148–159. [Google Scholar] [CrossRef]
- Irwin, M.R.; Olmstead, R.; Carroll, J.E. Sleep disturbance, sleep duration, and inflammation: A systematic review and me-ta-analysis of cohort studies and experimental sleep deprivation. Biol. Psychiatry 2016, 80, 40–52. [Google Scholar] [CrossRef]
- Heiser, P.; Dickhaus, B.; Opper, C.; Hemmeter, U.; Remschmidt, H.; Wesemann, W.; Krieg, J.-C.; Schreiber, W. Alterations of host defence system after sleep deprivation are followed by impaired mood and psychosocial functioning. World J. Biol. Psychiatry 2001, 2, 89–94. [Google Scholar] [CrossRef]
- Redwine, L.; Hauger, R.L.; Gillin, J.C.; Irwin, M. Effects of sleep and sleep deprivation on interleukin-6, growth hormone, cortisol, and melatonin levels in humans. J. Clin. Endocrinol. Metab. 2000, 85, 3597–3603. [Google Scholar] [CrossRef]
- Vgontzas, A.N.; Chrousos, G.P. Sleep, the hypothalamic-pituitary-adrenal axis, and cytokines: Multiple interactions and disturbances in sleep disorders. Endocrinol. Metab. Clin. N. Am. 2002, 31, 15–36. [Google Scholar] [CrossRef]
- Vgontzas, A.N.; Papanicolaou, D.A.; Bixler, E.O.; Lotsikas, A.; Zachman, K.; Kales, A.; Prolo, P.; Wong, M.-L.; Licinio, J.; Gold, P.W.; et al. Circadian interleukin-6 secretion and quantity and depth of sleep. J. Clin. Endocrinol. Metab. 1999, 84, 2603–2607. [Google Scholar] [CrossRef]
- Mosakhani, N.; Sarhadi, V.; Panula, P.; Partinen, M.; Knuutila, S. Narcolepsy patients’ blood-based miRNA expression profiling: miRNA expression differences with Pandemrix vaccination. Acta Neurol. Scand. 2017, 136, 462–469. [Google Scholar] [CrossRef]
- Wieczorek, S.; Gencik, M.; Rujescu, D.; Tonn, P.; Giegling, I.; Epplen, J.T.; Dahmen, N. TNFA promoter polymorphisms and narcolepsy. Tissue Antigens 2003, 61, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Huang, Y.S.; Chen, C.H. Increased plasma level of tumor necrosis factor α in patients with narcolepsy in Taiwan. Sleep Med. 2013, 14, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Mayeli, M.; Saghazadeh, A.; Rezaei, N. Cytokines in narcolepsy: A systematic review and meta-analysis. Cytokine 2020, 131, 155103. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef]
- Garcia-Garcia, F.; Yoshida, H.; Krueger, J.M. Interleukin-8 promotes non-rapid eye movement sleep in rabbits and rats. J. Sleep Res. 2004, 13, 55–61. [Google Scholar] [CrossRef]
- Bonvalet, M.; Ollila, H.M.; Ambati, A.; Mignot, E. Autoimmunity in narcolepsy. Curr. Opin. Pulm. Med. 2017, 23, 522–529. [Google Scholar] [CrossRef]
- Mahlios, J.; de la Herrán-Arita, A.K.; Mignot, E. The autoimmune basis of narcolepsy. Curr. Opin. Neurobiol. 2013, 23, 767–773. [Google Scholar] [CrossRef]
- Thannickal, T.C.; Nienhuis, R.; Siegel, J.M. Localized loss of hypocretin (orexin) cells in narcolepsy without cataplexy. Sleep 2009, 32, 993–998. [Google Scholar] [CrossRef]
- Barateau, L.; Lopez, R.; Arnulf, I.; Lecendreux, M.; Franco, P.; Drouot, X.; Leu-Semenescu, S.; Jaussent, I.; Dauvilliers, Y. Comorbidity between central disorders of hy-persomnolence and immune-based disorders. Neurology 2017, 88, 93–100. [Google Scholar] [CrossRef]
- Fronczek, R.; Verschuuren, J.; Lammers, G.J. Response to intravenous immunoglobulins and placebo in a patient with narcolepsy with cataplexy. J. Neurol. 2007, 254, 1607–1608. [Google Scholar] [CrossRef] [PubMed]
- Plazzi, G.; Poli, F.; Franceschini, C.; Parmeggiani, A.; Pirazzoli, P.; Bernardi, F.; Mignot, E.; Cicognani, A.; Montagna, P. Intravenous high-dose immunoglobulin treatment in recent onset childhood narcolepsy with cataplexy. J. Neurol. 2008, 255, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Valko, P.O.; Khatami, R.; Baumann, C.R.; Bassetti, C.L. No persistent effect of intravenous immunoglobulins in patients with narcolepsy with cataplexy. J. Neurol. 2008, 255, 1900–1903. [Google Scholar] [CrossRef] [PubMed]
- Dauvilliers, Y.; Abril, B.; Mas, E.; Michel, F.; Tafti, M. Normalization of hypocretin-1 in narcolepsy after intravenous immunoglobulin treatment. Neurology 2009, 73, 1333–1334. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Faraco, J.; Lin, L.; Hesselson, S.; Winkelmann, J.; Kawashima, M.; Mayer, G.; Plazzi, G.; Nevsimalova, S.; Bourgin, P.; et al. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat. Genet. 2009, 41, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Katzav, A.; Arango, M.T.; Kivity, S.; Tanaka, S.; Givaty, G.; Agmon-Levin, N.; Hondae, M.; Anayac, J.-M.; Chapmanab, J.; Shoenfeld, Y. Passive transfer of narcolepsy: Anti-TRIB2 autoantibody positive patient IgG causes hypothalamic orexin neuron loss and sleep attacks in mice. J. Autoimmun. 2013, 45, 24–30. [Google Scholar] [CrossRef]
- Black, J.L., III; Silber, M.H.; Krahn, L.E.; Fredrickson, P.A.; Pankratz, V.S.; Avula, R.; Walker, D.L.; Slocumb, N.L. Analysis of hypocretin (orexin) antibodies in patients with narcolepsy. Sleep 2005, 28, 427–431. [Google Scholar] [CrossRef]
- Deloumeau, A.; Bayard, S.; Coquerel, Q.; Déchelotte, P.; Bole-Feysot, C.; Carlander, B.; De Cock, V.C.; Fetissov, S.O.; Dauvilliers, Y. Increased Immune Complexes of Hypocretin Autoantibodies in Narcolepsy. PLoS ONE 2010, 5, e13320. [Google Scholar] [CrossRef]
- Knudsen, S.; Mikkelsen, J.D.; Bang, B.; Gammeltoft, S.; Jennum, P.J. Intravenous immunoglobulin treatment and screening for hypocretin neuron-specific autoantibodies in recent onset childhood narcolepsy with cataplexy. Neuropediatrics 2010, 41, 217–222. [Google Scholar] [CrossRef]
- Cvetkovic-Lopes, V.; Bayer, L.; Dorsaz, S.; Maret, S.; Pradervand, S.; Dauvilliers, Y.; Lecendreux, M.; Lammers, G.-J.; Donjacour, C.E.H.M.; Pasquier, R.A.D.; et al. Elevated Tribbles homolog 2-specific antibody levels in narcolepsy patients. J. Clin. Investig. 2010, 120, 713–719. [Google Scholar] [CrossRef]
- Kawashima, M.; Lin, L.; Tanaka, S.; Jennum, P.; Knudsen, S.; Nevsimalova, S.; Plazzi, G.; Mignot, E. Anti-tribbles homolog 2 (TRIB2) autoan-tibodies in narcolepsy are associated with recent onset of cataplexy. Sleep 2010, 33, 869–874. [Google Scholar] [CrossRef]
- Toyoda, H.; Tanaka, S.; Miyagawa, T.; Honda, Y.; Tokunaga, K.; Honda, M. Anti-tribbles homolog 2 autoantibodies in Japanese patients with narcolepsy. Sleep 2010, 33, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Lind, A.; Ramelius, A.; Olsson, T.; Arnheim-Dahlström, L.; Lamb, F.; Khademi, M.; Ambati, A.; Maeurer, M.; Nilsson, A.L.; Bomfim, I.L.; et al. A/H1N1 antibodies and TRIB2 auto-antibodies in narcolepsy patients diagnosed in conjunction with the Pandemrix vaccination campaign in Sweden 2009-2010. J. Autoimmun. 2014, 50, 99–106. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Volkmuth, W.; Duca, J.; Corti, L.; Pallaoro, M.; Pezzicoli, A.; Karle, A.; Rigat, F.; Rappuoli, R.; Narasimhan, V.; et al. Antibodies to influenza nucleoprotein cross-react with human hypocretin receptor 2. Sci. Transl. Med. 2015, 7, 294ra105. [Google Scholar] [CrossRef] [PubMed]
- Saariaho, A.H.; Vuorela, A.; Freitag, T.L.; Pizza, F.; Plazzi, G.; Partinen, M.; Vaarala, O.; Meri, S. Autoantibodies against ganglioside GM3 are associated with narcolepsy-cataplexy developing after Pandemrix vaccination against 2009 pandemic H1N1 type influenza virus. J. Autoimmun. 2015, 63, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.; Adori, C.; Vas, S.; Kai-Larsen, Y.; Sarkanen, T.; Cederlund, A.; Agerberth, B.; Julkunen, I.; Horvath, B.; Kostyalik, D.; et al. Narcolepsy patients have antibodies that stain distinct cell populations in rat brain and influence sleep patterns. Proc. Natl. Acad. Sci. USA 2014, 111, E3735–E3744. [Google Scholar] [CrossRef] [PubMed]
- Zandian, A.; Forsström, B.; Häggmark-Månberg, A.; Schwenk, J.M.; Uhlén, M.; Nilsson, P.; Ayoglu, B. Whole-Proteome Peptide Microarrays for Profiling Autoantibody Repertoires within Multiple Sclerosis and Narcolepsy. J. Proteome Res. 2017, 16, 1300–1314. [Google Scholar] [CrossRef] [PubMed]
- Sadam, H.; Pihlak, A.; Kivil, A.; Pihelgas, S.; Jaago, M.; Adler, P.; Vilo, J.; Vapalahti, O.; Neuman, T.; Lindholm, D.; et al. Prostaglandin D2 Receptor DP1 Antibodies Predict Vaccine-induced and Spontaneous Narcolepsy Type 1: Large-scale Study of Antibody Profiling. eBioMedicine 2018, 29, 47–59. [Google Scholar] [CrossRef]
- Dauvilliers, Y.; Bauer, J.; Rigau, V.; Lalloyer, N.; Labauge, P.; Carlander, B.; Liblau, R.; Peyron, C.; Lassmann, H. Hypothalamic Immunopathology in Anti-Ma–Associated Diencephalitis With Narcolepsy-Cataplexy. JAMA Neurol. 2013, 70, 1305–1310. [Google Scholar] [CrossRef]
- Overeem, S.; Dalmau, J.; Bataller, L.; Nishino, S.; Mignot, E.; Verschuuren, J.; Lammers, G.J. Hypocretin-1 CSF levels in anti-Ma2 associated encephalitis. Neurology 2004, 62, 138–140. [Google Scholar] [CrossRef]
- Fontana, A.; Gast, H.; Reith, W.; Recher, M.; Birchler, T.; Bassetti, C.L. Narcolepsy: Autoimmunity, effector T cell activation due to infection, or T cell independent, major histocompatibility complex class II induced neuronal loss? Brain 2010, 133, 1300–1311. [Google Scholar] [CrossRef]
- Kornum, B.R.; Kawashima, M.; Faraco, J.; Lin, L.; Rico, T.J.; Hesselson, S.; Axtell, R.C.; Kuipers, H.; Weiner, K.; Hamacher, A.; et al. Common variants in P2RY11 are associated with narcolepsy. Nat. Genet. 2011, 43, 66–71. [Google Scholar] [CrossRef] [PubMed]
- De La Herrán-Arita, A.K.; Garcia-Garcia, F. Narcolepsy as an Immune-Mediated Disease. Sleep Disord. 2014, 2014, 792687. [Google Scholar] [CrossRef] [PubMed]
- Ramberger, M.; Högl, B.; Stefani, A.; Mitterling, T.; Reindl, M.; Lutterotti, A. CD4+ T-Cell Reactivity to Orexin/Hypocretin in Patients With Narcolepsy Type 1. Sleep 2017, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, C.L.A.; Adamantidis, A.; Burdakov, D.; Han, F.; Gay, S.; Kallweit, U.; Khatami, R.; Koning, F.; Kornum, B.R.; Lammers, G.J.; et al. Narcolepsy—Clinical spectrum, aetiopathophysiology, diagnosis and treatment. Nat. Rev. Neurol. 2019, 15, 519–539. [Google Scholar] [CrossRef] [PubMed]
- Liblau, R.; Vassalli, A.; Seifinejad, A.; Tafti, M. Hypocretin (orexin) biology and the pathophysiology of narcolepsy with cataplexy. Lancet Neurol. 2015, 14, 318–328. [Google Scholar] [CrossRef]
- España, R.A.; Scammell, T.E. Sleep Neurobiology from a Clinical Perspective. Sleep 2011, 34, 845–858. [Google Scholar] [CrossRef]
- Scammell, T.E.; Jackson, A.C.; Franks, N.; Wisden, W.; Dauvilliers, Y. Histamine: Neural circuits and new medications. Sleep 2019, 1, 42. [Google Scholar] [CrossRef]
- Schwartz, M.D.; Kilduff, T.S. The Neurobiology of Sleep and Wakefulness. Psychiatr. Clin. N. Am. 2015, 38, 615–644. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, S.; Chao, H.H.; Li, C.-S.R. Resting-State Functional Connectivity of the Locus Coeruleus in Humans: In Comparison with the Ventral Tegmental Area/Substantia Nigra Pars Compacta and the Effects of Age. Cereb. Cortex 2016, 26, 3413–3427. [Google Scholar] [CrossRef]
- Drissi, N.M.; Szakács, A.; Witt, S.T.; Wretman, A.; Ulander, M.; Ståhlbrandt, H.; Darin, N.; Hallböök, T.; Landtblom, A.-M.; Engström, M. Altered Brain Microstate Dynamics in Adolescents with Narcolepsy. Front. Hum. Neurosci. 2016, 10, 369. [Google Scholar] [CrossRef]
- Tondelli, M.; Pizza, F.; Vaudano, A.E.; Plazzi, G.; Meletti, S. Cortical and Subcortical Brain Changes in Children and Adolescents With Narcolepsy Type 1. Sleep 2018, 1, 41. [Google Scholar] [CrossRef]
- Bican, A.; Bora, I.; Algn, O.; Hakyemez, B.; Özkol, V.; Alper, E. Neuroimaging in Narcolepsy. In Novel Frontiers of Advanced Neu-Roimaging; InTech: Rijeka, Croatia, 2013. [Google Scholar] [CrossRef]
- Joo, E.Y. Updates on Structural Neuroimaging of Narcolepsy with Cataplexy. Precis. Futur. Med. 2019, 3, 1–9. [Google Scholar] [CrossRef]
- Scherfler, C.; Frauscher, B.; Schocke, M.; Nocker, M.; Gschliesser, V.; Ehrmann, L.; Niederreiter, M.; Esterhammer, R.; Seppi, K.; Brandauer, E.; et al. White and Gray Matter Abnormalities in Narcolepsy with Cataplexy. Sleep 2012, 35, 345–351. [Google Scholar] [CrossRef]
- Siegel, J.M. Hypocretin (OREXIN): Role in Normal Behavior and Neuropathology. Annu. Rev. Psychol. 2004, 55, 125–148. [Google Scholar] [CrossRef]
- Nittur, N.; Konofal, E.; Dauvilliers, Y.; Franco, P.; Leu-Semenescu, S.; De Cock, V.C.; Inocente, C.O.; Bayard, S.; Scholtz, S.; Lecendreux, M.; et al. Mazindol in narcolepsy and idiopathic and symptomatic hypersomnia refractory to stimulants: A long-term chart review. Sleep Med. 2013, 14, 30–36. [Google Scholar] [CrossRef]
- Dye, T.J.; Gurbani, N.; Simakajornboon, N. Epidemiology and Pathophysiology of Childhood Narcolepsy. Paediatr. Respir. Rev. 2018, 25, 14–18. [Google Scholar] [CrossRef]
- Cho, J.R.; Treweek, J.B.; Robinson, J.E.; Xiao, C.; Bremner, L.R.; Greenbaum, A.; Gradinaru, V. Dorsal Raphe Dopamine Neurons Modulate Arousal and Promote Wakefulness by Salient Stimuli. Neuron 2017, 94, 1205–1219.e8. [Google Scholar] [CrossRef]
- Dyavanapalli, J.; Byrne, P.; Mendelowitz, D. Activation of D2-like dopamine receptors inhibits GABA and glycinergic neurotransmission to pre-motor cardiac vagal neurons in the nucleus ambiguus. Neuroscience 2013, 247, 213–226. [Google Scholar] [CrossRef]
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the Nervous System. Physiol. Rev. 2008, 88, 1183–1241. [Google Scholar] [CrossRef]
- Scammell, T.E.; Arrigoni, E.; Lipton, J.O. Neural Circuitry of Wakefulness and Sleep. Neuron 2017, 93, 747–765. [Google Scholar] [CrossRef]
- John, J.; Thannickal, T.C.; McGregor, R.; Ramanathan, L.; Ohtsu, H.; Nishino, S.; Sakai, N.; Yamanaka, A.; Stone, C.; Cornford, M.; et al. Greatly increased numbers of histamine cells in human narcolepsy with cataplexy. Ann. Neurol. 2013, 74, 786–793. [Google Scholar] [CrossRef]
- Valko, P.O.; Gavrilov, Y.V.; Yamamoto, M.; Reddy, H.; Haybaeck, J.; Mignot, E.; Baumann, C.R.; Scammell, T.E. Increase of histaminergic tuberomammillary neurons in narcolepsy. Ann. Neurol. 2013, 74, 794–804. [Google Scholar] [CrossRef]
- Martinez-Orozco, F.J.; Vicario, J.L.; De Andres, C.; Fernandez-Arquero, M.; Peraita-Adrados, R. Comorbidity of Narcolepsy Type 1 With Autoimmune Diseases and Other Immunopathological Disorders: A Case-Control Study. J. Clin. Med. Res. 2016, 8, 495–505. [Google Scholar] [CrossRef]
- Cavaliere, C.; Longarzo, M.; Fogel, S.; Engström, M.; Soddu, A. Neuroimaging of Narcolepsy and Primary Hypersomnias. Neuroscientist 2020, 26, 310–327. [Google Scholar] [CrossRef]
- Dauvilliers, Y.; Billiard, M.; Montplaisir, J. Clinical aspects and pathophysiology of narcolepsy. Clin. Neurophysiol. 2003, 114, 2000–2017. [Google Scholar] [CrossRef]
- Cavallero, C.; Cicogna, P.C.; Natale, V.; Occhionero, M.; Zito, A. Slow Wave Sleep Dreaming. Sleep 1992, 15, 562–566. [Google Scholar] [CrossRef]
- Katsuki, H.; Akaike, A. Excitotoxic degeneration of hypothalamic orexin neurons in slice culture. Neurobiol. Dis. 2004, 15, 61–69. [Google Scholar] [CrossRef]
- Terao, A.; Peyron, C.; Ding, J.; Wurts, S.W.; Edgar, D.M.; Heller, H.C.; Kilduff, T.S. Prepro-hypocretin (Prepro-Orexin) Expression is Unaffected by Short-Term Sleep Deprivation in Rats and Mice. Sleep 2000, 23, 867–874. [Google Scholar] [CrossRef]
- Roberts, H.J. The syndrome of narcolepsy and diabetogenic hyperinsulinism in the american negro: Its relationship to the pathogenesis of diabetes mellitus, obesity, dysrhythmias, and accelerated cardiovascular disease. J. Natl. Med. Assoc. 1964, 56, 18–42. [Google Scholar] [PubMed]
- Sarkanen, T.; Huutoniemi, A.; Partinen, M. Narcolepsy with coexisting type 1 diabetes: A case report. Sleep Med. 2013, 14, e258. [Google Scholar] [CrossRef]
- Mohammadi, S.; Dolatshahi, M.; Zare-Shahabadi, A.; Rahmani, F. Untangling narcolepsy and diabetes: Pathomechanisms with eyes on therapeutic options. Brain Res. 2019, 1718, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Roizenblatt, S.; Neto, N.S.R.; Tufik, S. Sleep Disorders and Fibromyalgia. Curr. Pain Headache Rep. 2011, 15, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Scarola, R.; Montemurro, N.; Ferrara, E.; Corsalini, M.; Converti, I.; Rapone, B. Temporomandibular Disorders and Fibromyalgia: A Narrative Review. Open Access Maced. J. Med. Sci. 2021, 9, 106–112. [Google Scholar] [CrossRef]
- Caillat-Zucman, S. Molecular mechanisms of HLA association with autoimmune diseases. Tissue Antigens 2009, 73, 1–8. [Google Scholar] [CrossRef]
- Lernmark, Å. Environmental factors in the etiology of type 1 diabetes, celiac disease, and narcolepsy. Pediatr. Diabetes 2016, 17, 65–72. [Google Scholar] [CrossRef]
- Adeghate, E.; Fernandez-Cabezudo, M.; Hameed, R.; El-Hasasna, H.; El Wasila, M.; Abbas, T.; Al-Ramadi, B. Orexin-1 Receptor Co-Localizes with Pancreatic Hormones in Islet Cells and Modulates the Outcome of Streptozotocin-Induced Diabetes Mellitus. PLoS ONE 2010, 5, e8587. [Google Scholar] [CrossRef]
- Kukkonen, J.P. Orexin/Hypocretin Signaling. In Current Topics in Behavioral Neurosciences; Springer: Bern, Switzerland, 2016; Volume 33, pp. 17–50. [Google Scholar] [CrossRef]
- Adeghate, E.; Hameed, R. Mechanism of Orexin B-Stimulated Insulin and Glucagon Release From the Pancreas of Normal and Diabetic Rats. Pancreas 2011, 40, 131–136. [Google Scholar] [CrossRef]
- Cai, X.J.; Evans, M.L.; Lister, C.A.; Leslie, R.A.; Arch, J.R.S.; Wilson, S.; Williams, G. Hypoglycemia Activates Orexin Neurons and Selectively Increases Hypothalamic Orexin-B Levels: Responses inhibited by feeding and possibly mediated by the nucleus of the solitary tract. Diabetes 2001, 50, 105–112. [Google Scholar] [CrossRef]
- Skrzypski, M.; Billert, M.; Nowak, K.W.; Strowski, M.Z. The role of orexin in controlling the activity of the adipo-pancreatic axis. J. Endocrinol. 2018, 238, R95–R108. [Google Scholar] [CrossRef]
- Goforth, P.B.; Myers, M.G. Roles for Orexin/Hypocretin in the Control of Energy Balance and Metabolism. Curr. Top. Behav. Neurosci. 2017, 33, 137–156. [Google Scholar] [CrossRef]
- Rani, M.; Kumar, R.; Krishan, P. Role of orexins in the central and peripheral regulation of glucose homeostasis: Evidences & mechanisms. Neuropeptides 2018, 68, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Suo, L.; Chang, X.; Zhao, Y. The Orexin-A-Regulated Akt/mTOR Pathway Promotes Cell Proliferation Through Inhibiting Apoptosis in Pancreatic Cancer Cells. Front. Endocrinol. 2018, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Liu, Y.; Zhao, Y.; Sun, X.; Fan, D.; Guo, L. Orexin A affects HepG2 human hepatocellular carcinoma cells glucose metabolism via HIF-1α-dependent and -independent mechanism. PLoS ONE 2017, 12, e0184213. [Google Scholar] [CrossRef]
- Xu, W.-W.; Zhang, Y.-Y.; Su, J.; Liu, A.-F.; Wang, K.; Li, C.; Liu, Y.-E.; Zhang, Y.-Q.; Lv, J.; Jiang, W.-J. Ischemia Reperfusion Injury after Gradual versus Rapid Flow Restoration for Middle Cerebral Artery Occlusion Rats. Sci. Rep. 2018, 8, 1638. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, Y.; Guo, L. Orexin A induces autophagy in HCT-116 human colon cancer cells through the ERK signaling pathway. Int. J. Mol. Med. 2016, 37, 126–132. [Google Scholar] [CrossRef]
- Xu, T.-R.; Yang, Y.; Ward, R.; Gao, L.; Liu, Y. Orexin receptors: Multi-functional therapeutic targets for sleeping disorders, eating disorders, drug addiction, cancers and other physiological disorders. Cell. Signal. 2013, 25, 2413–2423. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.; Chaurasia, B.; Fiorindi, A.; Umana, G.E.; Lu, B.; Montemurro, N. Ischemic Stroke and SARS-CoV-2 Infection: The Bidirectional Pathology and Risk Morbidities. Neurol. Int. 2022, 14, 391–405. [Google Scholar] [CrossRef]
- Kang, S.K.; Lee, D.H.; Bae, Y.C.; Kim, H.K.; Baik, S.Y.; Jung, J.S. Improvement of neurological deficits by intracerebral transplantation of human adipose tissue-derived stromal cells after cerebral ischemia in rats. Exp. Neurol. 2003, 183, 355–366. [Google Scholar] [CrossRef]
- De Luca, P.; Camaioni, A.; Marra, P.; Salzano, G.; Carriere, G.; Ricciardi, L.; Pucci, R.; Montemurro, N.; Brenner, M.J.; Di Stadio, A. Effect of Ultra-Micronized Palmitoylethanolamide and Luteolin on Olfaction and Memory in Patients with Long COVID: Results of a Longitudinal Study. Cells 2022, 11, 2552. [Google Scholar] [CrossRef]
- Lizana, J.; Aliaga, N.; Marani, W.; Escribano, A.; Montemurro, N. Spinal Vascular Shunts: Single-Center Series and Review of the Literature of Their Classification. Neurol. Int. 2022, 14, 581–599. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Montemurro, N.; Murrone, D.; Romanelli, B.; Ierardi, A. Postoperative Textiloma Mimicking Intracranial Rebleeding in a Patient with Spontaneous Hemorrhage: Case Report and Review of the Literature. Case Rep. Neurol. 2020, 12, 7–12. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Maida, C.; Maugeri, R.; Iacopino, D.; Pinto, A. Relationship between Diabetes and Ischemic Stroke: Analysis of Diabetes- Related Risk Factors for Stroke and of Specific Patterns of Stroke Associated with Diabetes Mellitus. J. Diabetes Metab. 2015, 6, 544–551. [Google Scholar] [CrossRef]
- Filchenko, I.A.; Sviryaev, Y.V.; Vlasov, T.D. Neuroprotective activity of orexin system in ischemic stroke. Reg. Blood Circ. Microcirc. 2018, 17, 4–11. [Google Scholar] [CrossRef]
- Lizana, J.; Reinoso, C.M.D.; Aliaga, N.; Marani, W.; Montemurro, N. Bilateral central retinal artery occlusion: An exceptional complication after frontal parasagittal meningioma resection. Surg. Neurol. Int. 2021, 12, 397. [Google Scholar] [CrossRef] [PubMed]
- Nishino, S.; Kanbayashi, T. Symptomatic narcolepsy, cataplexy and hypersomnia, and their implications in the hypothalamic hypocretin/orexin system. Sleep Med. Rev. 2005, 9, 269–310. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, N.; Perrini, P.; Lawton, M.T. Unsuccessful bypass and trapping of a giant dolichoectatic thrombotic basilar trunk aneurysm. What went wrong? Br. J. Neurosurg. 2022, 17, 1–4. [Google Scholar] [CrossRef]
- Lizana, J.; Montemurro, N.; Aliaga, N.; Marani, W.; Tanikawa, R. From textbook to patient: A practical guide to train the end-, to-side microvascular anastomosis. Br. J. Neurosurg. 2021, 7, 1–5. [Google Scholar] [CrossRef]
- Dohi, K.; Ripley, B.; Fujiki, N.; Ohtaki, H.; Shioda, S.; Aruga, T.; Nishino, S. CSF hypocretin-1/orexin-A concentrations in patients with subarachnoid hemorrhage (SAH). Peptides 2005, 26, 2339–2343. [Google Scholar] [CrossRef]
- Nakamachi, T.; Endo, S.; Ohtaki, H.; Yin, L.; Kenji, D.; Kudo, Y.; Funahashi, H.; Matsuda, K.; Shioda, S. Orexin-1 receptor expression after global ischemia in mice. Regul. Pept. 2005, 126, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.; Fujita-Hamabe, W.; Tokuyama, S. Effect of Orexin-A on Post-ischemic Glucose Intolerance and Neuronal Damage. J. Pharmacol. Sci. 2011, 115, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.-B.; Dong, H.-L.; Zhang, H.-P.; Zhao, R.-N.; Gong, G.; Chen, X.-M.; Zhang, L.-N.; Xiong, L. Neuroprotective Effect of Orexin-A Is Mediated by an Increase of Hypoxia-inducible Factor-1 Activity in Rat. Anesthesiology 2011, 114, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Sokołowska, P.; Urbańska, A.; Namiecińska, M.; Biegańska, K.; Zawilska, J.B. Orexins promote survival of rat cortical neurons. Neurosci. Lett. 2012, 506, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; White, R.E.; Xu, L.; Yang, L.; Sun, X.; Zou, B.; Pascual, C.; Sakurai, T.; Giffard, R.G.; Xie, X. Mitigation of Murine Focal Cerebral Ischemia by the Hypocretin/Orexin System is Associated With Reduced Inflammation. Stroke 2013, 44, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Sohail, S.; Yu, L.; Schneider, J.A.; Bennett, D.A.; Buchman, A.S.; Lim, A.S.P. Sleep fragmentation and Parkinson’s disease pathology in older adults without Parkinson’s disease. Mov. Disord. 2017, 32, 1729–1737. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Q.; Ji, B.; Pan, Y.; Xu, C.; Cheng, B.; Bai, B.; Chen, J. The Orexin/Receptor System: Molecular Mechanism and Therapeutic Potential for Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 220. [Google Scholar] [CrossRef]
- Fronczek, R.; van Geest, S.; Frölich, M.; Overeem, S.; Roelandse, F.W.; Lammers, G.J.; Swaab, D. Hypocretin (orexin) loss in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1642–1650. [Google Scholar] [CrossRef]
- Gabelle, A.; Jaussent, I.; Hirtz, C.; Vialaret, J.; Navucet, S.; Grasselli, C.; Robert, P.; Lehmann, S.; Dauvilliers, Y. Cerebrospinal fluid levels of orexin-A and histamine, and sleep profile within the Alzheimer process. Neurobiol. Aging 2017, 53, 59–66. [Google Scholar] [CrossRef]
- Liguori, C.; Romigi, A.; Nuccetelli, M.; Zannino, S.; Sancesario, G.; Martorana, A.; Albanese, M.; Mercuri, N.B.; Izzi, F.; Bernardini, S.; et al. Orexinergic System Dysregulation, Sleep Impairment, and Cognitive Decline in Alzheimer Disease. JAMA Neurol. 2014, 71, 1498–1505. [Google Scholar] [CrossRef]
- Urrestarazu, E.; Iriarte, J. Clinical management of sleep disturbances in Alzheimer’s disease: Current and emerging strategies. Nat. Sci. Sleep 2016, 8, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.H.; Jiang, H.; Finn, M.B.; Stewart, F.R.; Mahan, T.; Cirrito, J.R.; Heda, A.; Snider, B.J.; Li, M.; Yanagisawa, M.; et al. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J. Exp. Med. 2014, 211, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-β Dynamics Are Regulated by Orexin and the Sleep-Wake Cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Kallweit, U.; Hidalgo, H.; Engel, A.; Baumann, C.R.; Bassetti, C.L.; Dahmen, N. Post H1N1 vaccination narcolepsy–cataplexy with decreased CSF beta-amyloid. Sleep Med. 2012, 13, 323. [Google Scholar] [CrossRef][Green Version]
- Scammell, T.; Matheson, J.; Honda, M.; Thannickal, T.; Siegel, J. Coexistence of narcolepsy and Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1318–1319. [Google Scholar] [CrossRef]
- Berteotti, C.; Liguori, C.; Pace, M. Dysregulation of the orexin/hypocretin system is not limited to narcolepsy but has far-reaching implications for neurological disorders. Eur. J. Neurosci. 2021, 53, 1136–1154. [Google Scholar] [CrossRef]
- Chunduri, A.; Crusio, W.E.; Delprato, A. Narcolepsy in Parkinson’s disease with insulin resistance. F1000Research 2020, 9, 1361. [Google Scholar] [CrossRef]
- Haq, I.Z.; Naidu, Y.; Reddy, P.; Chaudhuri, K.R. Narcolepsy in Parkinson’s disease. Expert Rev. Neurother. 2010, 10, 879–884. [Google Scholar] [CrossRef]
- Montemurro, N.; Benet, A.; Lawton, M.T. Julius Caesar’s Epilepsy: Was It Caused by A Brain Arteriovenous Malformation? World Neurosurg. 2015, 84, 1985–1987. [Google Scholar] [CrossRef]
- Arnulf, I.; Leu, S.; Oudiette, D. Abnormal sleep and sleepiness in Parkinson’s disease. Curr. Opin. Neurol. 2008, 21, 472–477. [Google Scholar] [CrossRef]
- Montemurro, N.; Herbet, G.; Duffau, H. Right Cortical and Axonal Structures Eliciting Ocular Deviation During Electrical Stimulation Mapping in Awake Patients. Brain Topogr. 2016, 29, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Rapone, B.; Ferrara, E.; Montemurro, N.; Converti, I.; Loverro, M.; Loverro, M.T.; Gnoni, A.; Scacco, S.; Siculella, L.; Corsalini, M.; et al. Oral Microbiome and Preterm Birth: Correlation or Coincidence? A Narrative Review. Open Access Maced. J. Med. Sci. 2020, 8, 123–132. [Google Scholar] [CrossRef]
- Ylikoski, A.; Martikainen, K.; Sarkanen, T.; Partinen, M. Parkinson’s disease and narcolepsy-like symptoms. Sleep Med. 2015, 16, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Morgenthaler, T.I.; Kapur, V.K.; Brown, T.M.; Swick, T.J.; Alessi, C.; Aurora, R.N.; Boehlecke, B.; Chesson, A.L.; Friedman, L.; Maganti, R.; et al. Practice Parameters for the Treatment of Narcolepsy and other Hypersomnias of Central Origin: An American Academy of Sleep Medicine report. Sleep 2007, 30, 1705–1711. [Google Scholar] [CrossRef]
- Carskadon, M.A.; Dement, W.C.; Mitler, M.M.; Roth, T.; Westbrook, P.R.; Keenan, S. Guidelines for the Multiple Sleep Latency Test (MSLT): A Standard Measure of Sleepiness. Sleep 1987, 9, 519–524. [Google Scholar] [CrossRef]
- Montemurro, N.; Aliaga, N.; Graff, P.; Escribano, A.; Lizana, J. New Targets and New Technologies in the Treatment of Parkinson’s Disease: A Narrative Review. Int. J. Environ. Res. Public Health 2022, 19, 8799. [Google Scholar] [CrossRef]
- Omokawa, M.; Ayabe, T.; Nagai, T.; Imanishi, A.; Omokawa, A.; Nishino, S.; Sagawa, Y.; Shimizu, T.; Kanbayashi, T. Decline of CSF orexin (hypocretin) levels in Prader-Willi syndrome. Am. J. Med. Genet. Part A 2016, 170, 1181–1186. [Google Scholar] [CrossRef]
- Kato, T.; Kanbayashi, T.; Yamamoto, K.; Nakano, T.; SfflMIZU, T.; Hashimoto, T.; Ikeda, S.-I. Hypersomnia and Low CSF Hypocretin-1 (Orexin-A) Concentration in a Patient with Multiple Sclerosis Showing Bilateral Hypothalamic Lesions. Intern. Med. 2003, 42, 743–745. [Google Scholar] [CrossRef][Green Version]
- Han, F.; Faraco, J.; Dong, X.S.; Ollila, H.; Lin, L.; Li, J.; An, P.; Wang, S.; Jiang, K.W.; Gao, Z.C.; et al. Genome Wide Analysis of Narcolepsy in China Implicates Novel Immune Loci and Reveals Changes in Association Prior to Versus After the 2009 H1N1 Influenza Pandemic. PLoS Genet. 2013, 9, e1003880. [Google Scholar] [CrossRef]
- Krahn, L.E.; Pankratz, V.S.; Oliver, L.; Boeve, B.F.; Silber, M.H. Hypocretin (Orexin) Levels in Cerebrospinal Fluid of Patients with Narcolepsy: Relationship to Cataplexy and HLA DQB1*0602 Status. Sleep 2002, 25, 733–736. [Google Scholar] [CrossRef][Green Version]
- Filardi, M.; Pizza, F.; Martoni, M.; Vandi, S.; Plazzi, G.; Natale, V. Actigraphic assessment of sleep/wake behavior in central disorders of hypersomnolence. Sleep Med. 2015, 16, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Thorpy, M.J.; Shapiro, C.; Mayer, G.; Corser, B.C.; Emsellem, H.; Plazzi, G.; Chen, D.; Carter, L.P.; Wang, H.; Lu, Y.; et al. A randomized study of solriamfetol for excessive sleepiness in narcolepsy. Ann. Neurol. 2019, 85, 359–370. [Google Scholar] [CrossRef]
- Siegel, J.M.; Moore, R.; Thannickal, T.; Nienhuis, R. A Brief History of Hypocretin/Orexin and Narcolepsy. Neuropsychopharmacology 2001, 25, S14–S20. [Google Scholar] [CrossRef]
- Mieda, M.; Sakurai, T. Orexin (Hypocretin) Receptor Agonists and Antagonists for Treatment of Sleep Disorders: Rationale for development and current status. CNS Drugs 2013, 27, 83–90. [Google Scholar] [CrossRef]
- Forster, A.; Smith, J.; Young, J.; Knapp, P.; House, A.; Wright, J. Information provision for stroke patients and their caregivers. Cochrane Database Syst Rev. 2021, 11, CD001919. [Google Scholar] [CrossRef]
- Bagai, K.; Malow, B.A. A Novel Approach to Treating Morning Sleep Inertia in Narcolepsy. J. Clin. Sleep Med. 2010, 6, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Zaharna, M.; Dimitriu, A.; Guilleminault, C. Expert opinion on pharmacotherapy of narcolepsy. Expert Opin. Pharmacother. 2010, 11, 1633–1645. [Google Scholar] [CrossRef]
- Parmentier, R.; Kolbaev, S.; Klyuch, B.P.; Vandael, D.; Lin, J.-S.; Selbach, O.; Haas, H.L.; Sergeeva, O.A. Excitation of Histaminergic Tuberomamillary Neurons by Thyrotropin-Releasing Hormone. J. Neurosci. 2009, 29, 4471–4483. [Google Scholar] [CrossRef]
- Pizza, F.; Barateau, L.; Dauvilliers, Y.; Plazzi, G. The orexin story, sleep and sleep disturbances. J. Sleep Res. 2022, 31, e13665. [Google Scholar] [CrossRef]
- Barateau, L.; Pizza, F.; Plazzi, G.; Dauvilliers, Y. Narcolepsy. J. Sleep Res. 2022, 31, e13631. [Google Scholar] [CrossRef]
- Veneruso, M.; Pizza, F.; Finotti, E.; Amore, G.; Vandi, S.; Filardi, M.; Antelmi, E.; Nobili, L.; Cassio, A.; Pession, A.; et al. Child Neurology: A Case Series of Heterogeneous Neuropsychiatric Symptoms and Outcome in Very Early-Onset Narcolepsy Type 1. Neurology 2022, 98, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Caporali, L.; Moresco, M.; Pizza, F.; La Morgia, C.; Fiorini, C.; Strobbe, D.; Zenesini, C.; Kashani, B.H.; Torroni, A.; Pagotto, U.; et al. The role of mtDNA haplogroups on metabolic features in narcolepsy type 1. Mitochondrion 2022, 63, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Bhatia, M. iPSC Technology: Platform for Drug Discovery. Clin. Pharmacol. Ther. 2011, 89, 639–641. [Google Scholar] [CrossRef]
- Golicki, D.; Bala, M.M.; Niewada, M.; Wierzbicka, A. Modafinil for narcolepsy: Systematic review and meta-analysis. Med. Sci. Monit. 2020, 16, 177–186. [Google Scholar]
- Rosenberg, R.; Bogan, R. Armodafinil in the treatment of excessive sleepiness. Nat. Sci. Sleep 2010, 2, 95–105. [Google Scholar] [CrossRef]
- Sheikhina, N.; Najafi, M.R.; Chitsaz, A.; Ghadimi, K. Evaluation of the effectiveness of methylphenidate and modafinil in the treatment of daily drowsiness in patients with refractory epilepsy and their comparison with the control group. Am. J. Neurodegener. Dis. 2021, 10, 69–75. [Google Scholar]
- Abad, V.C.; Guilleminault, C. New developments in the management of narcolepsy. Nat. Sci. Sleep 2017, 9, 39–57. [Google Scholar] [CrossRef]
- Iturburu, A.; Vela, E.P.; Cruz, C.; Yepez, M.; Ortiz, J.F.; Krishna, K.; Peña, G.; Cordova, S.; Khurana, M.; Bandarupalli, P. Solriamfetol for the Use of Narcolepsy: A Systematic Review. Cureus 2022, 14, e24937. [Google Scholar] [CrossRef]
- AlShaikh, M.K.; Tricco, A.C.; Tashkandi, M.; Mamdani, M.; Straus, S.E.; Bahammam, A.S. Sodium Oxybate for Narcolepsy with Cataplexy: Systematic Review and Meta-Analysis. J. Clin. Sleep Med. 2012, 8, 451–458. [Google Scholar] [CrossRef]
- Elliott, L.; Swick, T. Treatment paradigms for cataplexy in narcolepsy: Past, present, and future. Nat. Sci. Sleep 2015, 7, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Mignot, E.J.M. A Practical Guide to the Therapy of Narcolepsy and Hypersomnia Syndromes. Neurotherapeutics 2012, 9, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Findling, R.L.; Candler, S.A.; Nasser, A.F.; Schwabe, S.; Yu, C.; Garcia-Olivares, J.; O’Neal, W.; Newcorn, J.H. Viloxazine in the Management of CNS Disorders: A Historical Overview and Current Status. CNS Drugs 2021, 35, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Larrosa, O.; De La Llave, Y.; Barrio, R.S.; Granizo, J.J.; Garcia-Borreguero, D. Stimulant and Anticataplectic Effects of Reboxetine in Patients with Narcolepsy: A Pilot Study. Sleep 2001, 24, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, H.D.A.; Lopes, D.A.; Pereira, D.; Sguillar, D.; Lopes, E.; Behrens, N.S.C.D.S.; Lima, T.F.D.A.; Pradella-Hallinan, M.; Castro, J.; Tufik, S.; et al. The use of citalopram for the treatment of cataplexy. Sleep Sci. 2014, 7, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Kansagra, S.; Walter, R.; Vaughn, B. Nocturnal Temazepam in the Treatment of Narcolepsy. J. Clin. Sleep Med. 2013, 9, 499–500. [Google Scholar] [CrossRef]
- Bhattarai, J.; Sumerall, S. Current and Future Treatment Options for Narcolepsy: A Review. Sleep Sci. 2017, 10, 19–27. [Google Scholar] [CrossRef]
- Fabara, S.P.; Ortiz, J.F.; Sohail, A.A.; Hidalgo, J.; Altamimi, A.; Tama, B.; Patel, U.K. Efficacy of Pitolisant on the Treatment of Narcolepsy: A Systematic Review. Cureus 2021, 13, e16095. [Google Scholar] [CrossRef]
- Black, S.W.; Schwartz, M.D.; Chen, T.-M.; Hoener, M.; Kilduff, T.S. Trace Amine-Associated Receptor 1 Agonists as Narcolepsy Therapeutics. Biol. Psychiatry 2017, 82, 623–633. [Google Scholar] [CrossRef]
- Papaconstantinou, E.; Cancelliere, C.; Verville, L.; Wong, J.J.; Connell, G.; Yu, H.; Shearer, H.; Timperley, C.; Chung, C.; Porter, B.J.; et al. Effectiveness of non-pharmacological interventions on sleep characteristics among adults with musculoskeletal pain and a comorbid sleep problem: A systematic review. Chiropr. Man. Ther. 2021, 29, 23. [Google Scholar] [CrossRef]
- Luo, G.; Ambati, A.; Lin, L.; Bonvalet, M.; Partinen, M.; Ji, X.; Maecker, H.T.; Mignot, E.J.-M. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc. Natl. Acad. Sci. USA 2018, 115, E12323–E12332. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chavda, V.; Chaurasia, B.; Umana, G.E.; Tomasi, S.O.; Lu, B.; Montemurro, N. Narcolepsy—A Neuropathological Obscure Sleep Disorder: A Narrative Review of Current Literature. Brain Sci. 2022, 12, 1473. https://doi.org/10.3390/brainsci12111473
Chavda V, Chaurasia B, Umana GE, Tomasi SO, Lu B, Montemurro N. Narcolepsy—A Neuropathological Obscure Sleep Disorder: A Narrative Review of Current Literature. Brain Sciences. 2022; 12(11):1473. https://doi.org/10.3390/brainsci12111473
Chicago/Turabian StyleChavda, Vishal, Bipin Chaurasia, Giuseppe E. Umana, Santino Ottavio Tomasi, Bingwei Lu, and Nicola Montemurro. 2022. "Narcolepsy—A Neuropathological Obscure Sleep Disorder: A Narrative Review of Current Literature" Brain Sciences 12, no. 11: 1473. https://doi.org/10.3390/brainsci12111473
APA StyleChavda, V., Chaurasia, B., Umana, G. E., Tomasi, S. O., Lu, B., & Montemurro, N. (2022). Narcolepsy—A Neuropathological Obscure Sleep Disorder: A Narrative Review of Current Literature. Brain Sciences, 12(11), 1473. https://doi.org/10.3390/brainsci12111473